

Abstract
Camptothecin (CPT) exhibits very strong antitumor effects by inhibiting the activity of DNA topoisomerase I, but its application is greatly limited due to its low solubility and the instability of the active lactone form. To overcome these shortcomings, in the present study, we prepared novel camptothecin nanocolloids based on N,N,N-trimethyl chitosan (CPT-TMC) to efficiently and safely administer CPT systemically. Herein, we investigated the antitumor activity of CPT-TMC against a murine Balb/c myeloma model. Our results showed that CPT-TMC more effectively inhibited tumor growth and prolonged survival time than CPT in vivo, but no statistical difference was observed in vitro between CPT-TMC and CPT. These findings suggest that N,N,N-trimethyl chitosan could increase the stability and the antitumor effect of CPT and CPT-TMC is a potential approach for the effective treatment of multiple myeloma.
Keywords: camptothecin; N,N,N-trimethyl chitosan; myeloma; antitumor
Introduction
Multiple myeloma (MM), a malignant plasma cell disorder, accounts for about 10% of all hematological cancer cases (1,2). The annual incidence of MM varies between 1 to 5 cases per 100,000 persons worldwide (3). As the second most frequent malignancy of the blood in the USA after non-Hodgkin’s disease, about 19,900 new cases of MM and 10,790 deaths occurred in the United States (4). Since the introduction of alkylating agents and melphalan in the 1960s, the median survival of patients with MM has improved (5), but the treatment outcome is far from satisfactory, and novel drugs are in urgent demand to better combat this malignancy.
Camptothecin (CPT) was first isolated by Monroe E. Wall and Mansukh C. Wani in 1958 from extracts of Camptotheca acuminata, a deciduous tree native to China and Tibet, which has been extensively used in traditional Chinese medicine (6). CPT represents an important class of agents useful in the treatment of cancer, which show a broad spectrum of antitumor activity, including lung, ovarian, breast, pancreas, stomach and leukemia (7–11). CPT exhibits antitumor effects by inhibiting the activity of DNA topoisomerase I, which is required for replication and transcription of the cell cycle and stabilization of the DNA-topoisomerase complex, thereby resulting in single-strand DNA breaks to induce the apoptosis of cancer cells (12–15). Cancer cells often overexpress topoisomerase 1 (Topo-1), and they are usually more susceptible to CPT than normal cells (16). It is suggested that CPT may be a promising anticancer agent that warrants further investigation.
However, there are some drawbacks of CPT significantly restricting its clinical use. CPT, similarly to a number of other potent anticancer agents of plant origin, is extremely water insoluble and can only be solubilized in dimethylsulfoxide (DMSO), dichloromethane: methanol (1:1) (v:v) and chloroform: methanol (4:1) (v:v). The serious side effects of co-solvents and bioavailability problems have hampered the usage of CPT in vivo (13). The lactone ring in CPT plays an important role in the drug’s biological activity but it opens at a physiological or higher pH values, making this drug much less active and highly toxic, and precluding its clinical use (Fig. 1) (17).
Figure 1.
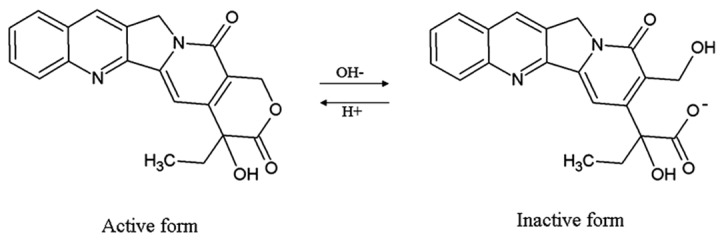
Camptothecin structure and equilibrium between the active lactone form and the inactive carboxylate form.
To overcome these drawbacks of aqueous solubility and stability, two strategies have been introduced. One is to synthesize water-soluble analogues, pro-drugs, and derivatives of CPT. Thus far many compounds have been reported, such as CPT-11, SN-38 and DX-8951f (18–22). However, these compounds are not stable enough in vivo and have lower activity than CPT (23). Alternatively, the development of adequate drug carrier systems to improve the solubility and stability of CPT is gaining attention. There are many reports about utilization of CPT in cancer therapy by using drug delivery systems, such as liposomes, polymer micelles, microemulsions, and microspheres (17,24–28). Unfortunately, these carriers are unsatisfactory due to the poor biocompatibility and biodegradability.
Chitosan is an aminoglucopyran composed of N-acetylglucosamine and glucosamine residues and has excellent properties, such as biocompatibility, biodegradability, and is nontoxic (29). However, the application of the polymer in medicine as a drug carrier in vivo is difficult to be achieved due to its insolubility. Thus, there is a need for chitosan derivatives with increased solubility, especially at neutral pH values, to aid in the delivery of therapeutic compounds (30,31). Trimethyl chitosan has been proven to be a derivative of chitosan with superior solubility compared to chitosan (32). N-Methylated chitosan with the hydrophilic groups N+(CH3)3 and the hydrophobic groups N(CH3)2 is amphiphilic and water-soluble in character at physiological pH and can be self-assembled to vesicles. N-Methylated chitosan has been previously used as a carrier for the delivery of small drug molecules due to its properties (33–35).
In the present study, we chose N,N,N-trimethyl chitosan (TMC) as a carrier to encapsulate CPT and to overcome its drawbacks. The anticancer efficacy of CPT encapsulated with N,N,N-trimethyl chitosan (CPT-TMC) was examined in vivo and in vitro.
Materials and methods
Materials
N,N,N-Trimethylated chitosan (TMC), camptothecin (CPT), propidium iodide (PI), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), RNase A and dimethyl sulfoxide (DMSO) were purchased from Sigma Chemical Co. (St. Louis, MO). All the chemicals employed in this study were of analytical purity and of culture grade. The in situ cell death detection kit was purchased from Roche Co. (Promega, Madison, WI).
Cell culture and tumor model
The murine Balb/c myeloma cell line MPC-11 was purchased from the American Type Culture Collection (ATCC, Manassas, VA) and cells were grown in RPMI-1640 (Life Technologies, Bedford, MA) containing 10% heat-inactivated FCS, 100 units/ml penicillin, and 100 units/ml streptomycin in a humid chamber at 37˚C under 5% CO2.
The MPC-11 tumor model was established in 8-week-old female BALB/c mice. Briefly, these BALB/c mice were inoculated subcutaneously with MPC-11 cells (2×105) in the dorsal area. All these mice were purchased from the Sichuan University Animal Center (Sichuan, Chengdu, China). All studies involving mice were approved by the Institute’s Animal Care and Use Committee.
Preparation of CPT-TMC nanocolloid
According to a previous study (36), CPT-TMC nanocolloid was successfully prepared by a combination of microprecipitation and sonication. Briefly, 6 mg/ml of CPT was first prepared by dissolving 30 mg CPT into 5 ml DMSO. Then TMC was dissolved in water solution at the concentration of 5 mg/ml. Subsequently, 0.1 ml of CPT solution was added in a dropwise fusion into 2 ml of TMC solution at 4˚C. The obtained colloid solution was ultrasonicated for 10 min keeping the temperature at 4˚C. Finally, the colloid solution was dialyzed against water using a membrane with a molecular weight cut-off of 8,000–14,000 (Solarbio, China). After dialysis for 3 days, the solution was centrifuged at 10,000 × g for 10 min to remove insoluble CPT. The amount of CPT in the TMC solution was measured by HPLC.
In vitro cytotoxicity assay
The growth-inhibitory activity of CPT-TMC on the MPC-11 cell lines was evaluated by MTT assay. Briefly, the MPC-11 cells (4–5×103) were seeded in 96-well plates and cultured for 24 h, followed by exposure to various doses of free CPT or CPT-TMC with equivalent doses of CPT for 48 h. A volume of 10 μl of 10 mg/ml MTT was added per well and incubated for another 4 h at 37˚C, then the supernatant fluid was removed and 150 μl/well DMSO was added for 15–20 min. The light absorption values (OD) were measured at 570 nm with the SpectraMAX M5 microplate spectrophotometer (Molecular Devices). The viability of cells was measured by the absorbance at 570 nm.
To assess the effect of CPT-TMC on cell apoptosis and the cell cycle, flow cytometric analysis was performed to measure the percentage of sub-G1 cells after PI staining in hypotonic buffer as previously described (37,38). Briefly, cells were suspended in 1 ml hypotonic fluorochrome solution containing 50 μg/ml PI in 0.1% sodium citrate plus 0.1% Triton X-100 and the cells were analyzed by a flow cytometer (ESP Elite, Beckman-Coulter, Miami, FL). Apoptotic cells appeared in the cell cycle distribution as cells with a DNA content of less than that of G1 cells and were estimated with the Listmode software.
For morphological analysis, the cells were fixed using 70% of ethanol following rinsing with PBS. Morphological analysis of apoptosis was performed after staining with PI (1 μg/ml, in PBS) under fluorescence microscopy (Axiovert 200, Zeiss, Germany) or under light microscopy without staining.
The pattern of DNA cleavage was analyzed as previously described (39). Briefly, cells (3×106) were lysed with 0.5 ml lysis buffer [5 mM Tris-HCl (pH 8.0), 0.25% Nonidet P-40, and 1 mM EDTA], followed by the addition of RNase A at a final concentration of 200 μg/ml, and incubated for 1 h at 37˚C. Cells were then treated with 300 μg/ml proteinase K for an additional 1 h at 37˚C. After addition of 4 μl loading buffer, 20 μl samples in each lane were subjected to electrophoresis on a 1.5% agarose gel at 50 V for 3 h. DNA was stained with ethidium bromide.
In vivo antitumor activity
MPC-11-bearing Balb/c mice were coded and divided into four groups (n=10 for each group). Each group was respectively treated with intravenous injections of CPT-TMC (2.5 mg/kg), CPT (2.5 mg/kg), TMC (25 mg/kg) or 0.9% NS. Treatment was initiated when the tumor volume was 90 mm3. Treatments were given every 3 days for 15 days and survival time and tumor volumes were observed. Tumor size was determined by caliper measurement of the largest and perpendicular diameters every three days. Tumor volumes were calculated according to the formula: V=axb2×0.52, where a is the largest superficial diameter and b is the smallest superficial diameter. The mice were sacrificed when they became moribund. The date of sacrifice was recorded to calculate the survival time. For further investigation, tumors tissues were excised and fixed in 10% formalin.
Apoptosis analysis in tumor tissues
Tumor tissues in paraffin blocks were cut into sections of 3–5 μm thickness. Apoptosis analysis was performed by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling staining using the DeadEnd™ Fluorometric TUNEL system (Promega) following the manufacturer’s protocol. Four equal-sized fields from the tissue sections were randomly chosen and analyzed. The positive-stained cells were visualized and analyzed under a fluorescence microscope (Olympus, BX60). The apoptotic index was calculated as a ratio of the positive cell number to the total tumor cell number based on the mean value from four high-power fields.
Statistical analysis
Data were assayed by ANOVA and the Student’s t-test. For the survival time, Kaplan-Meier curves were established for each group, and the survivals were compared by the log-rank test. All data were presented as mean ± SD. Experiments were performed at least in duplicate. All data were analyzed using the SPSS software (SPSS for Windows, version 17.0; SPSS, Chicago, IL). In all statistical analyses, P<0.05 denoted significant differences.
Results
Inhibition of cell viability
According to the results of the MTT assay, exposure to CPT-TMC and CPT for 48 h, significantly inhibited the cell viability of MPC-11 cells. The results directly suggest that CPT-TMC and CPT inhibited cell viability in a concentration-dependent manner. No statistical difference was observed between the CPT-TMC and the CPT group (P>0.05, Fig. 2).
Figure 2.
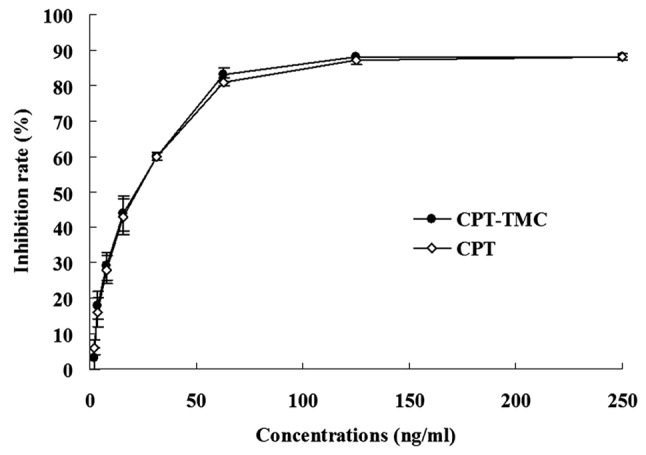
Dose-dependent inhibition of the viability of MPC-11 cells treated with CPT-TMC and CPT. MPC-11 cells were treated with various doses of CPT-TMC and CPT for 48 h. During the assays, the CPT dose in the CPT-TMC was always the same as that of free CPT. Cell viability was detected by MTT assays. Data are expressed as mean ± SD. No significant difference was observed between the CPT-TMC and the CPT group (P>0.05).
In vivo antitumor activity
MPC-11-bearing Balb/c mice were treated with CPT-TMC (2.5 mg/kg), CPT (2.5 mg/kg), TMC (25 mg/kg) and NS, respectively. No differences were observed between the TMC group and the NS group in terms of tumor growth (P>0.05), and it was proven that TMC did not have antitumor activity. On the contrary, CPT and CPT-TMC were found to have antitumor efficiency in inhibiting tumor progress. The inhibition rate of the tumor volume treated with CPT-TMC was 83% compared with the NS group (P<0.05). It was interesting that, as shown in Fig. 3A, the antitumor efficiency of CPT-TMC was better than that of CPT by 55% (P<0.05). However, no complete reaction was found in all these groups.
Figure 3.
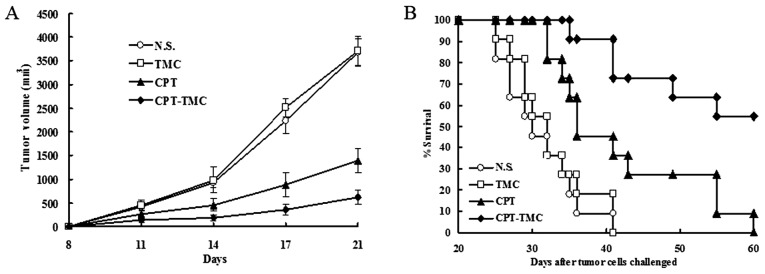
Tumor suppression and survival advantage in mice. (A) The CPT-TMC group shows significant differences in the tumor volume compared with NS, TMC and the CPT groups (P<0.05) from day 11. (B) Survival time was significantly prolonged by CPT-TMC compared with the other three control groups (log-rank test, P<0.05). Data shown are mean ± SD.
To further investigate the antitumor effects of CPT-TMC in vivo, we assayed the lifespan of the mice. Our results show that the groups treated with NS survived 31 days on average, and there was not significant difference between the TMC and NS groups (P>0.05). In contrast, systemic therapy with CPT-TMC significantly prolonged the survival time vs. the NS (P<0.05) and CPT group (P<0.05). When the study was terminated at 60 days after inoculation, more than half of the animals in the CPT-TMC group had survived (Fig. 3B). These data indicate that the animals significantly benefited from CPT-TMC treatment.
Induction of cell apoptosis in vitro and in vivo
The effect of apoptosis induction of CPT-TMC in vitro was measured by flow cytometric analysis, DNA ladder, morphological analysis and the TUNEL assay.
By the use of flow cytometry, we could assess the number of sub-G1 cells, which can be used to indirectly estimate the number of apoptotic cells. The results obtained with flow cytometry strongly show that the CPT-TMC treatment led to MPC-11 cell death by inducing apoptosis in a dose-dependent manner. After exposure to CPT-TMC for 48 h, apoptosis could be observed at 12.5 ng/ml. The increased number of apoptotic cells was detected and the apoptosis rate reached 72.9% at 50 ng/ml (Fig. 4).
Figure 4.
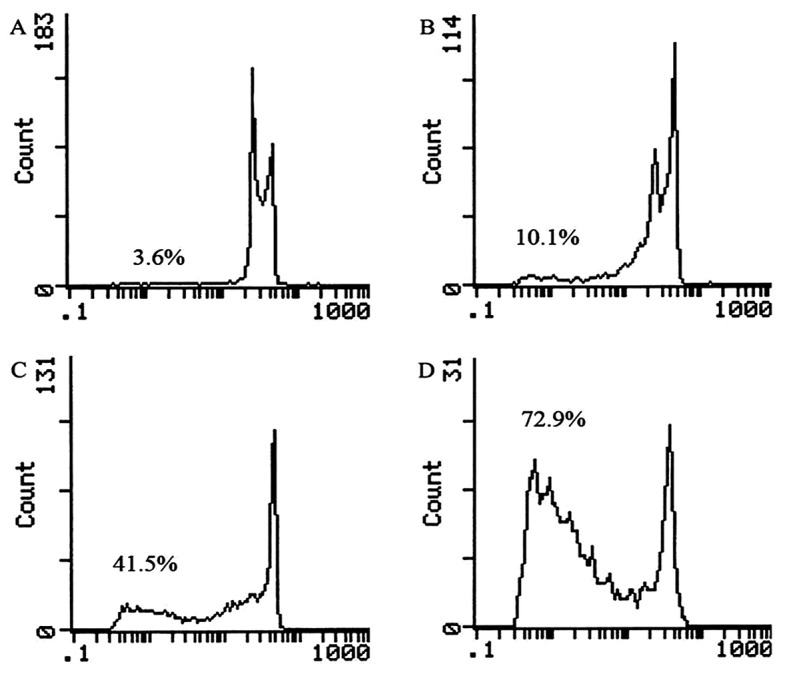
Representative DNA fluorescence histograms of propidium iodide (PI)-stained cells. MPC-11 cells were (A) untreated or treated with various doses of CPT-TMC [(B) 12.5 ng/ml, (C) 25 ng/ml, (D) 50 ng/ml] for 48 h. The cells in the sub-G1 phase were considered as apoptotic cells. The apoptosis rates in non-treated and CPT-TMC-treated cells were (A) 3.6%, (B) 10.1%, (C) 41.5%, (D) 72.9% as assessed by flow cytometry.
To further confirm the apoptosis induction effect of CPT-TMC, the pattern of DNA cleavage was analyzed after treatment with CPT-TMC demonstrated a ladder-like pattern of DNA fragments consisting of multiples of approximately 180–200 base pairs, consistent with internucleosomal DNA fragmentation (Fig. 5).
Figure 5.
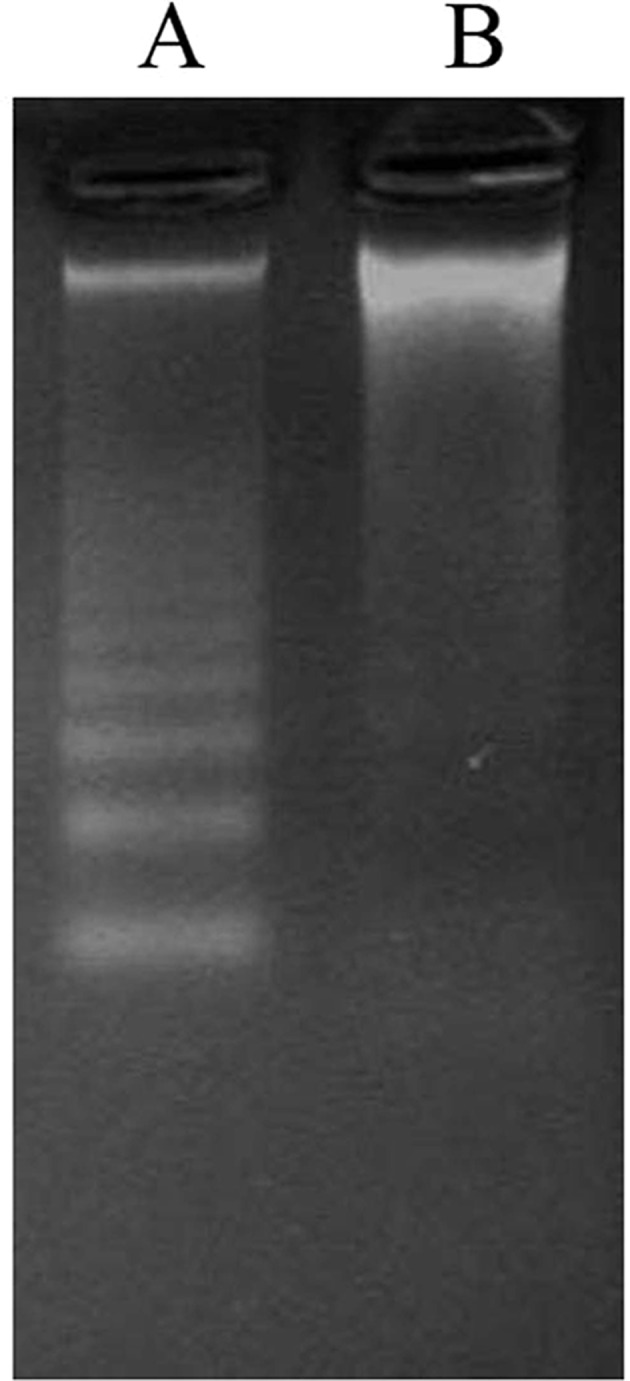
Agarose gel electrophoretic patterns of DNA. Agarose gel electrophoretic patterns of DNA isolated from MPC-11 cells (A) treated and (B) untreated with CPT-TMC (50 ng/ml) for 24 h.
Treatment with CPT-TMC also resulted in morphological changes consistent with apoptosis. Compared with the control, cells treated with CPT-TMC for 48 h were vacuolated, had shrunk and gradually showed increased membrane blebbing as the CPT-TMC concentration increased. After PI staining, the morphological changes were also characteristic of apoptosis: brightly red, condensed nuclei (intact or fragmented) were observed by fluorescence microscopy (Fig. 6).
Figure 6.

Morphological changes induced by CPT-TMC. Bright-field microscopy images (C and D) and fluorescence microscopic appearance of PI-stained nuclei (A and B) of MPC-11 cells untreated or treated with CPT-TMC (25 ng/ml) for 48 h (original magnification, ×400).
To investigate the apoptosis induction effect of CPT-TMC in vivo, the tumor tissues were subjected to terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling assays for respective determination of the apoptotic index. The results suggest that there were almost no TUNEL-positive nuclei in the NS and TMC groups. Both the CPT group and the CPT-TMC group had a higher apoptosis rate of tumor cells compared with the NS control (P<0.05). The apoptosis rate of the CPT-TMC group was much higher than that of the CPT group (P<0.05) (Fig. 7).
Figure 7.

TUNEL assay. (A) Only a few positive nuclei were observed in tumor tissues of NS and TMC controls (A and B), and there were many more positive nuclei in the CPT-TMC group than in the CPT group (C and D). (B) Percent apoptosis in each group revealed that CPT-TMC exhibited a more effective induction of apoptosis compared with the other three controls. Values are presented as the mean ± SD. (*P<0.05).
Discussion
Multiple myeloma (MM) is a chronic hematological disease affecting terminally differentiated B cells, for which there is currently no cure. In the last decades, there is a noticeable improvement in the treatment of MM due to the introduction of new therapeutic strategies and new agents, such as ASCT (autologous stem cell transplantation), thalidomide, bortezomib, and lenalidomide (40). But in fact, the median survival is 4.4–7.1 years in spite of all available therapies for relapse and drug resistance (41,42). For this reason, searching for new and more effective agents is necessary.
Camptothecin (CPT), a plant alkaloid, is a potent antitumor agent, which acts by inhibiting the nuclear enzyme topoisomerase I and inhibits the growth of a wide range of tumors. Some analogues of CPT, such as topotecan, have been ultilized as chemotherapy agent in MM (43–45). However, there is limited information of CPT itself in MM, because the major drawbacks of the drug, water insolubility and lactone instability, hamper its medical use. To overcome these drawbacks, some delivery systems were developed to increase the solubility and the stability of the drug, such as liposome and polymeric micelles (17,46). But all these approaches are far from satisfactory due to poor biocompatibility, biodegradability, or bioadhesivity. Therefore, we studied the effect of CPT on murine multiple myeloma cells MPC-11 in vitro and in vivo and attempted to increase its solubility and stability.
Chitin, a linear cationic heteropolymer of randomly distributed N-acetylglucosamine and glucosamine residues, is one of the most abundant polysaccharides in nature and is mostly derived from the exoskeleton of crustaceans (47). Chitosan, a polymer obtained by deacetylation of chitin is widely studied for its pharmaceutical and non-pharmaceutical applications. For example, chitosan has been extensively evaluated for its mucoadhesive and absorption enhancement properties. But chitosan is not soluble in a medium except below pH 5.6 and this property limits its use as a drug delivery system. Therefore, there is a need for chitosan derivatives with increased solubility at physiological pH values. N,N,N-Trimethyl chitosan (TMC) is a promising derivativ. TMC is soluble either in an acidic, basic or neutral medium (pH range 1–9 up to 10% w/v concentration) and has mucoadhesive and permeation enhancement properties like native chitosan (48,49). It has been reported that TMC could enhance the transport of small compounds, large molecules, peptide drugs and DNA and has shown promising results as a drug delivery agent as well as a DNA delivery agent (50). Hence, TMC was selected as a carrier to delivery CPT which is insoluble in water.
In the present study, we investigated the antitumor effect of CPT on the murine myeloma cell line MPC-11 and we demonstrated that CPT inhibited the growth of MPC-11 cells in vitro and in vivo. To further improve the antitumor activity, we chose TMC as a carrier to encapsulate and deliver CPT (CPT-TMC). The results of the MTT assay showed that both CPT and CPT-TMC significantly inhibited the growth of MPC-11 cells in vitro and there was a significant difference between their effects. TMC itself was not cytotoxic and TMC could not improve the antitumor activity of CPT in vitro. However, interestingly, the results of in vivo assays were different from those of in vitro assays. Compared with CPT, in murine models, we demonstrated that CPT-TMC more efficiently suppressed tumor growth in murine models. Moreover, the TUNEL assay showed a significant increase of the apoptotic index in the CPT-TMC group. A further study showed that the survival time of animals treated with CPT-TMC was significantly prolonged compared with the NS, TMC and CPT groups. According to these results, it was suggested that the TMC delivery system efficiently improved the antitumor activity in vivo, but not in vitro. The mechanism of the antitumor effects of CPT-TMC may be the prolonged blood circulation time or the accumulation of CPT in tumor tissue. In light of the encouraging results presented herein, delineation of the potential chemotherapeutic effects of CPT-TMC and its precise mechanism of actions warrants further investigation.
In conclusion, we demonstrated that CPT has powerful antitumor activity through induing apoptosis in murine multiple myeloma models, and that the TMC delivery system can efficiently improve the effects of CPT. Thereby, our finding may provide a strategy to permit utilization of CPT in clinical practice. CPT-TMC may be a new effective agent to better combat multiple myeloma.
Acknowledgements
This study was financially supported by the Chinese Key Basic Research Program (2010CB529906), the Technology Project of Changzhou Social Development (CS20102016) and the Natural Science Funds for Young Teacher of Soochow University (Q3124943).
References
- 1.Kyle RA, Rajkumar SV. Multiple myeloma. N Engl J Med. 2004;351:1860–1873. doi: 10.1056/NEJMra041875. [DOI] [PubMed] [Google Scholar]
- 2.Rajkumar SV, Kyle RA. Multiple myeloma: diagnosis and treatment. Mayo Clin Proc. 2005;80:1371–1382. doi: 10.4065/80.10.1371. [DOI] [PubMed] [Google Scholar]
- 3.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 4.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 5.Munshi NC. Plasma cell disorders: an historical perspective. Hematology Am Soc Hematol Educ Program. 2008;2008:297. doi: 10.1182/asheducation-2008.1.297. [DOI] [PubMed] [Google Scholar]
- 6.Wall ME, Wani MC, Cook CE, Palmer KH, McPhail AT, Sim GA. Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from Camptotheca acuminata. J Am Chem Soc. 1966;88:3888–3890. [Google Scholar]
- 7.Hsiang YH, Hertzberg R, Hecht S, Liu LF. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem. 1985;260:14873–14878. [PubMed] [Google Scholar]
- 8.Potmesil M. Camptothecins: from bench research to hospital wards. Cancer Res. 1994;54:1431–1439. [PubMed] [Google Scholar]
- 9.Takimoto CH, Wright J, Arbuck SG. Clinical applications of the camptothecins. Biochim Biophys Acta. 1998;1400:107–119. doi: 10.1016/s0167-4781(98)00130-4. [DOI] [PubMed] [Google Scholar]
- 10.Topcu Z. DNA topoisomerases as targets for anticancer drugs. J Clin Pharm Ther. 2001;26:405–416. doi: 10.1046/j.1365-2710.2001.00368.x. [DOI] [PubMed] [Google Scholar]
- 11.Bredholt T, Dimba EA, Hagland HR, et al. Camptothecin and khat (Catha edulis Forsk.) induced distinct cell death phenotypes involving modulation of c-FLIPL, Mcl-1, procaspase-8 and mitochondrial function in acute myeloid leukemia cell lines. Mol Cancer. 2009;8:101. doi: 10.1186/1476-4598-8-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia-Carbonero R, Supko JG. Current perspectives on the clinical experience, pharmacology, and continued development of the camptothecins. Clin Cancer Res. 2002;8:641–661. [PubMed] [Google Scholar]
- 13.Hatefi A, Amsden B. Camptothecin delivery methods. Pharm Res. 2002;19:1389–1399. doi: 10.1023/a:1020427227285. [DOI] [PubMed] [Google Scholar]
- 14.Kehrer DF, Soepenberg O, Loos WJ, Verweij J, Sparreboom A. Modulation of camptothecin analogs in the treatment of cancer: a review. Anticancer Drugs. 2001;12:89–105. doi: 10.1097/00001813-200102000-00002. [DOI] [PubMed] [Google Scholar]
- 15.Bodley AL, Wani MC, Wall ME, Shapiro TA. Antitrypanosomal activity of camptothecin analogs. Structure-activity correlations. Biochem Pharmacol. 1995;50:937–942. doi: 10.1016/0006-2952(95)00215-l. [DOI] [PubMed] [Google Scholar]
- 16.Haluska P, Rubin E, Verschraegen CF. Topoisomerase-I inhibitors in gynecologic tumors. Hematol Oncol Clin North Am. 1999;13:43–61. vii–viii. doi: 10.1016/s0889-8588(05)70153-2. [DOI] [PubMed] [Google Scholar]
- 17.Kawano K, Watanabe M, Yamamoto T, et al. Enhanced antitumor effect of camptothecin loaded in long-circulating polymeric micelles. J Control Release. 2006;112:329–332. doi: 10.1016/j.jconrel.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 18.Sun FX, Tohgo A, Bouvet M, et al. Efficacy of camptothecin analog DX-8951f (Exatecan Mesylate) on human pancreatic cancer in an orthotopic metastatic model. Cancer Res. 2003;63:80–85. [PubMed] [Google Scholar]
- 19.Zu YG, Li QY, Fu YJ, Wang W. Synthesis and cytotoxicity of water soluble quaternary salt derivatives of camptothecin. Bioorg Med Chem Lett. 2004;14:4023–4026. doi: 10.1016/j.bmcl.2004.05.032. [DOI] [PubMed] [Google Scholar]
- 20.van Hattum AH, Pinedo HM, Schluper HM, Erkelens CA, Tohgo A, Boven E. The activity profile of the hexacyclic camptothecin derivative DX-8951f in experimental human colon cancer and ovarian cancer. Biochem Pharmacol. 2002;64:1267–1277. doi: 10.1016/s0006-2952(02)01297-2. [DOI] [PubMed] [Google Scholar]
- 21.Akimoto K, Kawai A, Ohya K. Kinetic studies of the hydrolysis and lactonization of camptothecin and its derivatives, CPT-11 and SN-38, in aqueous aolution. Chem Pharm Bulletin. 1994;42:2135–2138. [Google Scholar]
- 22.Hanson BA, Schowen RL, Stella VJ. A mechanistic and kinetic study of the E-ring hydrolysis and lactonization of a novel phosphoryloxymethyl prodrug of camptothecin. Pharm Res. 2003;20:1031–1038. doi: 10.1023/a:1024410322870. [DOI] [PubMed] [Google Scholar]
- 23.Tanizawa A, Fujimori A, Fujimori Y, Pommier Y. Comparison of topoisomerase I inhibition, DNA damage, and cytotoxicity of camptothecin derivatives presently in clinical trials. J Natl Cancer Inst. 1994;86:836–842. doi: 10.1093/jnci/86.11.836. [DOI] [PubMed] [Google Scholar]
- 24.Burke TG, Staubus AE, Mishra AK, Malak H. Liposomal stabilization of camptothecin’s lactone ring. J Am Chem Soc. 1992;114:8318–8319. [Google Scholar]
- 25.Cortesi R, Esposito E, Maietti A, Menegatti E, Nastruzzi C. Formulation study for the antitumor drug camptothecin: liposomes, micellar solutions and a microemulsion. Int J Pharm. 1997;159:95–103. [Google Scholar]
- 26.Watanabe M, Kawano K, Yokoyama M, Opanasopit P, Okano T, Maitani Y. Preparation of camptothecin-loaded polymeric micelles and evaluation of their incorporation and circulation stability. Int J Pharm. 2006;308:183–189. doi: 10.1016/j.ijpharm.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 27.Shenderova A, Burke TG, Schwendeman SP. The acidic microclimate in poly(lactide-co-glycolide) microspheres stabilizes camptothecins. Pharm Res. 1999;16:241–248. doi: 10.1023/a:1018876308346. [DOI] [PubMed] [Google Scholar]
- 28.Tong W, Wang L, D’Souza MJ. Evaluation of PLGA microspheres as delivery system for antitumor agent-camptothecin. Drug Dev Ind Pharm. 2003;29:745–756. doi: 10.1081/ddc-120021774. [DOI] [PubMed] [Google Scholar]
- 29.Chandy T, Sharma CP. Chitosan-as a biomaterial. Biomater Artif Cells Artif Organs. 1990;18:1–24. doi: 10.3109/10731199009117286. [DOI] [PubMed] [Google Scholar]
- 30.Kotzé AF, Lueßen HL, de Boer AG, Verhoef JC, Junginger HE. Chitosan for enhanced intestinal permeability: prospects for derivatives soluble in neutral and basic environments. Eur J Pharm Sci. 1998;7:145–151. doi: 10.1016/s0928-0987(98)00016-5. [DOI] [PubMed] [Google Scholar]
- 31.Kotze AF, Luessen HL, de Leeuw BJ, de Boer BG, Verhoef JC, Junginger HE. N-trimethyl chitosan chloride as a potential absorption enhancer across mucosal surfaces: in vitro evaluation in intestinal epithelial cells (Caco-2) Pharm Res. 1997;14:1197–1202. doi: 10.1023/a:1012106907708. [DOI] [PubMed] [Google Scholar]
- 32.Jintapattanakit A, Mao S, Kissel T, Junyaprasert VB. Physicochemical properties and biocompatibility of N-trimethyl chitosan: effect of quaternization and dimethylation. Eur J Pharm Biopharm. 2008;70:563–571. doi: 10.1016/j.ejpb.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 33.Peng X, Zhang L. Surface fabrication of hollow microspheres from N-methylated chitosan cross-linked with gultaraldehyde. Langmuir. 2005;21:1091–1095. doi: 10.1021/la047689w. [DOI] [PubMed] [Google Scholar]
- 34.Peng X, Zhang L, Kennedy JF. Release behavior of microspheres from cross-linked N-methylated chitosan encapsulated ofloxacin. Carbohydr Polym. 2006;65:288–295. [Google Scholar]
- 35.Zhilei L, Steenekamp JH, Hamman JH. Cross-linked cationic polymer microparticles: effect of N-trimethyl chitosan chloride on the release and permeation of ibuprofen. Drug Dev Ind Pharm. 2005;31:311–317. doi: 10.1081/ddc-52079. [DOI] [PubMed] [Google Scholar]
- 36.Li X, Kong X, Zhang J, et al. A novel composite hydrogel based on chitosan and inorganic phosphate for local drug delivery of camptothecin nanocolloids. J Pharm Sci. 2011;100:232–241. doi: 10.1002/jps.22256. [DOI] [PubMed] [Google Scholar]
- 37.Yuan ZP, Chen LJ, Fan LY, et al. Liposomal quercetin efficiently suppresses growth of solid tumors in murine models. Clin Cancer Res. 2006;12:3193–3199. doi: 10.1158/1078-0432.CCR-05-2365. [DOI] [PubMed] [Google Scholar]
- 38.Muther RS, Bennett WM. Effects of dimethyl sulfoxide on renal function in man. JAMA. 1980;244:2081–2083. [PubMed] [Google Scholar]
- 39.Gorczyca W, Gong J, Ardelt B, Traganos F, Darzynkiewicz Z. The cell cycle related differences in susceptibility of HL-60 cells to apoptosis induced by various antitumor agents. Cancer Res. 1993;53:3186–3192. [PubMed] [Google Scholar]
- 40.Rajkumar SV. Multiple myeloma. Curr Probl Cancer. 2009;33:7–64. doi: 10.1016/j.currproblcancer.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sirohi B, Powles R. Multiple myeloma. Lancet. 2004;363:875–887. doi: 10.1016/S0140-6736(04)15736-X. [DOI] [PubMed] [Google Scholar]
- 42.Hideshima T, Anderson KC. Molecular mechanisms of novel therapeutic approaches for multiple myeloma. Nat Rev Cancer. 2002;2:927–937. doi: 10.1038/nrc952. [DOI] [PubMed] [Google Scholar]
- 43.Donato ML, Aleman A, Champlin RE, et al. High-dose topotecan, melphalan and cyclophosphamide (TMC) with stem cell support: a new regimen for the treatment of multiple myeloma. Leuk Lymphoma. 2004;45:755–759. doi: 10.1080/10428190310001603957. [DOI] [PubMed] [Google Scholar]
- 44.Kraut EH, Crowley JJ, Wade JL, et al. Evaluation of topotecan in resistant and relapsing multiple myeloma: a Southwest Oncology Group study. J Clin Oncol. 1998;16:589–592. doi: 10.1200/JCO.1998.16.2.589. [DOI] [PubMed] [Google Scholar]
- 45.Kraut EH, Young D, Farag S, James AG, Solove RJ. Phase II study of topotecan and cyclophosphamide in patients with relapsed and refractory multiple myeloma. Leuk Res. 2005;29:1233–1234. doi: 10.1016/j.leukres.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 46.Watanabe M, Kawano K, Toma K, Hattori Y, Maitani Y. In vivo antitumor activity of camptothecin incorporated in liposomes formulated with an artificial lipid and human serum albumin. J Control Release. 2008;127:231–238. doi: 10.1016/j.jconrel.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 47.Rinaudo M. Chitin and chitosan: properties and applications. Prog Polym Sci. 2006;31:603–632. [Google Scholar]
- 48.Cardile V, Frasca G, Rizza L, et al. Improved adhesion to mucosal cells of water-soluble chitosan tetraalkylammonium salts. Int J Pharm. 2008;362:88–92. doi: 10.1016/j.ijpharm.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 49.Snyman D, Hamman JH, Kotze AF. Evaluation of the mucoadhesive properties of N-trimethyl chitosan chloride. Drug Dev Ind Pharm. 2003;29:61–69. doi: 10.1081/ddc-120016684. [DOI] [PubMed] [Google Scholar]
- 50.Mourya VK, Inamdar NN. Trimethyl chitosan and its applications in drug delivery. J Mater Sci Mater Med. 2009;20:1057–1079. doi: 10.1007/s10856-008-3659-z. [DOI] [PubMed] [Google Scholar]