

Abstract
Drugs targeting the orphan receptor GPR35 have potential therapeutic application in a number of disease areas, including inflammation, metabolic disorders, nociception, and cardiovascular disease. Currently available surrogate GPR35 agonists identified from pharmacologically relevant compound libraries have limited utility due to the likelihood of off-target effects in vitro and in vivo and the variable potency that such ligands exhibit across species. We sought to identify and characterize novel GPR35 agonists to facilitate studies aimed at defining the physiologic role of GPR35. PathHunter β-arrestin recruitment technology was validated as a human GPR35 screening assay, and a high-throughput screen of 100,000 diverse low molecular weight compounds was conducted. Confirmed GPR35 agonists from five distinct chemotypes were selected for detailed characterization using both β-arrestin recruitment and G protein-dependent assays and each of the human, mouse, and rat GPR35 orthologs. These studies identified 4-{(Z)-[(2Z)-2-(2-fluorobenzylidene)-4-oxo-1,3-thiazolidin-5-ylidene]methyl}benzoic acid (compound 1) as the highest potency full agonist of human GPR35 yet described. As with certain other GPR35 agonists, compound 1 was markedly selective for human GPR35, but displayed elements of signal bias between β-arrestin-2 and G protein-dependent assays. Compound 1 also displayed competitive behavior when assessed against the human GPR35 antagonist, ML-145 (2-hydroxy-4-[4-(5Z)-5-[(E)-2-methyl-3-phenylprop-2-enylidene]-4-oxo-2-sulfanylidene-1,3-thiazolidin-3-yl]butanoylamino]benzoic acid). Of the other chemotypes studied, compounds 2 and 3 were selective for the human receptor, but compounds 4 and 5 demonstrated similar activity at human, rat, and mouse GPR35 orthologs. Further characterization of these compounds and related analogs is likely to facilitate a better understanding of GPR35 in health and disease.
Introduction
The class A orphan receptor GPR35 has been described as a potentially novel drug target (MacKenzie et al., 2011) based upon reports describing the activation of this receptor in a variety of cell types (Leonard et al., 2005; Wang et al., 2006; Ohshiro et al., 2008; Fallarini et al., 2010), the potential genetic linkage of GPR35 to disease (Shrimpton et al., 2004; Vander Molen et al., 2005; Ellinghaus et al., 2012), and the phenotypic profiling of a GPR35 knockout mouse line (Min et al., 2010). This has resulted in speculation that GPR35 may hold therapeutic potential for inflammatory disorders (Wang et al., 2006; Lattin et al., 2008; Barth et al., 2009), central nervous system function (Shrimpton et al., 2004; Lein et al., 2007; Guo et al., 2008), nociception (Ohshiro et al., 2008; Zhao et al., 2010; Cosi et al., 2011), metabolic disorders (Leonard et al., 2005; Vander Molen et al., 2005), hypertension, and cardiovascular disease (Sun et al., 2008; Min et al., 2010). The physiologic and pathophysiologic functions of this receptor, however, remain to be fully determined (Milligan, 2011).
The endogenously produced molecules kynurenic acid (Wang et al., 2006) and lysophosphatidic acid (Oka et al., 2010) have been reported as candidate GPR35 agonists; however, the nature of the cognate ligand(s) remains controversial (Jenkins et al., 2011). As a consequence, many studies examining GPR35 activation and intracellular signaling have been performed using the first reported surrogate ligand, zaprinast (Taniguchi et al., 2006), and this has become widely used as a reference compound. Although more widely known as a moderately potent phosphodiesterase type V and VI inhibitor, the GPR35 agonist activity of zaprinast appears to be unrelated to its phosphodiesterase inhibitory activity (Taniguchi et al., 2006).
Work from several laboratories, using a range of screening assays including calcium mobilization, β-arrestin-based receptor interaction and translocation assays, and so called “label free” technologies, have continued to identify an increasing number of structurally diverse surrogate ligands (MacKenzie et al., 2011; Milligan, 2011), not least the potent, but human ortholog selective, agent pamoic acid {(4-[(3-carboxy-2-hydroxynaphthalen-1-yl)methyl]-3-hydroxynaphthalene-2-carboxylic acid} from the Prestwick chemical library of US Food and Drug Administration-approved drugs (Jenkins et al., 2010; Zhao et al., 2010). Although approaches based on agonist-induced interactions between GPR35 and β-arrestin-2 have been central to these efforts, GPR35 is also able to activate signal transduction through pertussis toxin-sensitive Gi family G proteins and, very selectively, via the G protein G13 (MacKenzie et al., 2011; Milligan, 2011). Agonist ligands with the ability to stabilize receptor conformations that selectively promote engagement of one signal transduction pathway in preference to another are generally referred to as biased-ligands (Rajagopal et al., 2011). There is growing interest in the concept that such ligands may offer therapeutic benefit in situations where activation of multiple signals via a receptor can result in both useful and contraindicated outcomes (Dewire and Violin, 2011). As such, no matter the primary screen used, it is imperative to assess the activity of ligands across the range of signal pathways, both G protein-dependent and –independent, that the receptor is able to modulate.
The availability of structurally diverse, pharmacologically relevant surrogate ligands may facilitate early investigation into receptor pharmacology in multiple cell types; however, many of the current ligands with GPR35 activity also have prominent off-target effects at other pathways, which may diminish their utility for understanding GPR35 signaling and for identifying potential therapeutic uses of GPR35.
To facilitate the investigation of GPR35 biology we now report key outcomes from a high-throughput screen (HTS) of 100,000 compounds to identify novel agonists, utilizing Chinese hamster ovary (CHO) cells stably expressing human GPR35 and DiscoveRx (Fremont, CA) PathHunter β-arrestin recruitment technology. Candidate hit compounds, including five novel ligands that we detail herein, were profiled and characterized in both β-arrestin-2 and G protein-coupled read outs to explore potential ligand bias and against rodent orthologs of GPR35 to define potential species selectivity.
Materials and Methods
Materials.
Cell culture media were from Life Technologies (Paisley, UK). Cells were routinely passaged in complete growth medium (Dulbecco's modified Eagle's medium/F12 with GlutaMAX + 10% fetal bovine serum). Zaprinast was from Sigma-Aldrich (Gillingham, UK). ML-145 {2-hydroxy-4-[4-(5Z)-5-[(E)-2-methyl-3-phenylprop-2-enylidene]-4-oxo-2-sulfanylidene-1,3-thiazolidin-3-yl]butanoylamino]benzoic acid} was from Tocris Bioscience (Bristol, UK). Commercially available compound libraries were obtained from National Institute of Neurological Disorders and Stroke (NINDS) (National Institutes of Health, Bethesda, MD) and Sequoia Research Products Ltd. (Pangbourne, UK).
PathHunter β-Arrestin Recruitment Assay.
PathHunter β-arrestin recruitment assays were performed using a CHO–K1 cell line stably expressing human GPR35 and β-arrestin-2 (DiscoveRx). In this assay a receptor C-terminally tagged with a short fragment of the enzyme β-galactosidase is coexpressed alongside β-arrestin-2 tagged with the rest of the sequence of β-galactosidase (Eglen, 2007). Agonist-induced recruitment of β-arrestin-2 to GPR35 was monitored through β-galactosidase enzyme fragment complementation because this reflects proximity-induced complementation between the fragments of the enzyme. Prior to assay 5000 cells per well of a 384-well plate were seeded in 10 µl cell plating reagent 2 (DiscoveRx) and incubated overnight at 37°C. For primary screening, compounds were tested at a single final assay concentration of 10 µM (1.0% dimethylsulfoxide). For the determination of EC50 values compounds were tested across a 10-point concentration-response curve with a highest assay concentration of 100 µM. Cells were incubated with compounds at 37°C for 2.5 hours to facilitate complementation and enzyme action. This was followed by the addition of 10 µl PathHunter detection reagent (DiscoveRx) and incubation for 2.5 hours at room temperature in the dark. Chemiluminescence was measured on a PHERAstar FS plate reader (BMG Labtech, Ortenberg, Germany).
Intracellular Calcium Assay.
PathHunter β-arrestin GPR35 CHO–K1 cells were transfected with a chimeric Gαq-i5 G protein subunit plasmid (Jenkins et al., 2012) using lipofectamine (Life Technologies) and incubated overnight at 37°C. Cells were then reseeded in complete growth medium at 10,000 cells per well in 384-well black, clear bottom microplates and incubated overnight at 37°C. Cells were loaded with Fluo-4 direct calcium assay reagent (Life Technologies) for 45 minutes at 37°C + 15 minutes at room temperature in the presence of probenecid (5 mM). Compound addition and calcium mobilization were monitored on a Flex Station (Molecular Devices, Sunnyvale, CA).
Bioluminescence Resonance Energy Transfer-Based β-Arrestin-2 Recruitment Assay.
Human embryonic kidney (HEK)-293T cells, maintained as previously described (Jenkins et al., 2010), were transiently transfected with human, mouse, or rat orthologs of GPR35 C-terminally tagged with enhanced yellow fluorescent protein (eYFP) and β-arrestin-2-Renilla luciferase 6 using polyethylenimine (Polysciences, Inc, Warrington, PA) (Jenkins et al., 2010). Transfections with Renilla luciferase 6 construct and pcDNA3 expression vector alone were performed as controls. After 24 hours, cells were seeded at 50,000 cells per well in poly-d-lysine coated 96-well plates and incubated at 37°C overnight. After 24 hours, cells were washed twice with Hank’s balanced salt solution (pH 7.4) and incubated in the dark with 5 µM coelentrazine-h (Promega, Southampton, UK) for 10 minutes at 37°C. Following compound addition, the cells were incubated for a further 5 minutes at 37°C before sequential reading of emission signals on a PHERAstar FS plate reader (BMG Labtech) at 485 nm and 530 nm, which represent the Renilla luciferase 6 and eYFP signals, respectively. The bioluminescence resonance energy transfer (BRET) ratio was calculated as the BRET eYFP divided by Renilla luciferase 6. The net BRET ratio was obtained from the BRET ratio minus Renilla luciferase 6 control. This value was multiplied by 1000 to obtain mBRET units.
Inositol Monophosphate Accumulation Assay.
We used a homogeneous time-resolved fluorescence-based (HTRF) assay kit to detect inositol monophosphates derived from the breakdown of inositol 1,4,5, trisphosphate (Trinquet et al., 2011). HEK-293T cells were transiently cotransfected with human, mouse, or rat orthologs of FLAG-GPR35-eYFP and a chimeric G protein (Gαq-13) (Jenkins et al., 2012) using polyethylenimine. After 16– to 24–hour incubation at 37°C, cells were resuspended in IP-One stimulation buffer (10 mM HEPES, 1 mM CaCl2, 0.5 mM MgCl2, 4.2 mM KCl, 146 mM NaCl, 5.5 mM glucose, 50 mM LiCl, pH 7.4) and seeded at 10,000 cells per well in white, solid bottom 384-well plates. Ligands were diluted in IP-One stimulation buffer according to manufacturer’s instructions (HTRF IP-One Tb kit; Cisbio, Codolet, France). Cells were incubated with agonist for 2 hours at 37°C before addition of d2-conjugated inositol monophosphate (IP1) and anti-IP1 Lumi4-Tb cryptate diluted in lysis buffer per manufacturer’s instructions. After incubation at room temperature for 1 hour, HTRF was measured using a PHERAstar FS plate reader (BMG Labtech).
Data Analysis.
Curves were fitted by nonlinear regression analysis in GraphPad Prism (GraphPad, San Diego, CA).
Results
Screening NINDS and Sequoia Libraries Using PathHunter β-Arrestin-2 Recruitment.
Screening for GPR35 agonists within the Prestwick library of 1200 pharmacologically relevant compounds has previously been reported (Jenkins et al., 2010; Zhao et al., 2010). As an extension to this, initially we developed a 384-well human GPR35 agonist assay using the DiscoveRx PathHunter β-arrestin recruitment technology and performed a pilot screen using NINDS and Sequoia libraries of known drugs and pharmacologically active compounds, which show a degree of compound overlap with the Prestwick library. Zaprinast at an EC100 of 100 μM served as a positive control and 1.0% dimethylsulfoxide as a negative vehicle control on each assay plate. Assay performance was monitored and validated by statistical outcomes; the mean signal:background was 2.93, mean Z prime (Z′) was 0.66, mean %CV for positive and negative controls were 5.75 and 4.96, respectively, and the overall assay hit rate was 2.1% at a 30% activity of zaprinast cut-off. This resulted in selection of 29 compounds encompassing a variety of chemotypes that included both full and partial agonists with a range of potencies and efficacies (Fig. 1). The assay successfully identified zaprinast and other previously reported agonists including 5-nitro-2-(3-phenylpropylamino) benzoic acid (Taniguchi et al., 2008), dicumarol, cromolyn disodium, derivatives of pamoic acid (Zhao et al., 2010; Jenkins et al., 2010), furosemide (Yang et al., 2012), aspirin metabolites (Deng and Fang, 2012), and ellagic acid (Deng et al., 2012b). Novel agonists identified from the NINDS and Sequoia libraries included artemisinin, which behaved as a partial agonist with high potency, amlexanox, tannic acid, and 5-fluoroindole-2-carboxylic acid (Fig. 1). The identification of previously reported as well as novel agonists provided confidence in the validity of the assay and that a robust β-arrestin assay was configured and suitable for HTS.
Fig. 1.

Development of a HTS compatible β-arrestin recruitment assay for human GPR35. Concentration-response curves for agonist hits identified from a pilot screen of NINDS/Sequoia compound libraries were performed using the human GPR35 PathHunter β-arrestin recruitment assay. Data are expressed as % of the response to 100 µM zaprinast. pEC50 values for each compound are provided in parentheses. Data are means ± S.E.M. from two separate experiments performed in duplicate.
HTS of Medical Research Council Technology Diversity Compound Collection Using PathHunter β-Arrestin-2 Recruitment.
To gain greater diversity and potentially more chemically tractable ligands we performed a HTS against a diverse collection of 100,000 chemical compounds. This collection was selected to generate a high-quality set of compounds using drug-like filters based on physicochemical properties, together with analysis to maximize template diversity within the library. The compounds from the library were tested in “single shot” format at final assay concentration of 10 µM, and 753 positives were identified. A further assay run of these 753 positives resulted in a 73% hit confirmation rate. Analysis of the activity distribution profile revealed that only 5% of compounds were as or more active than zaprinast, with approximately 40% of the hits approaching only 20–30% of zaprinast activity (Fig. 2). Confirmed hits were profiled in full concentration-response curves, and the potency distribution of the pEC50 values is shown in Fig. 3. The largest proportion (38.9%) had pEC50 values within the range 4.5–5.0, whereas few were significantly more potent than zaprinast (pEC50 ~5.8–6.3), and only a small proportion (3.9%) displayed pEC50 ≥ 6.
Fig. 2.

Analysis of HTS data: efficacy of hits at human GPR35. A distribution of agonist activity recorded in the 100,000 compound library HTS is shown. Compound activity at 10 µM using a human GPR35 PathHunter β-arrestin recruitment assay is shown both as hit frequency, binned as % agonist activity versus zaprinast, with number of hits for each % activity indicated above each bar. The pie chart shows the percentage of hits within each % activity group.
Fig. 3.

Analysis of HTS data: potency of hits at human GPR35. Distribution of pEC50 values determined for 348 agonist compounds identified from a 100,000 compound library HTS performed using a human GPR35 PathHunter β-arrestin recruitment assay. Data are displayed as the percentage of these compounds within distinct pEC50 ranges.
Compound Profiling in Distinct β-Arrestin-2 and G Protein-Dependent Assay Formats.
To extend the initial screening data, a chemistry-based triage and assessment process highlighted a number of distinct chemical series among the hits. Seven hit families were prioritized, and 86 selected compounds were repurchased. All of the resupplied compounds were reassessed for activity and potency in a distinct GPR35-β-arrestin-2 interaction assay based on BRET between coexpressed β-arrestin-2-Renilla luciferase and C-terminally eYFP-tagged forms of each of human, mouse, or rat GPR35 (Jenkins et al., 2012) to explore possible species selectivity. The functionality of 30 compounds, each of which displayed pEC50 of at least 5.0 at one of the three orthologs is shown (Supplemental Table 1). Performed in 96-well plate format, these assays produced excellent assay performance with mean Z′ values of 0.83 for human, 0.75 for mouse, and 0.77 for rat GPR35 and mean signal:background of 8.15 for human, 7.98 for mouse, and 4.27 for the rat ortholog. Subsequently, a smaller number of compounds were selected as representatives of five different chemotypes and profiled in detail in a range of assays, including β-arrestin-2 interaction and both intracellular calcium mobilization (performed in the PathHunter human GPR35 CHO cell line following transient transfection with the chimeric G protein subunit Gαq-i5) (Jenkins et al., 2012) and an IP1 accumulation (performed in HEK-293T cells transiently transfected to express human, mouse, or rat GPR35 alongside the chimeric G protein subunit Gαq-13) assay (Jenkins et al., 2012). These studies were designed to explore if compounds might selectively modulate β-arrestin-2 over G protein-dependent signals. Figure 4 demonstrates that zaprinast was active in both PathHunter β-arrestin and Gαq-i5 Ca2+ elevation assays. A number of other compounds identified in the HTS including 4-{(Z)-[(2Z)-2-(2-fluorobenzylidene)-4-oxo-1,3-thiazolidin-5-ylidene] methyl} benzoic acid (compound 1), 2,2'-bithiophene-5,5′-dicarboxylic acid (compound 2), 4-chloro-3-(2,5-dimethyl-1H-pyrrol-1-yl)benzoic acid (compound 3), 4H-thieno[3,2-b]pyrrole-2-carboxylic acid (compound 4) and 9-benzyl-5-methyl-3-(4-methylphenyl)-5,9-dihydro-6H-[1,2,4]triazolo[4,3-e]purine-6,8(7H)-dione (compound 5) (see Table 1 for structures) displayed efficacy equivalent to zaprinast in both these assays (Fig. 4). Most notably, compound 1 was markedly more potent than zaprinast with pEC50 = 7.59 ± 0.05 in the PathHunter β-arrestin interaction assay and pEC50 = 8.36 ± 0.05 in the Gαq-i5 Ca2+assay (Fig. 4). It is thus the most potent agonist yet reported for human GPR35.
Fig. 4.

Comparison of agonist activity in β-arrestin and Ca2+ mobilization assays. Five selected compounds (1–5) resupplied following hit identification were compared with zaprinast for activity and potency in the human GPR35 PathHunter β-arrestin recruitment assay (A) and in intracellular calcium mobilization studies (B). Data in (A) are expressed as % of the maximum response to zaprinast and in (B) as fluorescence intensity units (FIU). Means ± S.E.M. from two separate experiments performed in duplicate.
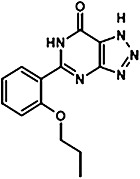
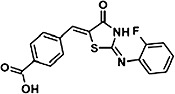
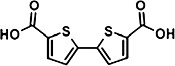
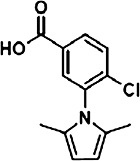
TABLE 1.
Characterization of novel GPR35 agonists
The chemical structures of compounds 1–5 are shown as are pEC50 and Emax values obtained in the various assays employed.
| Zaprinast | Compound 1 | Compound 2 | Compound 3 | Compound 4 | Compound 5 | |||
|---|---|---|---|---|---|---|---|---|
 |
 |
 |
 |
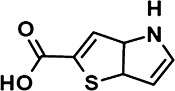 |
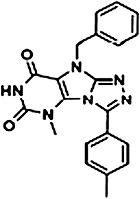 |
|||
| PathHunter β-arrestin | Hu GPR35 | pEC50 | 5.1 ± 0.01 | 7.6 ± 0.05 | 6.3 ± 0.05 | 5.4 ± 0.11 | 5.6 ± 0.01 | 4.9 ± 0.07 |
| Max | 100.0 ± 0.00 | 95.3 ± 2.24 | 98.3 ± 5.22 | 105.0 ± 9.95 | 101.8 ± 8.75 | 72.0 ± 5.58 | ||
| PathHunter (Gqi5) | Hu GPR35 | pEC50 | 6.0 ± 0.34 | 8.5 ± 0.05 | 6.0 ± 0.04 | 5.5 ± 0.10 | 4.9 ± 0.03 | 6.2 ± 0.09 |
| Max | 1.7 ± 0.05 | 1.6 ± 0.05 | 1.7 ± 0.02 | 1.8 ± 0.06 | 1.5 ± 0.03 | 1.6 ± 0.01 | ||
| BRET β-arrestin | Hu GPR35 | pEC50 | 5.6 ± 0,03 | 7.6 ± 0.03 | 5.5 ± 0.04 | 5.5 ± 0.03 | 4.9 ± 0.06 | 4.7 ± 0.04 |
| Max | 100 ± 1.35 | 100.2 ± 1.32 | 96.1 ± 1.81 | 123.0 ± 1.98 | 104.3 ± 4.25 | 134.4 ± 4.10 | ||
| Mu GPR35 | pEC50 | 6.6 ± 0.04 | 4.78 ± 0.05 | <4 | <4 | 4.7 ± 0.16 | 4.0 ± 0.24 | |
| Max | 101.1 ± 1.53 | 61.7 ± 2.52 | — | — | 68.1 ± 7.97 | 109.4 ± 32.7 | ||
| Rat GPR35 | pEC50 | 7.1 ± 0.04 | 5.1 ± 0.08 | <4 | <4 | 4.3 ± 0.09 | 4.9 ± 0.05 | |
| Max | 100.0 ± 1.67 | 36.0 ± 1.53 | — | — | 88.4 ± 8.00 | 116.9 ± 3.8 | ||
| Inositol phosphate (Gαqα13) | Hu GPR35 | pEC50 | 6.9 ± 0.06 | 8.1 ± 0.09 | 6.5 ± 0.06 | 6.8 ± 0.07 | 5.6 ± 0.10 | 6.0 ± 0.10 |
| Max | 99.0 ± 2.87 | 86.0 ± 3.53 | 100.9 ± 3.1 | 84.1 ± 4.63 | 96.8 ± 5.60 | 92.5 ± 5.18 | ||
| Mu GPR35 | pEC50 | 7.9 ± 0.60 | 5.6 ± 0.09 | 4.5 ± 0.14 | <4 | 6.0 ± 0.08 | 5.0 ± 0.16 | |
| Max | 99.3 ± 5.49 | 133.7 ± 6.08 | 197.0 ± 20.7 | — | 107.3 ± 3.80 | 101.4 ± 11.02 | ||
| Rat GPR35 | pEC50 | 8.3 ± 0.42 | 5.6 ± 0.10 | 4.5 ± 0.14 | <4 | 5.8 ± 0.11 | 5. ± 0.93 | |
| Max | 97.9 ± 6.36 | 110.6 ± 6.29 | 197.2 ± 21.3 | — | 92.9 ± 5.81 | 105.5 ± 4.96 |
Hu, human; Mu, mouse.
Given the high potency of compound 1 at human GPR35, we also examined the activity of a pair of closely related analogs. Compound 6 (4-{[-4-oxo-2-(phenylimino)-1,3-thiazolidin-5-ylidene]methyl}benzoic acid) and compound 7 (-[{2-[(4-fluorophenyl)imino]-4-oxo-1,3-thiazolidin-5-ylidene}methyl]benzoic acid) were also both potent agonists at human GPR35 using both PathHunter and BRET-based β-arrestin-2 recruitment assays, but neither displayed higher potency than compound 1 (Fig. 5).
Fig. 5.
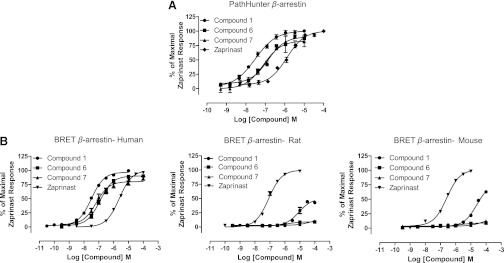
Analogs of compound 1 are also potent agonists of human GPR35. (A) The potency of compound 1 (circles) was compared with that of the related analogs, compounds 6 (squares) and 7 (triangles) as well as zaprinast (diamonds) in the human GPR35 PathHunter β-arrestin recruitment assay. (B) By using the BRET-based β-arrestin-2 interaction assay the potency of these ligands were compared at human, rat, and mouse GPR35. Data are means ± S.E.M., n = 3.
Agonist Activity at GPR35 Rodent Orthologs.
We previously demonstrated marked differences in potency of certain GPR35 agonists when assessed at species ortholog variants. We next assessed, therefore, this possibility for compounds 1–5. By using the BRET-based GPR35-β-arrestin-2 interaction assay, as reported previously (Jenkins et al., 2010, 2012), in this format zaprinast was substantially more potent at rat than human GPR35 and showed intermediate potency at the mouse ortholog (Fig. 6A). Although this assay confirmed the ability of compounds 1, 2, and 3 to recruit β-arrestin-2 via human GPR35, when using either rat or mouse GPR35, both compound 2 and compound 3 were close to inactive at concentrations up to 100 μM and, therefore, at least 100-fold less potent in this assay than at the human ortholog (Fig. 6A). Furthermore, although compound 1 displayed agonist activity at both rodent orthologs at concentrations of 10 μM and above, this compound was some 1000-fold more potent at the human receptor (Fig. 6A). As for compound 1, the related compounds 6 and 7 also displayed poor potency at the rat and mouse orthologs, being essentially inactive at concentrations up to 100 μM (Fig. 5). By contrast, the potency of compound 4 was similar at all three orthologs in this assay (Fig. 6A), whereas compound 5 displayed similar potency at the human and rat orthologs but some 10-fold lower potency at mouse GPR35 (Fig. 6A).
Fig. 6.

Comparison of agonist activity at GPR35 orthologs. Compounds 1–5 as well as zaprinast were employed in both BRET-based β-arrestin-2 recruitment assays (A) and in IP1 accumulation assays (B) conducted in HEK-293T cells transfected to transiently express human, mouse, or rat GPR35. Data are expressed as % of the maximum response to zaprinast. Data represent means ± S.E.M. from three separate experiments performed in duplicate.
The Gαq-i5 Ca2+ elevation assays (Fig. 4) provide a surrogate endpoint for GPR35-mediated activation of Gi-family G proteins (Jenkins et al., 2012). However, GPR35 is also known to selectively activate the G protein Gα13 (Jenkins et al., 2010). Thus to assess engagement of this pathway, we employed IP1 accumulation assays in HEK-293T cells transfected to express an ortholog of GPR35 and the chimeric G protein Gαq-13. This assay again confirmed the rank order of potency of zaprinast as highest at rat and lowest at human GPR35 (Fig. 6B) and, in general, produced results for the newly identified ligands in agreement with the BRET-based assays. For example, compound 4 was of similar potency at all three orthologs, compound 5 was somewhat less potent at mouse GPR35 than at either the human or rat orthologs (Fig. 6B), while compound 3 was some 1000-fold more potent at human than at either rodent ortholog (Fig. 6B).
Despite these similarities, some differences were noted. For example, although compound 2 remained substantially less potent at the rodent versions of GPR35 than at human (Fig. 6B), at the highest concentration tested it did display greater efficacy than zaprinast. It remains unclear if this might be an assay artifact however, as the production of IP1 did not display signs of saturation. Most interestingly, although a potential partial agonist compared with zaprinast at mouse and rat GPR35 in the BRET-based β-arrestin-2 interaction assay (Fig. 6A) and remaining less potent than at human GPR35, compound 1 clearly displayed greater efficacy than zaprinast at the rodent orthologs in the IP1 assay (Fig. 6B).
Mode of Action of Novel GPR35 Agonists.
ML-145 is a high affinity, but highly human selective, blocker of GPR35 (Jenkins et al., 2012). In BRET-based β-arrestin-2 interaction assays using human GPR35, increasing concentrations of ML-145 increased the concentration of compound 1 required to achieve half-maximal effect and did so in a fully surmountable fashion (Fig. 7A). Moreover, ML-145 displayed similar effects when coincubated with either compound 2 (Fig. 7B) or compound 3 (Fig. 7C), also consistent with competitive effects, although the moderate potency of compounds 2 and 3 at human GPR35 made it impossible to demonstrate unambiguously full surmountability of the effect of the antagonist. We also explored the issue of competitivity with ML-145 in the IP1 accumulation assay. Despite the data analysis being affected by the ligand-independent constitutive activity of human GPR35 in this assay (Jenkins et al., 2012) and this being reduced in a concentration-dependent fashion by ML-145 due to the inverse agonist nature of ML-145 (Jenkins et al., 2012), similar conclusions as to the competitive nature of interactions between compounds 1–3 and ML-145 were produced (Fig. 7, D–F).
Fig. 7.
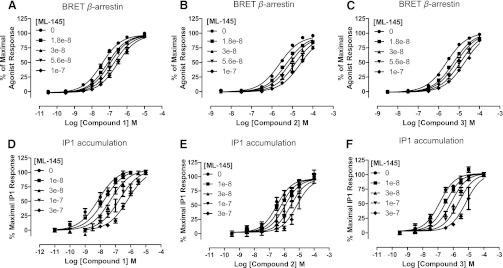
ML-145 is a competitive antagonist of compounds 1–3 at human GPR35. The ability of varying concentrations of ML-145 to alter concentration-response curves to compound 1 (A and D), compound 2 (B and E), and compound 3 (C and F) at human GPR35 was assessed in either BRET-based β-arrestin-2 recruitment (A–C) or IP1 accumulation (D–F) assays. In each case, fitting of the data was consistent with competitive interactions. Data represent means ± S.E.M., n = 3 in each case.
Discussion
Efforts to identify novel chemotypes of ligands at many targets for therapeutic drugs are initiated via HTS campaigns. The poorly characterized G protein-coupled receptor (GPCR) GPR35 has been suggested as a potential target in conditions ranging from cardiovascular disease and metabolic disorders to inflammation and nociception (MacKenzie et al., 2011; Milligan, 2011,). These suggestions have arisen from studies exploring each of tissue distribution, functional studies, and genetic associations of GPR35 with disease (Leonard et al., 2005; Vander Molen et al., 2005; Wang et al., 2006; Min et al., 2010; Ellinghaus et al., 2012). However, understanding the biology of GPR35 has been restricted due to a paucity of high potency/affinity and selective ligands. Furthermore, it remains unclear whether any of the proposed endogenous ligands, including kynurenic acid (Wang et al., 2006) and lysophosphatidic acid (Oka et al., 2010), actually function as the key activator of this receptor in vivo. Largely because of this, the surrogate GPR35 agonist zaprinast has become the standard reference ligand, as it is an effective and reasonably potent activator of GPR35 (Taniguchi et al., 2006; Jenkins et al., 2010, 2011, 2012). However, its use to define the biology of GPR35 is limited by its known action as a PDE V and VI inhibitor (Taniguchi et al., 2006). Moreover, other recently reported GPR35 agonists, although of reasonable potency, have been identified predominantly via screens of small libraries of known, pharmacologically relevant, drugs (Jenkins et al., 2010; Zhao et al., 2010) and/or have known activity at other targets (Yang et al., 2010, 2012; Deng and Fang, 2012; Deng et al., 2012a,b; Hu et al., 2012). Furthermore, certain of these ligands, including the currently described antagonists, display markedly different affinity at human and rodent orthologs of GPR35 (Jenkins et al., 2010, 2012), limiting their potential to define the function of GPR35 in rodent models and in cells derived from these species. To attempt to define novel, distinct chemotypes of GPR35 agonists, we therefore conducted a HTS campaign following initial demonstration that the PathHunter β-arrestin-2 recruitment assay could be used successfully in 384-well format to identify both previously reported and novel GPR35 agonists from a pair of small scale libraries. A 100,000 compound library produced some 550 confirmed actives and, following triage based on potency, chemical tractability, and availability, some 86 compounds within seven broad chemotypes were assessed further. Herein we detailed five distinct exemplars, compounds 1–5, and compared these to the standard ligand, zaprinast. Zaprinast is used routinely as the standard reference agonist ligand for GPR35 and, therefore herein, akin to the situation with well-defined endogenous agonists, efficacy of other ligands has been related to that of zaprinast. However, it should be noted that certain ligands generated greater signal than zaprinast in different assays and, therefore, it should not be assumed that zaprinast is intrinsically a full agonist at GPR35.
β-Arrestin-2 based cellular translocation and receptor interaction methodologies have become central to many HTS efforts at GPCRs (Hamdan et al., 2005; Verkaar et al., 2008), including screens that identified two distinct, but in retrospect highly human selective (Jenkins et al., 2012), GPR35 antagonists (Heynen-Genel et al., 2010). This reflects the suitability of GPR35 in such assays and a view that agonist occupancy of most GPCRs will generate a positive readout (Hamdan et al., 2005). Each of the five selected compounds displayed close to full agonism in the PathHunter β-arrestin-2 recruitment assay, with compound 1 acting as the most potent GPR35 full agonist yet reported. Importantly, each of compounds 1–5 was also able to stimulate intracellular Ca2+ elevation following transfection of the human GPR35 expressing CHO cell line with a Gαq-i5 chimeric G protein (Kostenis et al., 2005; Milligan, 2011), indicating the capacity of these ligands to stabilize a conformation of GPR35 that results in G protein activation as well as β-arrestin-2 interactions. Notably, however, in concentration-response studies, the Hill slope for each agonist was markedly lower in this assay than in the PathHunter β-arrestin-2 recruitment assay. Although we have not explored this in detail it may reflect the very different timings of the assays in which Ca2+ elevation is monitored rapidly and dynamically after ligand addition, a situation that is unlikely to allow ligand equilibrium to be achieved. By contrast the PathHunter β-arrestin-2 assay is based on enzyme-fragment complementation, a relative slow process, and measured 2.5 hours after ligand addition. However, it should be noted that although the BRET-based β-arrestin-2 assay also reports on induced-interactions between agonist-occupied GPR35 and β-arrestin-2 and the observed agonist-induced Hill slopes were very similar in the two β-arrestin-2 interaction assays, the BRET-based assay was conducted only 5 minutes after ligand addition and, hence, in a more similar time frame to the Ca2+ elevation studies. As such, the basis for the variation in Hill slope appears to reflect more than aspects of potential nonequilibrium conditions. Understanding of this will require further analysis but is clearly not related simply to G protein-dependent versus -independent signal pathways as the IP1 accumulations assays, which lie on the same pathway as Ca2+ elevation, also displayed Hill slopes similar to the β-arrestin-2 interaction assays.
To avoid focusing measures of GPR35 activation only toward recruitment of β-arrestin-2 we deliberately also developed and employed a pair of G protein-dependent assays. As we previously showed that GPR35 selectively activates G13 (Jenkins et al., 2011), we wished to monitor downstream effects from this interaction. Direct measures of activation of G13 are both limited and technically challenging (Jenkins et al., 2011). They are also poorly suited to high throughput or the production of concentration-response data. We therefore employed a chimeric G protein methodology. Such assays have been used widely in ligand screens and rely on the selective recognition of the GPCR by the extreme C-terminal α helix of a G protein α subunit (Kostenis et al., 2005). Linking this sequence to the body of a distinct α subunit, in this case Gαq, that engages an effector enzyme that generates an easy to detect and quantify secondary messenger allowed receptor activation of G13 to be converted to elevation of IP1 levels. In a similar vein, Ca2+ elevation as a surrogate monitor of GPR35 activation of Gi-family G proteins was achieved in an equivalent way. By their very nature these are indirect assays, and this must be considered in terms of the pharmacology observed. However, despite some clear examples where this has resulted in unexpected, and potentially confusing, pharmacology (see Kostenis et al., 2005 for review), these approaches are widely used and viewed generally as being robust. Although not the primary focus of the current studies, it is interesting to note potential variation in activity of certain GPR35 agonists in different functional assays, with compound 1 being a case in point. Although markedly less potent and efficacious at rat and mouse GPR35 than the human ortholog in the BRET-based β-arrestin-2 interaction assay, this compound was more efficacious at rat and mouse GPR35 than at human in the IP1 accumulation assay, and the difference in potency between species was substantially less. Such observations suggest that different conformations of the receptor may engage different signaling modalities and, hence, that ligand bias can be noted between different GPR35 agonist ligands. This idea is also supported by a recent comparison of various natural phenols that act as GPR35 agonists (Deng et al., 2012b). For example, when compared with zaprinast, although ellagic acid displayed as high efficacy in a label-free, dynamic mass redistribution assay, it displayed much lower efficacy in a β-arrestin-2 recruitment assay (Deng et al., 2012b). A number of other ligands showed a similar pattern, including niflumic acid, previously identified as a partial agonist at GPR35 in a distinct a β-arrestin-2 recruitment assay (Jenkins et al., 2010). Interestingly, although two further previously identified GPR35 partial agonists, luteolin and quercetin (Jenkins et al., 2010), were partial agonists in both dynamic mass redistribution and β-arrestin-2 recruitment assays (Deng et al., 2012b), no ligands were identified with the opposite bias.
As in other detailed studies (Jenkins et al., 2010, 2012), marked variation in agonist potency between species orthologs was observed for a number of the novel agonists identified, and, importantly, this was largely conserved between assays. As the HTS was then conducted against human GPR35, inherently, each of the five novel ligands selected for detailed profiling has at least moderate potency at human GPR35. However, the three most potent agonists at human GPR35 from the current series, compounds 1, 2, and 3, each displayed very modest, if even detectable, potency at either rat or mouse GPR35. A challenge in defining the biology of GPR35 has been to identify potent ligands, either agonists or antagonists, at the rat or mouse orthologs, and despite the inherent limitations of attempting to use zaprinast to interrogate GPR35 function, this remains the most potent ligand currently described for this pair of rodent orthologs. It is likely that more potent ligands at rat and/or mouse GPR35 could be identified by repeating the HTS but using the rodent orthologs as the target, because a number of nonoverlapping hits were identified previously when screening the Prestwick library against human and rat GPR35 (Jenkins et al., 2010). This may eventually be required. Although the number of atomic level structures of GPCRs has increased substantially in recent times and may be of considerable use in drug discovery (Congreve et al., 2011; Salon et al., 2011), none of these receptors is highly related to GPR35, and, therefore, confidence in homology modeling and possible virtual screening at this target remains low.
Overall these studies demonstrate the utility of HTS based on receptor-β-arrestin interaction technologies to identify novel and potent GPR35 agonists. However, the identification of potential bias in ligand function at this receptor means that initial identification at this level needs to be assessed further in a series of G protein-dependent assays. Moreover, as noted previously, the marked variation in potency and, indeed, even activity at rodent versus human orthologs means that considerable care must be taken to select ligands suitable for rodent-based studies. However, the growing number of reported ligands derived from distinct chemotypes and with potency and affinity differences at human and/or rodent orthologs will now provide an opportunity to define the ligand binding sites and to understand the molecular basis of this pharmacological variation.
Supplementary Material
Abbreviations
- BRET
bioluminescence resonance energy transfer
- CHO–K1
Chinese hamster ovary-K1 cell line
- compound 1
4-{(Z)-[(2Z)-2-(2-fluorobenzylidene)-4-oxo-1,3-thiazolidin-5-ylidene]methyl}benzoic acid
- compound 2
2,2'-bithiophene-5,5′-dicarboxylic acid
- compound 3
4-chloro-3-(2,5-dimethyl-1H-pyrrol-1-yl)benzoic acid
- compound 4
4H-thieno[3,2-b]pyrrole-2-carboxylic acid
- compound 5
9-benzyl-5-methyl-3-(4-methylphenyl)-5,9-dihydro-6H-[1,2,4]triazolo[4,3-e]purine-6,8(7H)-dione
- eYFP
enhanced yellow fluorescent protein
- GPCR
G protein-coupled receptor
- HEK
human embryonic kidney
- HTRF
homogeneous time-resolved fluorescence
- HTS
high-throughput screen
- IP1
inositol monophosphate
- ML-145
{2-hydroxy-4-[4-(5Z)-5-[(E)-2-methyl-3-phenylprop-2-enylidene]-4-oxo-2-sulfanylidene-1,3-thiazolidin-3-yl]butanoylamino]benzoic acid}
- NINDS
National Institute of Neurological Disorders and Stroke
Authorship Contributions
Participated in research design: Neetoo-Isseljee, Mackenzie, Southern, Jerman, McIver, Taylor, Milligan.
Conducted experiments: Neetoo-Isseljee, Mackenzie, Southern, Jerman, Harries.
Contributed new reagents or analytic tools: Southern, McIver, Taylor.
Performed data analysis: Neetoo-Isseljee, Mackenzie, Southern, Jerman, McIver, Harries, Milligan.
Wrote or contributed to the writing of the manuscript: Neetoo-Isseljee, Mackenzie, Southern, Milligan.
Footnotes
This study was supported by the Biotechnology and Biosciences Research Council [Grant 60788] (studentship to A.E.M.).
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Barth MC, Ahluwalia N, Anderson TJ, Hardy GJ, Sinha S, Alvarez-Cardona JA, Pruitt IE, Rhee EP, Colvin RA, Gerszten RE. (2009) Kynurenic acid triggers firm arrest of leukocytes to vascular endothelium under flow conditions. J Biol Chem 284:19189–19195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congreve M, Langmead C, Marshall FH. (2011) The use of GPCR structures in drug design. Adv Pharmacol 62:1–36 [DOI] [PubMed] [Google Scholar]
- Cosi C, Mannaioni G, Cozzi A, Carlà V, Sili M, Cavone L, Maratea D, Moroni F. (2011) G-protein coupled receptor 35 (GPR35) activation and inflammatory pain: Studies on the antinociceptive effects of kynurenic acid and zaprinast. Neuropharmacology 60:1227–1231 [DOI] [PubMed] [Google Scholar]
- Deng H, Fang Y. (2012) Aspirin metabolites are GPR35 agonists. Naunyn Schmiedebergs Arch Pharmacol 385:729–737 [DOI] [PubMed] [Google Scholar]
- Deng H, Hu H, Fang Y. (2012a) Multiple tyrosine metabolites are GPR35 agonists. Sci Rep 2:373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Hu H, Ling S, Ferrie AM, Fang Y. (2012b) Discovery of natural phenols as G protein-coupled receptor-35 (GPR35) agonists. ACS Med. Chem Lett 3:165–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWire SM, Violin JD. (2011) Biased ligands for better cardiovascular drugs: dissecting G-protein-coupled receptor pharmacology. Circ Res 109:205–216 [DOI] [PubMed] [Google Scholar]
- Eglen RM. (2007) Assessing GPCR activation using protein complementation: a novel technique for HTS. Biochem Soc Trans 35:746–748 [DOI] [PubMed] [Google Scholar]
- Ellinghaus D, Folseraas T, Holm K, Ellinghaus E, Melum E, Balschun T, Laerdahl JK, Shiryaev A, Gotthardt DN, Weismüller TJ, et al. (2012) Genome-wide association analysis in sclerosing cholangitis and ulcerative colitis identifies risk loci at GPR35 and TCF4. Hepatology DOI: [published ahead of print]. [DOI] [PubMed] [Google Scholar]
- Fallarini S, Magliulo L, Paoletti T, de Lalla C, Lombardi G. (2010) Expression of functional GPR35 in human iNKT cells. Biochem Biophys Res Commun 398:420–425 [DOI] [PubMed] [Google Scholar]
- Guo J, Williams DJ, Puhl HL, 3rd, Ikeda SR., Sr (2008) Inhibition of N-type calcium channels by activation of GPR35, an orphan receptor, heterologously expressed in rat sympathetic neurons. J Pharmacol Exp Ther 324:342–351 [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Audet M, Garneau P, Pelletier J, Bouvier M. (2005) High-throughput screening of G protein-coupled receptor antagonists using a bioluminescence resonance energy transfer 1-based beta-arrestin2 recruitment assay. J Biomol Screen 10:463–475 [DOI] [PubMed] [Google Scholar]
- Heynen-Genel S, Dahl R, Shi S, Sauer M, Hariharan S, Sergienko E, Dad S, Chung TDY, Stonich D, Su Y, et al.(2010) Selective GPR35 Antagonists - Probes 1 & 2. Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-. 2010 Feb 28 [updated 2010 Oct 4] [Google Scholar]
- Hu H, Deng H, Fang Y. (2012) Label-free phenotypic profiling identified D-luciferin as a GPR35 agonist. PLoS ONE 7:e34934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins L, Alvarez-Curto E, Campbell K, de Munnik S, Canals M, Schlyer S, Milligan G. (2011) Agonist activation of the G protein-coupled receptor GPR35 involves transmembrane domain III and is transduced via Gα₁₃ and β-arrestin-2. Br J Pharmacol 162:733–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins L, Brea J, Smith NJ, Hudson BD, Reilly G, Bryant NJ, Castro M, Loza MI, Milligan G. (2010) Identification of novel species-selective agonists of the G-protein-coupled receptor GPR35 that promote recruitment of β-arrestin-2 and activate Gα13. Biochem J 432:451–459 [DOI] [PubMed] [Google Scholar]
- Jenkins L, Harries N, Lappin JE, Mackenzie AE, Neetoo-Isseljee Z, Southern C, McIver EG, Nicklin SA, Taylor DL, Milligan G. (2012) Antagonists of GPR35display high species ortholog selectivity and varying modes of action. J Pharmacol Exp Ther 343:683–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostenis E, Waelbroeck M, Milligan G. (2005) Techniques: promiscuous Galpha proteins in basic research and drug discovery. Trends Pharmacol Sci 26:595–602 [DOI] [PubMed] [Google Scholar]
- Lattin JE, Schroder K, Su AI, Walker JR, Zhang J, Wiltshire T, Saijo K, Glass CK, Hume DA, Kellie S, et al. (2008) Expression analysis of G Protein-Coupled Receptors in mouse macrophages. Immunome Res 4:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. PE (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445:168–176 [DOI] [PubMed] [Google Scholar]
- Leonard JN, Chu ZL, Unett DJ, Gatlin JE, Gaidarov I, Qiu J, Skinner PJ, and Boatman D (2005), inventors. GPR35 and modulators thereof for the treatment of metabolic-related disorders. U.S. Patent Application G01N33/50.
- Mackenzie AE, Lappin JE, Taylor DL, Nicklin SA, Milligan G. (2011) GPR35 as a novel therapeutic target. Front Endocrinol (Lausanne) 2:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G. (2011) Orthologue selectivity and ligand bias: translating the pharmacology of GPR35. Trends Pharmacol Sci 32:317–325 [DOI] [PubMed] [Google Scholar]
- Min KD, Asakura M, Liao Y, Nakamaru K, Okazaki H, Takahashi T, Fujimoto K, Ito S, Takahashi A, Asanuma H, et al. (2010) Identification of genes related to heart failure using global gene expression profiling of human failing myocardium. Biochem Biophys Res Commun 393:55–60 [DOI] [PubMed] [Google Scholar]
- Ohshiro H, Tonai-Kachi H, Ichikawa K. (2008) GPR35 is a functional receptor in rat dorsal root ganglion neurons. Biochem Biophys Res Commun 365:344–348 [DOI] [PubMed] [Google Scholar]
- Oka S, Ota R, Shima M, Yamashita A, Sugiura T. (2010) GPR35 is a novel lysophosphatidic acid receptor. Biochem Biophys Res Commun 395:232–237 [DOI] [PubMed] [Google Scholar]
- Rajagopal S, Ahn S, Rominger DH, Gowen-MacDonald W, Lam CM, Dewire SM, Violin JD, Lefkowitz RJ. (2011) Quantifying ligand bias at seven-transmembrane receptors. Mol Pharmacol 80:367–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salon JA, Lodowski DT, Palczewski K. (2011) The significance of G protein-coupled receptor crystallography for drug discovery. Pharmacol Rev 63:901–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrimpton AE, Braddock BR, Thomson LL, Stein CK, Hoo JJ. (2004) Molecular delineation of deletions on 2q37.3 in three cases with an Albright hereditary osteodystrophy-like phenotype. Clin Genet 66:537–544 [DOI] [PubMed] [Google Scholar]
- Sun YV, Bielak LF, Peyser PA, Turner ST, Sheedy PF, 2nd, Boerwinkle E, Kardia SL. (2008) Application of machine learning algorithms to predict coronary artery calcification with a sibship-based design. Genet Epidemiol 32:350–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi Y, Tonai-Kachi H, Shinjo K. (2006) Zaprinast, a well-known cyclic guanosine monophosphate-specific phosphodiesterase inhibitor, is an agonist for GPR35. FEBS Lett 580:5003–5008 [DOI] [PubMed] [Google Scholar]
- Taniguchi Y, Tonai-Kachi H, Shinjo K. (2008) 5-Nitro-2-(3-phenylpropylamino)benzoic acid is a GPR35 agonist. Pharmacology 82:245–249 [DOI] [PubMed] [Google Scholar]
- Trinquet E, Bouhelal R, Dietz M. (2011) Monitoring Gq-coupled receptor response through inositol phosphate quantification with the IP-One assay. Expert Opin Drug Discov 6:981–994 [DOI] [PubMed] [Google Scholar]
- Vander Molen J, Frisse LM, Fullerton SM, Qian Y, Del Bosque-Plata L, Hudson RR, Di Rienzo A. (2005) Population genetics of CAPN10 and GPR35: implications for the evolution of type 2 diabetes variants. Am J Hum Genet 76:548–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkaar F, van Rosmalen JW, Blomenröhr M, van Koppen CJ, Blankesteijn WM, Smits JF, Zaman GJ. (2008) G protein-independent cell-based assays for drug discovery on seven-transmembrane receptors. Biotechnol Annu Rev 14:253–274 [DOI] [PubMed] [Google Scholar]
- Wang J, Simonavicius N, Wu X, Swaminath G, Reagan J, Tian H, Ling L. (2006) Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J Biol Chem 281:22021–22028 [DOI] [PubMed] [Google Scholar]
- Yang Y, Lu JY, Wu X, Summer S, Whoriskey J, Saris C, Reagan JD. (2010) G-protein-coupled receptor 35 is a target of the asthma drugs cromolyn disodium and nedocromil sodium. Pharmacology 86:1–5 [DOI] [PubMed] [Google Scholar]
- Yang Y, Fu A, Wu X, Reagan JD. (2012) GPR35 is a target of the loop diuretic drugs bumetanide and furosemide. Pharmacology 89:13–17 [DOI] [PubMed] [Google Scholar]
- Zhao P, Sharir H, Kapur A, Cowan A, Geller EB, Adler MW, Seltzman HH, Reggio PH, Heynen-Genel S, Sauer M, et al. (2010) Targeting of the orphan receptor GPR35 by pamoic acid: a potent activator of extracellular signal-regulated kinase and β-arrestin2 with antinociceptive activity. Mol Pharmacol 78:560–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.