

Abstract
Mitochondrial dysfunction and perturbed degradation of proteins have been implicated in Parkinson's disease (PD) pathogenesis. Mutations in the Parkin and PINK1 genes are a cause of familial PD. PINK1 is a putative kinase associated with mitochondria, and loss of PINK1 expression leads to mitochondrial dysfunction, which increases with time. Parkin is suggested to be downstream of PINK1 and also mediates the removal of damaged mitochondria by macroautophagy (mitophagy). We investigated whether mitochondrial dysfunction in dopaminergic SH-SY5Y cells following decreased PINK1 expression by RNAi may in part be due to the inhibition of mitophagy. Reduced flux through the macroautophagy pathway was found to be coincident with the inhibition of ATP synthesis following 12 days of PINK1 silencing. Overexpression of parkin in these cells restored both autophagic flux and ATP synthesis. Overexpression and RNAi studies also indicated that PINK1 and parkin were required for mitophagy following CCCP-induced mitochondrial damage. The ubiquitination of several mitochondrial proteins, including mitofusin 1 and mitofusin 2, were detected within 3 h of CCCP treatment. These post-translational modifications were reduced following the silencing of parkin or PINK1. The ubiquitination of mitochondrial proteins appears to identify mitochondria for degradation and facilitate mitophagy. PINK1 and parkin are thus required for the removal of damaged mitochondria in dopaminergic cells, and inhibition of this pathway may lead to the accumulation of defective mitochondria which may contribute to PD pathogenesis.
INTRODUCTION
The mechanisms contributing to the pathogenesis of the neurodegenerative disorder Parkinson's disease (PD) are still unclear. Mitochondrial dysfunction and the mishandling/perturbed degradation of proteins have both been implicated in the loss of dopaminergic neurons (1,2).
Deficiency of complex I activity of the mitochondrial electron transport chain (ETC) has been reported in the substantia nigra of PD brains (3), while complex I inhibitors such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and rotenone can induce parkinsonian features in humans and animal models (4,5). High levels of mitochondrial DNA deletions occur in the dopaminergic neurons of the substantia nigra of aged individuals and patients with parkinsonism (6).
The presence of protein inclusions known as Lewy bodies in the surviving dopaminergic neurons of the substantia nigra is a hallmark of PD. The predominant protein found in Lewy bodies is alpha-synuclein, and the perturbed degradation of this protein by the ubiquitin/proteasome system (UPS) and/or lysosomes via chaperone-mediated autophagy or macroautophagy has been proposed to contribute to the formation of these inclusions (2). Decline in the UPS and autophagy occurs with ageing (the greatest risk for developing PD), and inhibition of autophagy has been implicated in several neurodegenerative disorders including PD (2,7,8).
Genes associated with familial forms of PD further support the role of mitochondria and protein quality control in disease pathogenesis (9), in particular, the PINK1, Parkin and ATP13A2 genes, which encode for a mitochondrially targeted serine/threonine kinase, an E3 ubiquitin-protein ligase and a lysosomal ATPase, respectively (10–12).
Loss of PINK1 function in mammalian models has been reported to result in the inhibition of the ETC, increased oxidative stress, changes in mitochondrial calcium homeostasis, increased susceptibility to apoptosis and impaired dopamine release and synaptic plasticity (13–22). PINK1 deficiency in Drosophila also causes loss of mitochondrial integrity and dopaminergic neuronal loss (23,24). The use of PINK1 and Parkin Drosophila models indicated that parkin acts downstream of PINK1 to protect mitochondrial integrity and that they genetically interact with components responsible for the fission and fusion of mitochondria (23–26). These studies suggested that loss of PINK1/parkin function leads to increased mitochondrial fusion. The role of PINK1/parkin in mitochondrial fission and fusion in mammalian cell models is less clear, with reports suggesting increased fission upon loss of PINK1 (27–29) or that increased fission only occurs secondary to impaired oxidative phosphorylation (14,30). Conversely, increased mitochondrial aggregation in PINK1-deficient neurons was reported following proteasomal stress (22).
The presence of PINK1 and parkin in a common signalling pathway and subsequent reports suggesting that they directly interact (31,32) led to the hypothesis that parkin is not only involved in ubiquitinating cytosolic protein substrates for degradation by the UPS, but is also involved in mitochondrial homeostasis. The discovery that parkin translocates from the cytoplasm to depolarised mitochondria and promotes their degradation by the autophagy–lysosome pathway (also termed mitophagy) supported this notion (33). Subsequently, it has been established that PINK1 is required for the recruitment of parkin to depolarized mitochondria in mammalian (34–35) and Drosophila cells (36). The recruitment of parkin to the mitochondria has been reported to result in the ubiquitination of the mitochondrial proteins VDAC1 (35) and mitofusin (36), in mammalian and Drosophila cells, respectively.
Previously, we have reported that silencing of PINK1 in the human dopaminergic neuroblastoma SH-SY5Y cell line results in mitochondrial dysfunction and oxidative stress, which increases with time (13). Differentiated neurons with PINK1 deficiency also exhibit increasing oxidative stress with time and a significant increase in mitochondrial mass 30 days post-silencing (16). A progressive inhibition of mitochondrial respiration, accumulation of abnormal mitochondrial morphology and increased oxidative stress have also been reported in two mice PINK1 knockout models (20,22). Our hypothesis is that PINK1 deficiency results in the decreased removal of damaged mitochondria by mitophagy, causing mitochondria to accumulate, resulting in an increased production of reactive oxygen species that will further exacerbate mitochondrial damage.
We report here that the inhibition of ATP synthesis following long-term PINK1 silencing correlates with reduced flux through the autophagy–lysosome pathway. Overexpression of parkin restores both flux and ATP synthesis. The involvement of PINK1 and parkin in mitophagy was further investigated in an acute model of mitochondrial damage [carbonyl cyanide-m-chlorophenylhydrazone (CCCP) dissipation of mitochondrial membrane potential]. An increase in the ubiquitination of a number of mitochondrial proteins correlates with the induction of mitophagy, including the fusion factors of the outer membrane, mitofusins 1 (MFN-1) and 2 (MFN-2). These post-translational modifications are reduced following either PINK1 or parkin silencing. Our results indicate that the ubiquitination of MFN-1 and MFN-2 precedes the removal of damaged mitochondria and is thus an early event in mitophagy. The role of ubiquitination in facilitating mitophagy is discussed, including a putative effect on mitochondrial fission, which is required for mitophagy to proceed (37).
RESULTS
Silencing of PINK1 in SH-SY5Y cells has previously been shown to inhibit ATP synthesis (13). Since parkin has been reported to be downstream of PINK1, the effect of overexpression of parkin on ATP synthesis was investigated. PINK1 silencing in SH-SY5Y cells significantly decreased mitochondrial oxidative phosphorylation using all three substrates used in agreement with our previous findings (Fig. 1A; P < 0.05) (13). However, oxidative phosphorylation was not decreased in PINK1-silenced cells with increased parkin expression (Park OE cells). Similar results were achieved using a different combination of PINK1 siRNA sequences (Supplementary Material, Fig. S1). These results suggest that parkin is able to negate the effect of PINK1 deficiency consistent with parkin acting downstream of PINK1.
Figure 1.

ATP synthesis and autophagy flux after 12 days of PINK1 silencing. SH-SY5Y cells or Park OE cells were transfected with PINK1 siRNA #2 or control siRNA for 12 days. (A) Cells were harvested by trypsinization, permeabilized with digitonin and incubated with glutamate + malate (complexes I, III, IV), succinate + rotenone (complexes II, III, IV) or ascorbate + TMPD (complex IV) at 37°C to measure ATP synthesis. ATP was measured by a luciferase assay and normalized to cell number. Data are expressed as % control siRNA ATP synthesis (mean ± SEM; n = 5). *P < 0.05 versus control siRNA. (B) Cell lysates were prepared from untreated cells (basal) or cells treated with the lysosomal inhibitors E64d and pepstatin A (both 10 μg/ml) for 3 h. Western blots were probed with LC3 antibody and equal protein loading was assessed using an antibody against β-actin. The density of bands was measured and the LC3-II/LC3-I ratio was normalized to β-actin. Data are expressed as % of control siRNA-treated cell lines under basal conditions or in the presence of lysosomal inhibitors (n = 5). *P < 0.05 versus ParkSH + siRNA under basal conditions.
Since parkin has been shown to be involved in the removal of damaged mitochondria (33,34), the induction of macroautophagy was investigated in PINK1-silenced cells. Basal macroautophagy in SH-SY5Y cells treated with PINK1 siRNA for 12 days was assessed by measuring the post-translational modification of the LC3 protein. Upon activation of autophagy, LC3 undergoes cleavage to form LC3-I and then conjugation to phosphatidylethanolamine to form LC3-II, which then binds to the autophagosome that envelops the cargo for degradation. This process was followed by western blot and basal autophagy determined by measuring the LC3-II/LC3-I ratio normalized to β-actin (38). Following PINK1 silencing, the LC3-II/LC3-I ratio, and thus basal autophagy, was decreased by 35% (P < 0.05; n = 5), when compared with SH-SY5Y cells treated with scrambled control siRNA (Fig. 1B). Whereas cells overexpressing parkin (Park OE) had LC3-II/LC3-I ratios similar to control cells, PINK1 knockdown in these cells caused an even greater decrease (60%) in the LC3-II/LC3-I ratio level (P < 0.05; n = 6), when compared with SH-SY5Y cells overexpressing parkin (Park OE) treated with control siRNA (Fig. 1B). This suggests that the steady-state levels of LC3-II are decreased in both normal and parkin overexpressing cells with decreased PINK1 expression.
Although western blot analysis of LC3-II gives an indication of the level of autophagic vesicles, to understand the impact on the rate of autophagy, it is important to identify if the flux of LC3-II through the autophagy pathway has been altered. To measure the flux through the autophagy–lysosome pathway, LC3-II levels were measured in the presence of the lysosomal inhibitors pepstatin A and E64d (38). Under these conditions, LC3-II levels in Park OE cells with depleted PINK1 were now similar to control and were significantly increased (P < 0.05; n = 5), when compared with Park OE cells with PINK1-silencing under basal conditions (Fig. 1B). Conversely, treatment of PINK1-silenced SH-SY5Y cells with lysosomal inhibitors still resulted in decreased LC3-II levels, when compared with cells treated with control siRNA (Fig. 1B). These results indicate that PINK1 silencing leads to decreased autophagy flux and this phenomenon was reversed by the overexpression of parkin. Since ATP synthesis was not inhibited in PINK1-silenced cells overexpressing parkin, this implies that the decrease in autophagy in PINK1-silenced cells might be a result of the inhibition of mitophagy, which results in an accumulation of damaged mitochondria.
To investigate further the role of PINK1 and parkin in mitophagy, an acute model of mitochondrial damage was used. SH-SY5Y cells were treated with 10 μm CCCP to dissipate the mitochondrial membrane potential and induce mitophagy as previously described (33,34). The decrease in mitochondrial content over time was measured by assaying the activity of citrate synthase (CS), a citric acid cycle enzyme used to measure mitochondrial mass in cells and tissue (16,39–41). CS activity is generally unaffected upon significant mitochondrial dysfunction such as the inhibition of the ETC (40) or the depletion of mtDNA (41), provided there is no accompanying cell death. Therefore, any changes in the activity are typically indicative of mitochondrial content (16,39). Treatment with CCCP resulted in a significant decrease in mitochondrial mass in both normal SH-SY5Y cells containing an empty vector and parkin overexpressing cells (P < 0.01; n ≥ 3) (Fig. 2A). To confirm that the 79% decrease in CS activity in Park OE cells after 24 h of CCCP treatment was due to loss of mitochondria, the protein levels of two mitochondrial proteins were measured by western blot (Fig. 2B). Expression of both TFAM (encoded by the nucleus) and MTCOII (encoded by the mitochondria) were decreased.
Figure 2.

CCCP-induced mitophagy requires parkin and PINK1 expression. (A) SH-SY5Y cells containing an empty vector (SH) or Park OE cells were treated with vehicle [0.05% (v/v) ethanol] or 10 μm CCCP, lysed and CS activity assessed. Data are expressed as % CS activity of vehicle-treated cells (mean ± SEM; n = 5). *P < 0.05 versus vehicle and **P < 0.01 versus vehicle. (B) Cells were treated with vehicle or CCCP for 24 h, lysed and mitochondrial protein expression (TFAM, MTCOII) assessed by western blot. Equal loading was assessed by probing for GAPDH. Blot representative of four separate experiments. (C) Normal SH-SY5Y cells (control), parkin KD cells or Park OE cells were treated with vehicle or CCCP for 16 h, lysed and CS activity measured. Data are expressed as nmol/min/mg protein (mean ± SEM; n = 5). *P < 0.05 versus vehicle and **P < 0.01 versus vehicle. (D) SH-SY5Y cells or Park OE cells were treated with PINK1 siRNA or control siRNA for 72 h. For the last 16 h, cells were treated with vehicle or CCCP. CS activity was then measured. Data are expressed as nmol/min/mg protein (mean ± SEM; n = 5). *P < 0.05 versus control siRNA + CCCP and **P < 0.01 versus control siRNA + CCCP.
The influence of parkin levels on CCCP-induced mitochondrial degradation was studied in SH-SY5Y cells with decreased levels of endogenous parkin levels using the constitutive expression of a shRNA targeted against parkin (Parkin KD; Supplementary Material, Fig. S2), normal parkin levels (control) and cells overexpressing parkin (Park OE). After 16 h of exposure to CCCP, there was a clear progression in the decrease in mitochondrial content as parkin levels increased (Fig. 2C). Similar to that observed in parkin-depleted cells, the silencing of PINK1 expression for 72 h using two independent siRNA combinations partially decreased the CCCP-induced mitochondrial loss in normal SH-SY5Y cells (Fig. 3D, P < 0.05). This observation was more dramatic in Park OE cells with depleted PINK1 (P < 0.01; Fig. 2D), confirming that PINK1 plays a key role in parkin-mediated removal of depolarized mitochondria.
Figure 3.
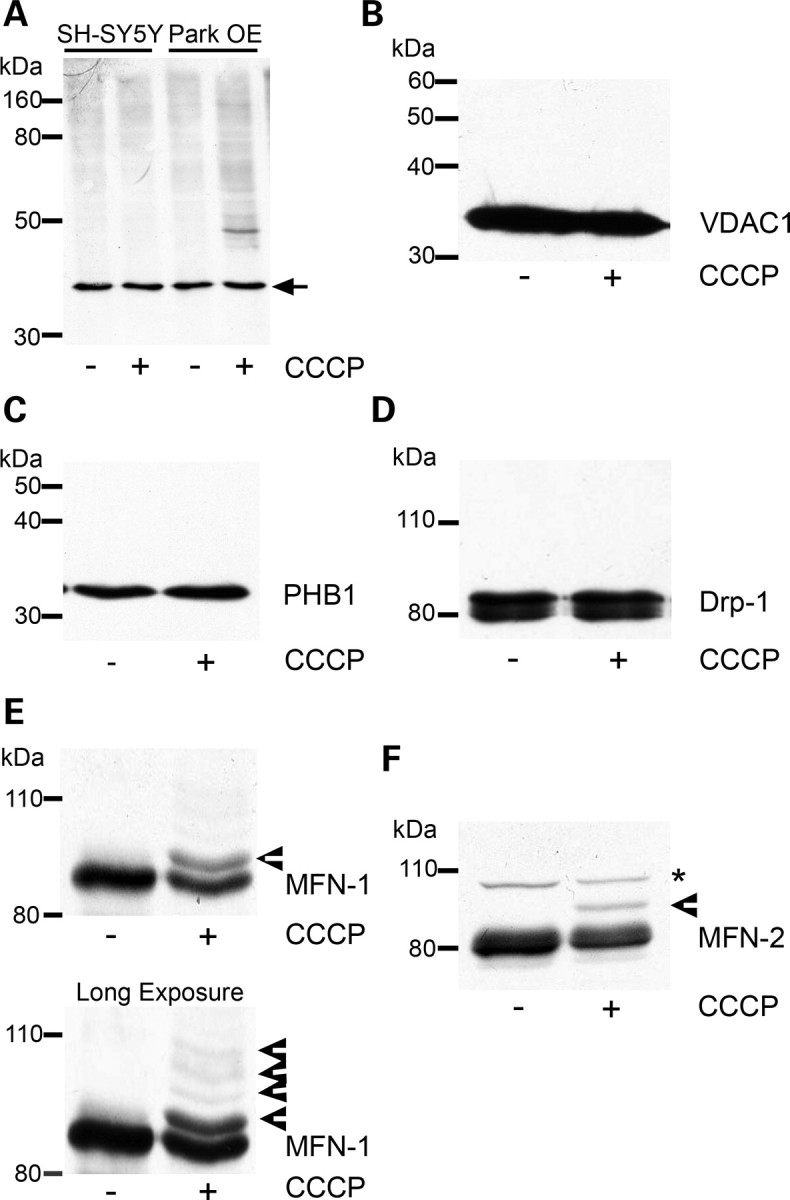
Ubiquitination of mitochondrial proteins following CCCP treatment. (A) SH-SY5Y cells containing an empty vector or Park OE cells were treated with vehicle [0.05% (v/v) ethanol] or 10 µm CCCP for 3 h, mitochondria isolated and a western blot performed. The western blot was probed with an antibody against ubiquitin. Equal loading was determined by probing for the 39 kDa subunit of complex I (arrow). Blot representative of four separate experiments. (B) Park OE cells were treated with CCCP for 3 h, mitochondria isolated and probed for VDAC1 or (C) prohibitin (PHB1) by western blot. (D) Park OE cells were treated with CCCP for 3 h and cell lysates probed for Drp1 by western blot. (E) Mitochondria isolated from Park OE cells treated with vehicle or CCCP for 3 h were probed for MFN-1 or (F) MFN-2 by western blot. Post-translationally modified proteins are indicated by arrowheads. Non-specific band is denoted by an asterisk. Blots are representative of at least four separate experiments.
Parkin is an E3-ubiquitin ligase and has been suggested to be recruited to damaged mitochondria. To investigate whether this is associated with the ubiquitination of specific proteins, mitochondria were isolated from SH-SY5Y containing an empty vector and Park OE cells following 3 h of CCCP treatment. This timepoint was chosen to allow the recruitment of parkin to mitochondria (33,34) but in the absence of significant mitochondrial loss (Fig. 2A). CCCP treatment increased the ubiquitination of mitochondrial proteins in Park OE cells (Fig. 3A), which was not apparent in normal cells. A prominent band was detected with a molecular mass of approximately 50 kDa. The identity of this band is unknown. Several candidates have been investigated by western blot including VDAC1 and prohibitin (Fig. 3B and C). No bands of higher molecular weight indicative of ubiquitination were detected for either protein following CCCP treatment for 3 h. The analysis of LC3-II levels in SH-SY5Y and Park OE cell lysates indicates that the autophagy–lysosomal pathway had been induced at this timepoint (Supplementary Material, Fig. S3).
The dynamics of fission and fusion of mitochondria are inter-related with mitochondrial turnover and under the control of a variety of proteins including Drp-1, MFN-1 and MFN-2 (37,42–44). As potential ubiquitination substrates during mitophagy, mitochondrial preparations from CCCP-treated Park OE cells were probed with antibodies against Drp-1, MFN-1 and MFN-2. Mitofusin (also known as Marf), the Drosophila homologue of MFN-1/2, has been reported to be ubiquitinated following CCCP treatment of Drosophila S2R+ cells (36). Although there was no evidence for bands representing modified Drp-1 (Fig. 3D), both MFN-1 and MFN-2 had additional bands of higher molecular weight (Fig. 3E and F, arrowheads). These band shifts are consistent with monoubiquitination of MFN-1 (∼ 8.5 kDa) and diubiquitination of MFN-2 (∼16 kDa). Longer exposures indicated further high-molecular-weight bands for MFN-1 (Fig. 3E), which suggests that MFN-1 does undergo multiple ubiquitination modifications upon mitophagy induction.
The putative ubiquitination of MFN-1 and MFN-2 upon CCCP treatment also occurred in SH-SY5Y cells containing an empty vector following CCCP treatment (Fig. 4A). A time course indicated that the putative ubiquitination of both MFN-1 and MFN-2 differed in cell lysates from SH-SY5Y cells containing an empty vector (Fig. 4A), when compared with Park OE cells (Fig. 4B). It should be noted that the multiple post-translational modifcation species of MFN-1 were difficult to differentiate from non-specific bands when cell lysates were used for western blots. Therefore, only the putative monoubiquitinated MFN-1 and diubiquitinated MFN-2 proteins are shown. For both MFN-1 and MFN-2, putative ubiquitination tended to peak after 1 h of CCCP treatment in Park OE cells and then diminish over time (Fig. 4B; Supplementary Material, Fig. S4A and B). Analysis of band density and calculation of the ratio of ubiquitinated MFN-1 or MFN-2 (MFN-1/2 ubq) to total MFN-1 or MFN-2 levels (unmodified + ubiquitinated MFN-1/2) confirmed this observation (Supplementary Material, Fig. S5). A similar trend was observed in a different SH-SY5Y clone overexpressing parkin (Supplementary Material, Fig. S4F). In SH-SY5Y cells containing an empty vector, the putative ubiquitination of MFN-1 and MFN-2 took longer to peak (2 h) and did not show a noticeable decrease after 3 h (Fig. 4A; Supplementary Material, Figs S4A and B and S5). Shorter exposures show that protein levels of unmodified MFN-1 and MFN-2 tended to decrease over time, particularly in Park OE cells (Supplementary Material, Fig. S4A and B). Measurement by western blotting of the mitochondrial protein succinate dehydrogenase subunit A (SDHA) indicated that the mitochondrial content was not noticeably decreased in these samples. Previously, post-translationally modified MFN-1 and MFN-2 were detected in mitochondria isolated from Park OE cells following 3 h of CCCP treatment (Fig. 3E and F). Comparison of mitochondria isolated from SH-SY5Y cells containing an empty vector and Park OE cells after 3 h of CCCP treatment confirmed that the putative ubiquitination of MFN-1 and MFN-2 in Park OE cells was lower than in SH-SY5Y cells containing an empty vector at this timepoint (Supplementary Material, Fig. S4D and E).
Figure 4.

Ubiquitination of MFN-1 and MFN-2 following CCCP treatment. (A) SH-SY5Y cells containing an empty vector or (B) Park OE cells were treated with 10 µm CCCP for 0–3 h, cell lysates prepared and MFN-1 and MFN-2 post-translational modifications assessed by western blot using antibodies against MFN-1 and MFN-2. Blots were re-probed with an antibody against the mitochondrial protein SDHA, to assess mitochondrial content in each lysate. (C) Mitochondria were isolated from SH-SY5Y cells containing an empty vector treated with vehicle or 10 µm CCCP for 2 h. Endogenous MFN-1 was immunoprecipitated from mitochondrial fractions and ubiquitinated species detected by western blotting. The blot was then re-probed with MFN-1 antibody. (D) MFN-2 was immunoprecipitated from mitochondrial fractions and ubiquitination detected as above. Blots are representative of at least three separate experiments. Ubiquitinated species are denoted by arrowheads.
To confirm that the post-translational modification observed following CCCP treatment was ubiquitination, MFN-1 and MFN-2 were immunoprecipitated from mitochondria and ubiquitinated protein detected by western blot. In accordance with previous findings (Fig. 3E and F), samples of immunoprecipitated MFN-1 displayed several species of greater molecular mass (Fig. 4C), whereas immunoprecipitated MFN-2 indicated two species of increased molecular mass (Fig. 4D). Ubiquitinated MFN-1 and MFN-2 species were exclusively detected in mitochondria isolated from CCCP-treated cells. The specificity of the immunoprecipitation was confirmed with MFN-1 and MFN-2 antibodies (Fig. 4C and D). MFN-1 and MFN-2 species with increased molecular mass were also detected by these antibodies and had a pattern similar to blots probed with the ubiquitin antibody. These results suggest that MFN-1 is subject to multiple ubiquitination modifications. Two ubiquitinated species were also detected for endogenous MFN-2 (Fig. 4D). Re-probing of the blot with an antibody against MFN-2 initially picked up the di-ubiquitinated species. Longer exposures indicated that the tri-ubiquitinated species was also detected (Supplementary Material, Fig. S6).
In order to confirm that the ubiquitination of MFN-1 and MFN-2 was parkin-dependent, SH-SY5Y cells or SH-SY5Y cells expressing parkin shRNA (Parkin KD) were treated with 10 μm CCCP for 2 h and western blots of cell lysates probed for MFN-1 (Fig. 5A) and MFN-2 (Fig. 5B). The intensity of the post-translational modified MFN-1 and MFN-2 was decreased in cells with parkin deficiency. The ubiquitination of MFN-1 and MFN-2 after 2 h in SH-SY5Y cells containing an empty vector precedes significant loss of mitochondria as demonstrated by the CS activity (Fig. 2A) and equal levels of SDHA (Fig. 5A and B).
Figure 5.

Parkin and PINK1 are required for ubiquitination of MFN-1 and MFN-2. (A) Control or Parkin KD cells were treated with vehicle or CCCP for 2 h, cell lysates prepared and MFN-1 or (B) MFN-2 ubiquitination assessed by western blot using antibodies against MFN-1 and MFN-2. (C) SH-SY5Y cells were untransfected (UT), transfected with PINK1 siRNA #1 or PINK1 siRNA #2 or transfected with control siRNA for 72 h. Untransfected cells and siRNA transfected cells were then treated with 10 µm CCCP for 2 h, cell lysates prepared and separated by western blot. Blots were probed with an antibody against MFN-1 or (D) MFN-2. Blots were re-probed with an antibody against SDHA for mitochondrial content. Blots are representative of at least three separate experiments. Ubiquitinated species are denoted by arrowheads.
Since PINK1 has been suggested to recruit parkin to depolarized mitochondria (34) and can inhibit mitophagy in SH-SY5Y cells following PINK1 silencing (Fig. 2D), we investigated whether the silencing of PINK1 decreased the parkin-dependent ubiquitination of MFN-1 and MFN-2. SH-SY5Y cells containing an empty vector were untransfected (UT), transfected with PINK1 siRNA #1 or PINK1 siRNA #2 or control siRNA for 72 h. Cells were then treated with 10 μm CCCP for 2 h. Ubiquitination of MFN-1 (Fig. 5C) or MFN-2 (Fig. 5D) was induced in untransfected cells and cells transfected with control siRNA. However, ubiquitination of MFN-1 and MFN-2 was significantly reduced when treated with either PINK1 siRNA combination. These results suggest that PINK1 is required for the ubiquitination of MFN-1 and MFN-2.
DISCUSSION
Mitochondrial dysfunction and impaired protein metabolism have been implicated in PD pathogenesis. In this paper, we report that PINK1 and parkin are involved in the degradation of damaged mitochondria in dopaminergic cells. Perturbation of this pathway by silencing of PINK1 led to mitochondrial dysfunction and decreased basal autophagy flux. Overexpression of parkin restored autophagy flux and mitochondrial ATP synthesis suggesting that the mitochondrial dysfunction observed in several PINK1 cell and animal models is connected, at least in part, with impaired mitophagy. The ubiquitination of a large number of mitochondrial proteins occurs soon after mitophagy induction. MFN-1 and MFN-2 were two such proteins, and their ubiquitination was dependent on the presence of PINK1 and parkin.
The involvement of both PINK1 and parkin in CCCP-induced mitophagy has been reported in a variety of cell models (33–36,45). In agreement with our findings, ubiquitination of mitochondrial proteins upon induction of mitophagy has been observed (45,46) including VDAC in mammalian cells (35) and mitofusin in Drosophila (36). Mammalian cells have two mitofusins, MFN-1 and MFN-2, and in this report, we observed that both of these proteins are ubiquitinated following CCCP treatment. To the authors’ knowledge, this is the first report of human mitofusins undergoing covalent modification. The speed of the post-translational modification of MFN-1 and MFN-2 was similar; however, the pattern of ubiquitination appears to differ. The ubiquitination of both these proteins was dependent on parkin and PINK1. These findings concur with Ziviani et al. (36) who reported that the parkin-dependent ubiquitination of the Drosophila homologue mitofusin resulted in the addition of one, three or four ubiquitin adducts. Overexpression of parkin also restored the decreased ubiquitination of mitofusin following PINK1 silencing (36). Immunoprecipitation studies have indicated that parkin binds mitofusin in Drosophila cells (36), even under basal conditions (47). In the latter study, mitofusin was found to be modified by three ubiquitin moieties and was dependent on both PINK1 and parkin (47). Despite evidence from both mammalian and Drosophila cells that the ubiquitination of MFN-1 and MFN-2 is parkin-dependent, in vitro ubiquitination studies with recombinant/purified proteins are needed to definitively confirm that MFN-1 and MFN-2 are direct substrates of parkin. Indeed, it is unclear to which extent the large number of mitochondrial proteins ubiquitinated following CCCP treatment reported here and by others (46) is a direct result of parkin or the activation of other cytosolic or mitochondrial ubiquitin ligases such as MARCH5 and MULAN (48).
The type of linkage and number of ubiquitin moieties added to a particular cargo determines whether the substrate is destined for degradation by the proteasome or autophagy (49,50). Typically, polyubiquitination linked via Lys48 of ubiquitin targets the protein to the proteasome, whereas monoubiquitination or oligomeric Lys63-linked ubiquitin chains target proteins to lysosomes. There is also growing evidence that ubiquitination of proteins may also affect protein function, similar to other post-translational modifications such as phosphorylation (44,48). Therefore, the type of ubiquitination and the function of the mitochondrial proteins most likely determine the role they play in mediating mitophagy. Further work is required to determine whether the post-translational modification of MFN-1 and MFN-2 is due to polyubiquitination or multiple monoubiquitination events. The ubiquitination of MFN-1 and MFN-2 may act as tags to identify damaged mitochondria (51,52) and recruit the ubiquitin-binding autophagic receptors p62 and HDAC6 (34,46), and subsequently autophagosome components such as LC3-II (49).
For mitophagy to occur, the mitochondrial reticular network needs to first undergo fission (37). An appealing hypothesis is that the ubiquitination of MFN-1 and MFN-2 could be a means of preventing mitochondrial fusion, either by promoting degradation of these proteins by the proteasome or by physically interfering with the formation of MFN dimers between mitochondria (51,52). The degradation of the yeast homologue Fzo1 is dependent on the UPS (53). Furthermore, loss of PINK1 or parkin in Drosophila resulted in an increased abundance of mitofusin (36,47). Unmodified MFN-1 and MFN-2 protein levels appear to be slightly decreased following 3 h of CCCP treatment, and this occurs before a significant loss of mitochondrial number is observed. This might suggest that the mitofusins are being degraded by the proteasome or that a fraction of the mitofusins have undergone multiple ubiquitination events. It should be noted that MFN-2 is modified by two or three ubiquitin molecules, whereas degradation of proteins by the proteasome generally requires more than four (54). Decreases in MFN-1 and MFN-2 protein levels seen at later timepoints are difficult to interpret as it is unclear whether this is due to degradation by the proteasome and/or loss of whole mitochondria by mitophagy.
Given the putative role of MFN-1/2 in mitophagy discussed above, the analysis of mitochondrial morphology and dynamics needs to be assessed upon the induction of mitophagy by CCCP or paraquat (33,36), in the absence or presence of PINK1/parkin. Similar studies are required for the 12-day PINK1 silencing cell models. Previously, no overt changes in the mitochondrial network (i.e. large-scale fragmentation of the mitochondrial network) were seen following transient transfection of PINK1 for 12 days (13), unlike other models (27,29). More quantitative analyses such as changes in mitochondrial branching and form factor might indicate whether fission/fusion is being affected in our cell models.
In addition to promoting fusion of mitochondria, several other cellular processes have been ascribed to MFN-2, and two of these could be relevant for mitophagy. First, MFN-2 connects mitochondria with the endoplasmic reticulum (55), and for mitophagy to occur, it will be necessary to sever this connection. Recently, MFN-2 has also been found to be necessary for the transport of mitochondria and interacts with the Miro/Milton complex (56). Mitochondria destined for degradation will need to be transported to lysosomes/aggresomes.
Drp-1 was not found to be ubiquitinated, which is consistent with previous reports (36,47). Indeed, the ubiquitination of Drp1 is thought to negatively regulate fission (44,48) and would thus impede mitophagy if modified. It should be noted that sumoylation of Drp1 promotes fission (48). Although PINK1/parkin-mediated mitophagy has focused on ubiquitination, other means of post-translational regulation (sumoylation, phosphorylation) of mitophagic proteins need to be investigated in the PINK1/parkin pathway.
In addition to their role in mitophagy, the function of PINK1 and parkin in macroautophagy as a whole should be considered. PINK1 has been reported to enhance basal and starvation-induced autophagy and interacts with the pro-autophagic protein Beclin-1 (57). Therefore, the reduced autophagic flux we observed in SH-SY5Y cells after PINK1 silencing might not be completely due to the inhibition of mitophagy. We also found that the induction of autophagy (LC3-II levels) in SH-SY5Y cells overexpressing parkin upon starvation was greater than in SH-SY5Y cells containing an empty vector. A recent study has reported that the stable knockdown of PINK1 by RNAi activates autophagy/mitophagy (28). However, in keeping with the results reported here and by others (33–36), parkin overexpression in this cell model did increase mitophagy. Perhaps, chronic PINK1 knockdown in mammalian cells activates compensatory pathways able to promote mitophagy in the absence of PINK1 protein, suggesting that other proteins can recruit parkin to depolarized mitochondria. Stable knockdown of PINK1 also caused a significant fragmentation of the mitochondrial network (28), a scenario we do not observe following 12 days of transient PINK1 transfection (13). Since fission is required for mitophagy (37), perhaps this is also a driving force for mitophagy in the stable knockdown model (28).
Mitophagy is likely to be particularly important in post-mitotic cells such as neurons, as the damaging effect of dysfunctional mitochondria cannot be diluted upon cell division as with proliferating cells. Instead, damaged mitochondria will accumulate, resulting in energy deficiency and increasing oxidative stress. These events will further damage mitochondria and other macromolecules, causing an ever-increasing spiral of damage. Both mitochondrial performance and efficient protein degradation diminish with age. Ageing is the greatest risk factor for PD. It is notable that chaperone-mediated autophagy markers, the predominant route for the degradation of alpha-synuclein, are decreased in PD substantia nigra and amygdala compared with matched control or Alzheimer disease brains (58). High levels of mitochondrial DNA deletions also occur in the dopaminergic neurons of the substantia nigra of aged individuals and patients with parkinsonism (6). Animal and cell models of PINK1 deficiency exhibit increasing mitochondrial damage and oxidative stress with time (13,20,22). We have shown that restoration of autophagy in PINK1-silenced cells by parkin reverses ATP synthesis inhibition and that the ubiquitination of MFN-1 and MFN-2 play a role in PINK1/parkin-mediated mitophagy.
MATERIALS and METHODS
Cell culture
The human neuroblastoma cell line SH-SY5Y was cultured in 1:1 (v/v) DMEM (no glucose):F12 (Ham) media (0.9 g/l glucose final concentration) supplemented with 10% fetal bovine serum, 1 mm pyruvate and penicillin–streptomycin. SH-SY5Y cells containing an empty vector or parkin cDNA (Park OE; pcDNA 3.1 vector with haemaglutinin tag; Invitrogen) were also used (13). To generate SH-SY5Y with constitutive parkin silencing (Parkin KD cells), cells were transfected with a shRNA vector (psi STRIKE; Promega) encoding Parkin shRNA (5′-CACCUGAUCGCAACAAAUUUCttcaagagaATTTGTTGCGATCAGGTGC-3′) and stable clones selected for by resistance to the antibiotic G418. Empty vector, Park OE and Parkin KD cells were cultured in the media described above supplemented with 20 μg/ml of G418.
Transient transfection of SH-SY5Y cells with PINK1 siRNA
SH-SY5Y cells or SH-SY5Y overexpressing parkin (1.8 × 105 cells/ml) were transfected with two different pairs of PINK1 siRNA [10 nM; PINK1 #1 (Dharmacon) or PINK1 #2 (Qiagen)] or 10 nm scrambled control siRNA (Ambion) using HiPerfect transfection reagent (Qiagen). As previously reported, PINK1 mRNA levels were depleted by approximately 80% after 72 h of silencing with either pair of PINK1 siRNAs (13). Cells were transfected with siRNA every 3 days, up to a maximum of 12 days.
CS activity
Following treatment with 10 μm CCCP, cells were washed once with PBS and lysed on the plate in 0.25% (v/v) Triton X-100 in PBS supplemented with protease and phosphatase inhibitors. Debris was removed by centrifugation and CS activity measured by following the oxidation of 5,5′-Dithiobis(2-nitrobenzoic acid) in a spectrophotometer (absorbance at 412 nm) over time at 30°C in the presence of acetyl co-enzyme A and oxaloacetate (59). Protein concentration in the same aliquot was measured using the BCA protein assay (Pierce) and enzyme activity expressed as nmol/min/mg protein.
Western blotting
SH-SY5Y cells were harvested with trypsin and lysed on ice in 1% (v/v) Triton X-100 in PBS supplemented with protease and phosphatase inhibitors. Triton soluble fractions (10 μg) were resolved by SDS–PAGE and transferred to Hybond P (GE Healthcare). Blots were probed with antibodies against the 39 kDa subunit of complex I (clone 20C11; Mitosciences), β-actin (ab8227; Abcam), GAPDH (clone 6C5; Abcam), LC3 (cat. # 2775; Cell Signaling Technology), SDHA (clone 2E3; Mitosciences), subunit MTCO2 of complex IV (clone 12C4; Mitosciences), parkin (clone PRK8; Santa-Cruz Biotechnology), ubiquitin (clone P4D1; Santa-Cruz Biotechnology), Drp1 (also known as DLP1; clone 8/DLP1; BD Biosciences) and TFAM (Clone K-18, Santa-Cruz Biotechnology). Rabbit polyclonal antibodies against MFN-1 (DUP antibody) and MFN-2 (CT antibody) have previously been described (60,61). Bands were detected with respective horse radish peroxidase-linked secondary antibodies (Dako) and enhance chemiluminescence (Pierce). The density of bands was determined by the AlphaDigiDoc software.
Isolation of mitochondria
Following CCCP treatment, SH-SY5Y cells (>30 × 106 cells) were harvested by trypsinization, washed in PBS supplemented with protease and phosphatase inhibitors and homogenized with a glass-teflon homogenizer in isolation medium (250 mm sucrose, 1 mm EDTA, 10 mm Tris, pH 7.4, supplemented with protease and phosphatase inhibitors). Nuclei were removed by centrifugation (1500g) and mitochondria pelleted at 11 800g and resuspended in isolation medium.
Immunoprecipitation of MFN-1 and MFN-2
Mitochondria were isolated from SH-SY5Y cells containing an empty vector treated with vehicle (ethanol) or 10 μm CCCP for 2 h. Mitochondria were lysed in 0.5% (v/v) Triton X-100 in PBS supplemented with protease inhibitors, MFN-1 or MFN-2 antibodies added and rotated at 4°C for 2 h. Protein A sepaharose (GE Healthcare) was then added and the mixture rotated at 4°C for a further 1.5 h. The antibody bound beads were washed three times with 1% (w/v) BSA in PBS, two times with PBS and then resuspended in 2× Laemmlli buffer containing 0.1 m DTT. The supernatant was then separated by SDS–PAGE and transferred to HybondP for western blotting.
ATP synthesis assay
SH-SY5Y cells or SH-SY5Y cells overexpressing parkin were treated with PINK1 siRNA or scrambled siRNA for 12 days, cells harvested with trypsin, permeabilized with digitonin and ATP synthesis measured in the presence of the electron donors glutamate and malate (complexes I, III, IV), succinate and rotenone (complexes II, III, IV) or ascorbate and N,N,N',N'-tetramethyl-p-phenylenediamine (TMPD; complex IV) as previously described (13). The ATP synthesized in the assay was quantified using the ATP Bioluminesence Assay kit HSII (Roche). Data were expressed as pmoles ATP synthesized/min/105 cells. ATP synthesized in the absence of substrates was subtracted from the data.
Statistical analyses
Data are expressed as the mean ± SEM of separate experiments (n). Statistical significance was determined by two-tailed t-test or one-way ANOVA followed by the Tukey HSD test where appropriate.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
This work was funded by The Brain Research Trust, the Wellcome/MRC Parkinson's Disease Consortium grant to University College London/Institute of Neurology, the University of Sheffield and the MRC Protein Phosphorylation Unit at the University of Dundee, and The Kattan Trust.
REFERENCES
- 1.Schapira A.H., Tolosa E. Molecular and clinical prodrome of Parkinson disease: implications for treatment. Nat. Rev. Neurol. 2010;6:309–317. doi: 10.1038/nrneurol.2010.52. doi:10.1038/nrneurol.2010.52. [DOI] [PubMed] [Google Scholar]
- 2.Cuervo A.M., Wong E.S., Martinez-Vicente M. Protein degradation, aggregation, and misfolding. Mov. Disord. 2010;25:S49–S54. doi: 10.1002/mds.22718. doi:10.1002/mds.22718. [DOI] [PubMed] [Google Scholar]
- 3.Schapira A.H., Cooper J.M., Dexter D., Jenner P., Clark J.B., Marsden C.D. Mitochondrial complex I deficiency in Parkinson's disease. Lancet. 1989;1:1269. doi: 10.1016/s0140-6736(89)92366-0. doi:10.1016/S0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- 4.Schapira A.H.V. Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Lancet Neurol. 2008;7:97–109. doi: 10.1016/S1474-4422(07)70327-7. doi:10.1016/S1474-4422(07)70327-7. [DOI] [PubMed] [Google Scholar]
- 5.Betarbet R., Sherer T.B., MacKenzie G., Garcia-Osuna M., Panov A.V., Greenamyre J.T. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat. Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. doi:10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 6.Bender A., Krishnan K.J., Morris C.M., Taylor G.A., Reeve A.K., Perry R.H., Jaros E., Hersheson J.S., Betts J., Klopstock T., et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006;38:515–517. doi: 10.1038/ng1769. doi:10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 7.Rubinsztein D.C., DiFiglia M., Heintz N., Nixon R.A., Qin Z.H., Ravikumar B., Stefanis L., Tolkovsky A. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy. 2005;1:11–22. doi: 10.4161/auto.1.1.1513. doi:10.4161/auto.1.1.1513. [DOI] [PubMed] [Google Scholar]
- 8.Pan T., Kondo S., Le W., Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson's disease. Brain. 2008;131:1969–1978. doi: 10.1093/brain/awm318. doi:10.1093/brain/awm318. [DOI] [PubMed] [Google Scholar]
- 9.Abou-Sleiman P.M., Muqit M.M., Wood N.W. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat. Rev. Neurosci. 2006;7:207–219. doi: 10.1038/nrn1868. doi:10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- 10.Valente E.M., Abou-Sleiman P.M., Caputo V., Muqit M.M., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A.R., Healy D.G., et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. doi:10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 11.Shimura H., Hattori N., Kubo S., Mizuno Y., Asakawa S., Minoshima S., Shimizu N., Iwai K., Chiba T., Tanaka K., et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000;25:302–305. doi: 10.1038/77060. doi:10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 12.Ramirez A., Heimbach A., Gründemann J., Stiller B., Hampshire D., Cid L.P., Goebel I., Mubaidin A.F., Wriekat A.L., Roeper J., et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. doi:10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 13.Gegg M.E., Cooper J.M., Schapira A.H., Taanman J.W. Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS ONE. 2009;4:e4756. doi: 10.1371/journal.pone.0004756. doi:10.1371/journal.pone.0004756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grünewald A., Gegg M.E., Taanman J.W., King R.H., Kock N., Klein C., Schapira A.H.V. Differential effects of PINK1 nonsense and missense mutations on mitochondrial function and morphology. Exp. Neurol. 2009;219:266–273. doi: 10.1016/j.expneurol.2009.05.027. doi:10.1016/j.expneurol.2009.05.027. [DOI] [PubMed] [Google Scholar]
- 15.Hoepken H.H., Gispert S., Morales B., Wingerter O., Del Turco D., Mulsch A., Nussbaum R.L., Muller K., Drose S., Brandt U., et al. Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol. Dis. 2006;25:401–411. doi: 10.1016/j.nbd.2006.10.007. doi:10.1016/j.nbd.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 16.Wood-Kaczmar A., Gandhi S., Yao Z., Abramov A.Y., Miljan E.A., Keen G., Stanyer L., Hargreaves I., Klupsch K., Deas E., et al. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE. 2008;3:e2455. doi: 10.1371/journal.pone.0002455. doi:10.1371/journal.pone.0002455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gandhi S., Wood-Kaczmar A., Yao Z., Plun-Favreau H., Deas E., Klupsch K., Downward J., Latchman D.S., Tabrizi S.J., Wood N.W., et al. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell. 2009;33:627–638. doi: 10.1016/j.molcel.2009.02.013. doi:10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marongiu R., Spencer B., Crews L., Adame A., Patrick C., Trejo M., Dallapiccola B., Valente E.M., Masliah E. Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson's disease by disturbing calcium flux. J. Neurochem. 2009;108:1561–1574. doi: 10.1111/j.1471-4159.2009.05932.x. doi:10.1111/j.1471-4159.2009.05932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang H.L., Chou A.H., Yeh T.H., Li A.H., Chen Y.L., Kuo Y.L., Tsai S.R., Yu S.T. PINK1 mutants associated with recessive Parkinson's disease are defective in inhibiting mitochondrial release of cytochrome c. Neurobiol. Dis. 2007;28:216–226. doi: 10.1016/j.nbd.2007.07.010. doi:10.1016/j.nbd.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 20.Gautier C.A., Kitada T., Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl Acad. Sci. USA. 2008;105:11364–11369. doi: 10.1073/pnas.0802076105. doi:10.1073/pnas.0802076105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitada T., Pisani A., Porter D.R., Yamaguchi H., Tscherter A., Martella G., Bonsi P., Zhang C., Pothos E.N., Shen J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl Acad. Sci. USA. 2007;104:11441–11446. doi: 10.1073/pnas.0702717104. doi:10.1073/pnas.0702717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gispert S., Ricciardi F., Kurz A., Azizov M., Hoepken H.H., Becker D., Voos W., Leuner K., Müller W.E., Kudin A.P., et al. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS ONE. 2009;4:e5777. doi: 10.1371/journal.pone.0005777. doi:10.1371/journal.pone.0005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park J., Lee S.B., Lee S., Kim Y., Song S., Kim S., Bae E., Kim J., Shong M., Kim J.M., et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. doi:10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 24.Clark I.E., Dodson M.W., Jiang C., Cao J.H., Huh J.R., Seol J.H., Yoo S.J., Hay B.A., Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. doi:10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 25.Poole A.C., Thomas R.E., Andrews L.A., McBride H.M., Whitworth A.J., Pallanck L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl Acad. Sci. USA. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. doi:10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Y., Ouyang Y., Yang L., Beal M.F., McQuibban A., Vogel H., Lu B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc. Natl Acad. Sci. USA. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. doi:10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Exner N., Treske B., Paquet D., Holmström K., Schiesling C., Gispert S., Carballo-Carbajal I., Berg D., Hoepken H.H., Gasser T., et al. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J. Neurosci. 2007;27:12413–12418. doi: 10.1523/JNEUROSCI.0719-07.2007. doi:10.1523/JNEUROSCI.0719-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dagda R.K., Cherra S.J., Kulich S.M., Tandon A., Park D., Chu C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 2009;284:13843–13855. doi: 10.1074/jbc.M808515200. doi:10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lutz A.K., Exner N., Fett M.E., Schlehe J.S., Kloos K., Lämmermann K., Brunner B., Kurz-Drexler A., Vogel F., Reichert A.S., et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 2009;284:22938–22951. doi: 10.1074/jbc.M109.035774. doi:10.1074/jbc.M109.035774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sandebring A., Thomas K.J., Beilina A., van der Brug M., Cleland M.M., Ahmad R., Miller D.W., Zambrano I., Cowburn R.F., Behbahani H., et al. Mitochondrial alterations in PINK1 deficient cells are influenced by calcineurin-dependent dephosphorylation of dynamin-related protein 1. PLoS ONE. 2009;4:e5701. doi: 10.1371/journal.pone.0005701. doi:10.1371/journal.pone.0005701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sha D., Chin L.S., Li L. Phosphorylation of parkin by Parkinson disease-linked kinase PINK1 activates parkin E3 ligase function and NF-kappaB signaling. Hum. Mol. Genet. 2010;19:352–363. doi: 10.1093/hmg/ddp501. doi:10.1093/hmg/ddp501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiba K., Arai T., Sato S., Kubo S., Ohba Y., Mizuno Y., Hattori N. Parkin stabilizes PINK1 through direct interaction. Biochem. Biophys. Res. Commun. 2009;383:331–335. doi: 10.1016/j.bbrc.2009.04.006. doi:10.1016/j.bbrc.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 33.Narendra D., Tanaka A., Suen D.F., Youle R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. doi:10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vives-Bauza C., Zhou C., Huang Y., Cui M., de Vries R.L., Kim J., May J., Tocilescu M.A., Liu W., Ko H.S., et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl Acad. Sci. USA. 2010;107:378–383. doi: 10.1073/pnas.0911187107. doi:10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geisler S., Holmström K.M., Skujat D., Fiesel F.C., Rothfuss O.C., Kahle P.J., Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. doi:10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 36.Ziviani E., Tao R.N., Whitworth A. J. Drosophila Parkin requires PINK1 for mitochondrial translocation and ubiquitinates Mitofusin. Proc. Natl Acad. Sci. USA. 2010;107:5018–5023. doi: 10.1073/pnas.0913485107. doi:10.1073/pnas.0913485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Twig G., Elorza A., Molina A.J., Mohamed H., Wikstrom J.D., Walzer G., Stiles L., Haigh S.E., Katz S., Las G., et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klionsky D.J., Abeliovich H., Agostinis P., Agrawal D.K., Aliev G., Askew D.S., Baba M., Baehrecke E.H., Bahr B.A., Ballabio A., et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watts J.A., Kline J.A., Thornton L.R., Grattan R.M., Brar S.S. Metabolic dysfunction and depletion of mitochondria in hearts of septic rats. J. Mol. Cell. Cardiol. 2004;36:141–150. doi: 10.1016/j.yjmcc.2003.10.015. doi:10.1016/j.yjmcc.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 40.Hargreaves I.P., Duncan A.J., Wu L., Agrawal A., Land J.M., Heales S.J. Inhibition of mitochondrial complex IV leads to secondary loss complex II–III activity: implications for the pathogenesis and treatment of mitochondrial encephalomyopathies. Mitochondrion. 2007;7:284–287. doi: 10.1016/j.mito.2007.02.001. doi:10.1016/j.mito.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 41.Bodnar A.G., Cooper J.M., Leonard J.V., Schapira A.H. Respiratory-deficient human fibroblasts exhibiting defective mitochondrial DNA replication. Biochem. J. 1995;305:817–822. doi: 10.1042/bj3050817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tolkovsky A.M. Mitophagy. Biochim. Biophys. Acta. 2009;1793:1508–1515. doi: 10.1016/j.bbamcr.2009.03.002. doi:10.1016/j.bbamcr.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 43.Zorzano A., Liesa M., Sebastián D., Segalés J., Palacín M. Mitochondrial fusion proteins: dual regulators of morphology and metabolism. Semin. Cell Dev. Biol. 2010 doi: 10.1016/j.semcdb.2010.01.002. in press. 10.1016/j.semcdb.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 44.Yonashiro R., Ishido S., Kyo S., Fukuda T., Goto E., Matsuki Y., Ohmura-Hoshino M., Sada K., Hotta H., Yamamura H., et al. A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 2006;25:3618–3626. doi: 10.1038/sj.emboj.7601249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsuda N., Sato S., Shiba K., Okatsu K., Saisho K., Gautier C.A., Sou Y.S., Saiki S., Kawajiri S., Sato F., et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010;189:211–221. doi: 10.1083/jcb.200910140. doi:10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee J.Y., Nagano Y., Taylor J.P., Lim K.L., Yao T.P. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J. Cell Biol. 2010;189:671–679. doi: 10.1083/jcb.201001039. doi:10.1083/jcb.201001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poole A.C., Thomas R.E., Yu S., Vincow E.S., Pallanck L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS ONE. 2010;5:e10054. doi: 10.1371/journal.pone.0010054. doi:10.1371/journal.pone.0010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Germain D. Ubiquitin-dependent and -independent mitochondrial protein quality controls: implications in ageing and neurodegenerative diseases. Mol. Microbiol. 2008;70:1334–1341. doi: 10.1111/j.1365-2958.2008.06502.x. doi:10.1111/j.1365-2958.2008.06502.x. [DOI] [PubMed] [Google Scholar]
- 49.Kirkin V., McEwan D.G., Novak I., Dikic I. A role for ubiquitin in selective autophagy. Mol. Cell. 2009;34:259–269. doi: 10.1016/j.molcel.2009.04.026. doi:10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 50.Chin L.S., Olzmann J.A., Li L. Parkin-mediated ubiquitin signalling in aggresome formation and autophagy. Biochem. Soc. Trans. 2010;38:144–149. doi: 10.1042/BST0380144. doi:10.1042/BST0380144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Whitworth A.J., Pallanck L.J. The PINK1/Parkin pathway: a mitochondrial quality control system? J. Bioenerg. Biomembr. 2009;41:499–503. doi: 10.1007/s10863-009-9253-3. doi:10.1007/s10863-009-9253-3. [DOI] [PubMed] [Google Scholar]
- 52.Ziviani E., Whitworth A.J. How could Parkin-mediated ubiquitination of mitofusin promote mitophagy. Autophagy. 2010;6:660–662. doi: 10.4161/auto.6.5.12242. doi:10.4161/auto.6.5.12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cohen M.M., Leboucher G.P., Livnat-Levanon N., Glickman M.H., Weissman A.M. Ubiquitin–proteasome-dependent degradation of a mitofusin, a critical regulator of mitochondrial fusion. Mol. Biol. Cell. 2008;19:2457–2464. doi: 10.1091/mbc.E08-02-0227. doi:10.1091/mbc.E08-02-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sorokin A.V., Kim E.R., Ovchinnikov L.P. Proteasome system of protein degradation and processing. Biochemistry (Mosc.) 2009;74:1411–1442. doi: 10.1134/s000629790913001x. doi:10.1134/S000629790913001X. [DOI] [PubMed] [Google Scholar]
- 55.Hailey D.W., Rambold A.S., Satpute-Krishnan P., Mitra K., Sougrat R., Kim P.K., Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. doi:10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Misko A., Jiang S., Wegorzewska I., Milbrandt J., Baloh R.H. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 2010;30:4232–4240. doi: 10.1523/JNEUROSCI.6248-09.2010. doi:10.1523/JNEUROSCI.6248-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Michiorri S., Gelmetti V., Giarda E., Lombardi F., Romano F., Marongiu R., Nerini-Molteni S., Sale P., Vago R., Arena G., et al. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ. 2010;17:962–974. doi: 10.1038/cdd.2009.200. doi:10.1038/cdd.2009.200. [DOI] [PubMed] [Google Scholar]
- 58.Alvarez-Erviti L., Rodriguez-Oroz M.C., Cooper J.M., Caballero C., Ferrer I., Obeso J.A., Schapira A.H.V. Chaperone-mediated autophagy markers are abnormal in Parkinson's disease brain. Arch. Neurol. 2010 doi: 10.1001/archneurol.2010.198. in press. 10.1001/archneurol.2010.198. [DOI] [PubMed] [Google Scholar]
- 59.Lai J.C., Clark J.B. Preparation and properties of mitochondria derived from synaptosomes. Biochem. J. 1976;154:423–432. doi: 10.1042/bj1540423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yoon Y.S., Yoon D.S., Lim I.K., Yoon S.H., Chung H.Y., Rojo M., Malka F., Jou M.J., Martinou J.C., Yoon G. Formation of elongated giant mitochondria in DFO-induced cellular senescence: involvement of enhanced fusion process through modulation of Fis1. J. Cell Physiol. 2006;209:468–480. doi: 10.1002/jcp.20753. doi:10.1002/jcp.20753. [DOI] [PubMed] [Google Scholar]
- 61.Rojo M., Legros F., Chateau D., Lombès A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J. Cell Sci. 2002;115:1663–1674. doi: 10.1242/jcs.115.8.1663. [DOI] [PubMed] [Google Scholar]