

Abstract
Anthrax lethal toxin (LeTx) is a virulence factor of Bacilillus anthracis that is a bivalent toxin, containing lethal factor (LF) and protective Ag proteins, which causes cytotoxicity and altered macrophage function. LeTx exposure results in early K+ efflux from macrophages associated with caspase-1 activation and increased IL-1β release. The mechanism of this toxin-induced K+ efflux is unknown. The goals of the current study were to determine whether LeTx-induced K+ efflux from macrophages is mediated by toxin effects on specific K+ channels and whether altered K+-channel activity is involved in LeTx-induced IL-1β release. Exposure of macrophages to LeTx induced a significant increase in the activities of two types of K+ channels that have been identified in mouse macrophages: Ba2+-sensitive inwardly rectifying K+ (Kir) channels and 4-aminopyridine–sensitive outwardly rectifying voltage-gated K+ (Kv) channels. LeTx enhancement of both Kir and Kv required the proteolytic activity of LF, because exposure of macrophages to a mutant LF-protein (LFE687C) combined with protective Ag protein had no effect on the currents. Furthermore, blocking Kir and Kv channels significantly decreased LeTx-induced release of IL-1β. In addition, retroviral transduction of macrophages with wild-type Kir enhanced LeTx-induced release of IL-1β, whereas transduction of dominant-negative Kir blocked LeTx-induced release of IL-1β. Activation of caspase-1 was not required for LeTx-induced activation of either of the K+ channels. These data indicate that a major mechanism through which LeTx stimulates macrophages to release IL-1β involves an LF-protease effect that enhances Kir and Kv channel function during toxin stimulation.
Anthrax is caused by Bacillus anthracis, a Gram-positive spore-forming bacterium that has multiple virulence factors: a polyglutamic acid capsule and two anthrax toxins, lethal toxin (LeTx) and edema toxin, that act in synergy (1). The binary toxins are formed by combinations of a receptor-binding moiety, protective Ag (PA), that binds with two catalytic moieties: lethal factor (LF), a zinc metalloproteinase (2, 3) that binds PA to form LeTx, and edema factor, a calmodulin-dependent adenylate cyclase that binds PA to form edema toxin (2, 4). LeTx induces macrophages to release IL-1β (5), which is a proinflammatory cytokine involved in the systemic response to anthrax infection (6–8). Before release of the fully processed IL-1β, the inactive precursor (pro–IL-1β) is cleaved by caspase-1 protease (9), a process that is triggered by depletion of in-tracellular K+ ions (e.g., Refs. 10–12). K+ efflux is also an early event that is triggered by LeTx treatment of macrophages (13). Inhibition of ion flux by elevated levels of extracellular KCl blocks LeTx-induced caspase-1 activation, cell lysis, and maturation of IL-1β (14). These observations suggest that LeTx-induced efflux of K+ is a key early step in the toxin-induced release of selected cytokines by macrophages.
A key mechanistic question is whether LeTx-induced K+ efflux is the result of a nonspecific increase in the permeability of the membrane lipid bilayer or specific alteration in the activity of a K+ channel. Earlier studies have shown that macrophages express two types of K+ channels, inwardly rectifying K+ channels (Kir 2.1), and voltage-gated K+ channels (Kv1.3 and Kv1.5) (15–18). In this study, we report that exposure to LeTx results in a significant increase in the activity of both inwardly-rectifying K+ (Kir) and voltage-gated K+ (Kv) channels in alveolar macrophages and that toxin-induced macrophage release of IL-1β critically depends on the LeTx activation of these channels.
Materials and Methods
Cell culture and reagents
MH-S is an alveolar macrophage cell line, derived by SV40 transformation of macrophages from BALB/c mice, that retains morphological and functional traits of normal alveolar macrophages (19); MH-S was obtained from American Type Culture Collection (Manassas, VA). Peritoneal macrophages were harvested by lavage from adult (>8 wk old), female BALB/c mice (National Cancer Institute, Fredrick Cancer Research Center). Cells were cultured in RPMI 1640 medium (Cellgro; Mediatech, Herndon, VA) supplemented with 10% FBS (HyClone, Logan, UT), 2 mM L-glutamine (Cellgro; Mediatech), 100 U/ml penicillin G, and 100 μg/ml streptomycin sulfate (Cellgro; Mediatech) and maintained at 5% CO2 in a humidified atmosphere. All other reagents were obtained from Sigma-Aldrich (St. Louis, MO), unless specified otherwise.
Toxin protein purification
Wild-type (WT) and mutant LF and WT PA proteins were purified as described (38). Purified toxin protein preparations were passed through a final column (Detoxi-gel endotoxin removing gel; Thermo Fisher, Rockford, IL) to remove any endotoxin and were tested using a limulus amebocyte lysate assay (Endochrome; Charles River Laboratories, Charleston, SC) to ensure that they were endotoxin free (<1 pg/ml at working concentrations). The final purified products were >99% pure as judged by SDS-PAGE.
Isolation of peritoneal and bone marrow-derived macrophages
Adult 25–30 g BALB/c female mice (>8 wk old) were purchased from the National Cancer Institute (Fredrick Cancer Research Center). Mice were housed under pathogen-free conditions in the animal facility at the University of Illinois at Chicago and given free access to food and water. A 1-ml suspension of 3% thioglycollate was injected i.p. to elicit an inflammatory response. Mice were rested for 4 d after injection, and then peritoneal exudate cells were harvested sterilely by flushing the peritoneal cavities with 7 ml RPMI 1640 medium (Invitrogen). Peritoneal exudate cells were washed twice with RPMI media, treated with ammonium chloride RBC lysis buffer containing 10% FBS, washed twice with RPMI 1640 medium, and suspended in RPMI 1640 complete medium (10% FBS + antibiotics). Bone marrow-derived macrophages (BMDMs) were obtained from C57BL6/DBA background mice (3–6 wk old) following the method of Celada et al. (20) with modifications. After mice were killed, bone marrow was flushed from the femurs. The cells were washed and resuspended in DMEM, with 10% endotoxin-free FBS and 10% (v/v) L929 cell-conditioned medium as a biological source of M-CSF. The medium was replenished at day 4, and nonadherent cells weare removed. The cells were used for the experiments after days 7–9.
Electrophysiology
For patching, cells were plated overnight in complete media on sterile glass coverslips at a density of 1.5 × 103 cells per well in 24-well plates (Sarstedt). On the day of the experiment, cells were incubated with or without LeTx (PA at 100 ng/ml plus LF at 5, 25, or 100 ng/ml) for 1 h. For K+ channel blocking experiments, cells were exposed to 300 μM barium (Ba2+) or 3 mM 4- aminopyridine (4-AP) added at the same time as the toxins and left for the duration of the experiment. Ionic currents were measured using whole-cell configurations of the standard patch clamp techniques. Pipettes were pulled (SG10 glass, 1.20 mm internal diameter, 1.60 mm outer diameter; part number FPENNU1.20ID1.60OD; Richland Glass, Richland, NJ) to give a final resistance of 2–6 MΩ, allowing for high-resistance seals without fire polishing. A saturated salt agar bridge was used as the reference electrode. Currents were recorded using an EPC9 amplifier (HEKA Electronik, Lambrecht, Germany) and accompanying acquisition and analysis software (Pulse & PulseFit; HEKA Electronik). The external recording solution contained (mM): 150 NaCl, 6 KCl, 10 HEPES, 1.5 CaCl2, 1 MgCl2, and 1 EGTA at pH 7.3. The pipette solutions contained (mM): 145 KCl, 10 HEPES, 1 MgCl2, 4 ATP, and 1 EGTA at pH 7.3. Currents were elicited by 500 ms linear voltage ramps from −100 mV to +60 mV at an interpulse interval of 5 s. The holding potential was −60 mV. Pipette and whole-cell capacitances were automatically compensated. Whole-cell capacitance and series resistance were compensated and monitored throughout the recording. Rs was compensated by 60–90% with the compensation being limited by the stability of the patch. The experiments were conducted at room temperature. A total of 10–20 cells was recorded for each experimental condition in 3–5 independent experiments. All recordings were performed within a 1–3 h window after exposing the cells to the toxin.
Generation of retroviral constructs of dominant-negative and WT Kir (WT-Kir2.1) channels
The pWPI lentivirus vector plasmid and the packaging plasmids (psPAX2 and pMD2.G) were obtained from Didier Trono’s laboratory (http://tronolab.epfl.ch/; Ecole Polytechnique Federale de Lausanne, Lausanne, Switzerland) through Addgene (Addgene plasmids 12254, 12260, and 12259). WT- and dominant-negative (DN)-Kir2.1 cDNAs were subcloned in the pWPI lentivirus vector plasmid between PacI and PmeI sites. All plasmids were amplified in Escherichia coli DH5α and purified by Nucleobond anion exchange columns (Macherey-Nagel) following the manufacturer’s instructions. The HEK293T/17 cell line used for lentivirus production was obtained from American Type Culture Collection (Manassas, VA) and were grown in DMEM (Invitrogen) supplemented with 10% FCS (Life Technologies). Lentiviruses were produced by following the protocols available from Didier Trono’s laboratory. Briefly, a subconfluent monolayer of HEK293T/17 cells was transfected with a mixture of the pWPI-Kir2.1 vector and the psPAX2 and pMD2.G packaging plasmids by the calcium-phosphate precipitation method. Twelve to 14 h after transfection, the culture medium was replaced by fresh culture medium. The culture supernatant containing the viruses was harvested 36–48 h post-transfection. The harvested medium was clarified from cell debris by low-speed centrifugation and filtration using a 0.45-μm pore size syringe-attachable filter. Virus particles were precipitated by ultracentrifugation at 47000 × g (relative centrifugal force average) for 2 h, resuspended in sterile PBS containing 10% BSA, and stored at −80°C in aliquots. WT Kir2.1 construct was a generous gift of Dr. Carol Vandenberg. DN-Kir2.1 was generated and characterized in our previous studies (21).
Cytokine release assay
Bio-plex assay system
Release of IL-1β, IL-1α, TNF-α, and IL-6 was determined using a fourplex bead-based immunoassay kit (Bio-Rad, Hercules, CA) and read using a Luminex 100 analyzer (Bio-Rad). Cells were seeded at a density of 5 × 105 cells per well in 24-well plates and incubated overnight. On the day of the experiment, cells were preincubated for 1 h with or without 300 μM Ba2+ or 3 mM 4-AP in 500 μl fresh RPMI 1640 plus 10% FCS media, and LeTx was added at a final concentration of 100 ng/ml (LF and PA) for 3 h. Cells not treated with LeTx were used as controls. Cell supernatants were collected, centrifuged, and frozen at −80°C until assayed. Bio-plex assays were performed according to the manufacturer’s protocol (Bio-Rad). Briefly, a filter-bottom 96-well microplate was prewetted with assay buffer, following which the buffer was removed using vacuum filtration. Fifty microliters polystyrene beads precoated with Abs against the four cytokines was added to each well. Beads were washed twice with buffer provided in the kit, and the buffer was removed by vacuum filtration. Cytokine standards (10–1000 pg/ml) were prepared in RPMI 1640 media, and samples were added and allowed to incubate on a shaker for 30 min. After washing three times with buffer to remove unbound protein, 25 μl Bio-Plex detection Ab specific for a different epitope on the cytokine was added and allowed to incubate for 30 min. After further washings (three times), 50 μl streptavidin-PE was added and allowed to incubate for 10 min. The plate was washed (three times) to remove any unbound dye and suspended in 125 μl assay buffer and analyzed on a Luminex 100 analyzer, set to run a 50 μl sample and a minimum of 100 events per cytokine. All incubations were performed at room temperature and with assay plates covered with sealing tape and aluminum foil while shaking (300 rpm). After each wash step, the underside of the plate was blotted with a paper towel. Data were calculated using Bio-Plex Manager software (Bio-Rad) using a five-parameter logistic regression along with weighting model (used when six or more standards are found to be within 70–130% of expected value). Cytokine concentrations in standards, positive controls, and samples were expressed as picograms per milliliter. All concentrations were rounded to the next whole digits. Values <0.5 were taken as zero and represented as not detected. Results are presented as mean (SD) of three incubations for each treatment. Alternatively, IL-1β was measured by ELISA (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions.
Statistical analysis
Data were analyzed using Student t test and Microsoft Excel software (Microsoft).
Results
LeTx activates K+ channels in alveolar macrophages
Alveolar macrophages are likely to be the first immune cells exposed to inhalational anthrax infection. We first tested, therefore, the effect of LeTx on K+ channels in the BALB/c alveolar macrophage cell line, MH-S, that has been used as a model of alveolar macrophage function (19, 22, 23). As expected, two components of K+ current were observed in MH-S cells, inwardly rectifying K+ channels that are open at hyperpolarizing potentials and voltage-gated K+ channels that are open at depolarizing potentials (Fig. 1A). Exposure of MH-S cells to 5–100 ng/ml LeTx resulted in a dose-dependent increase in Kir current density (Fig. 1B), as well as Kv current density (Fig. 1C). This concentration range of LeTx was chosen because LeTx-induced IL-1β release in these cells reached a maximal level at a LeTx dose of 100 ng/ml with no further increase observed when LeTx concentration was increased to up to 1000 ng/ml (Supplemental Fig. 1). An increase in macrophage K+ currents was not accompanied by any change in the reversal potential (−65 ± 2 mV versus −64 ± 2 mV for control and LeTx-treated cells, respectively), indicating that the toxin-induced increase in K+ current is due to the opening of specific K+ selective ion channels and does not result from an increase in a nonspecific membrane leak.
FIGURE 1.
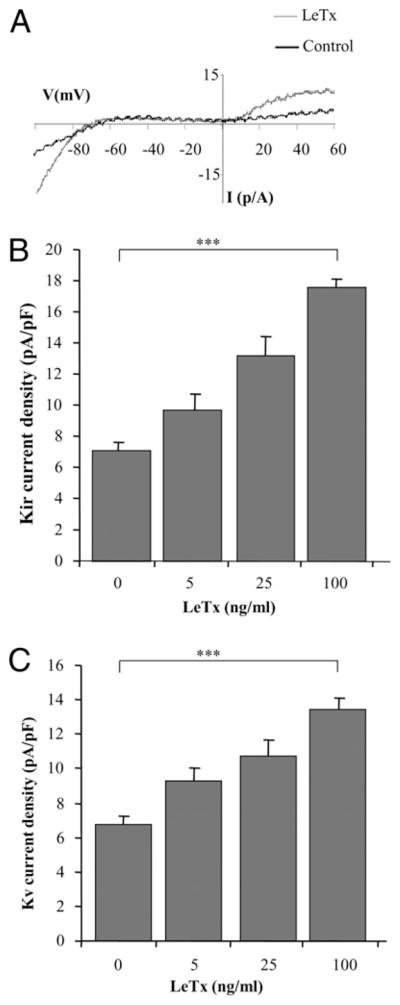
LeTx activates Kir and Kv channels in an alveolar macrophage cell line. A, Typical current recordings from MH-S cells under control and LeTx 100 ng/ml (LF and PA) treatment. B, Dose-response effect of LeTx on Kir current density. The currents were measured at −95 mV. C, Dose-response effect of LeTx on Kv currents measured at +60 mV. Each data set represents 4–15 independent experiments with 12–45 cells per condition and is expressed as mean ± SEM. The Kir and Kv current density was significantly increased in cells treated with LF and PA at 100 ng/ml each, when compared with the untreated control. ***p < 0.001.
To test further the specificity of the LeTx effects on Kir and Kv currents, the cells were exposed either to Ba2+, a blocker of Kir channels, or to 4-AP, a blocker of voltage-gated K+ channels (Fig. 2). As expected, exposing the cells to 300 μM Ba2+ blocked the Kir component of the current but had no effect on the Kv component in the same cells (compare typical traces in the upper panel and in the middle panel of Fig. 2A). Conversely, exposing cells to 3 mM 4-AP significantly blocked the Kv component of the current but had no effect on Kir (compare the upper and lower panels of Fig. 2A). LeTx-induced increase in Kir was also completely blocked by Ba2+ but not by 4-AP, whereas LeTx-induced increase in Kv current was inhibited by 4-AP but not by Ba2+ (Fig. 2B, 2C). These observations indicate that LeTx induces activation of Kir and Kv channels independently.
FIGURE 2.

LeTx-induced activation of Kir and Kv channels is specifically blocked by Ba2+ and by 4-AP, respectively. A, Overlays of typical current recordings from MH-S cells in control cells and in cells treated with 100 ng/ml LeTx (LF and PA) recorded in the absence of channel blockers (upper panel), in the presence of 300 μM Ba2+ to block Kir channels (middle panel), and in the presence of 3 mM 4-AP to block Kv channels (lower panel). B, Mean Kir current densities (−95 mV) in the six experimental cell populations described above. C, Mean Kv current densities (+60 mV) in the same experimental cell populations. Each data set is expressed as mean ± SEM. All experiments were repeated with three to five independent cell populations with 5–30 cells per condition. ***p < 0.001.
LeTx activates K+ channels in peritoneal macrophages
To determine whether the effect of LeTx on the activity of K+ channels is also observed in primary macrophages, K+ currents were recorded in peritoneal macrophages isolated from BALB/c mice, a commonly used model to study macrophage function in response to LeTx (24, 25). Similarly to MH-S cells, K+ current in peritoneal macrophages had two distinct components, a Ba2+-sensitive Kir component and a 4-AP–sensitive Kv component (Fig. 3A, upper panel). The currents were, however, significantly lower than in MH-S cells, particularly the Kv component (compare the y-axis amplitudes of Fig. 1B and 1C for MH-S cells with Fig. 3B and 3C for peritoneal macrophages). Similarly to MH-S cells, exposing peritoneal macrophages to 100 ng/ml LeTx (i.e., LF and PA both at 100 ng/ml) significantly increased the Kir current (Fig. 3A, 3B). Also, as described above for the MH-S cells, LeTx-induced increase in Kir current was completely abrogated by blocking the channels with Ba2+, indicating that LeTx activates Kir channels (Fig. 3B). There was also a slight, but not significant, increase in the Kv current (Fig. 3C).
FIGURE 3.

LeTx activates K+ channels in peritoneal macrophages. A, Typical current traces recorded from BALB/c peritoneal macrophages treated with Ba2+ (300 μM), 3 mM 4-AP, LeTx (LF and PA, 100 ng/ml), or Ba2+ followed by LeTx, as compared with an untreated control cell. B, Mean peak Kir current densities for the cell populations described above were measured at −95 mV, with 6–33 cells per condition in three to five independent experiments, and are expressed as the mean ± SEM. C, Mean peak Kv current densities for the cell populations treated with LeTx and untreated control were measured at +60 mV, with 5–20 cells per condition in three to five independent experiments, and are expressed as the mean ± SEM.
Requirement for LeTx metalloprotease activity for increased K+ channel activity
LF (one of the two proteins of LeTx) is a 776 aa protein that contains a zinc-binding site at residues 686–690, which is characteristic of metalloproteases (26). LF is inactivated by mutations in this region, such as a cysteine substitution for glutamic acid at E-687 (E687C) (26). We therefore tested the effect of the LF mutant protein E687C on K+ channel activity. Treatment with mutant LF protein LFE687C combined with PA (mLeTx) had no significant effect on Kir or Kv currents in MH-S cells, indicating that metalloprotease activity is required for the activation of both types of K+ channels (Fig. 4A–C). Similarly, mLeTx had no significant effect on Kir currents in peritoneal macrophages (Fig. 4D, 4E). No significant effect of either LeTx or mLeTx on Kv currents was observed in peritoneal macrophages (Fig. 4F).
FIGURE 4.

Requirement for LF protein metalloprotease activity for Kir and Kv channel activation in alveolar and peritoneal macrophages. A, Overlays of typical current recordings from alveolar macrophages (MH-S) under three experimental conditions: control untreated, treated with LeTx (LF and PA, 100 ng/ml), or treated with metalloprotease-inactive mutant LF (LFE687C) plus PA (100 ng/ml each) (mLeTx). Mean Kir (B) and Kv (C) current densities recorded at −95 mV or +60 mV, respectively, for each of the experimental cell populations described above. D, Overlays of typical current recordings from peritoneal macrophages under the same three experimental conditions. Mean Kir (E) and Kv (F) current densities measured in peritoneal macrophages. Each data set is shown as means ± SEM (n = 8–33 cells per condition). ***p < 0.001.
LeTx-induced activation of macrophage K+ channels does not require caspase-1 activation
Next, we tested whether LeTx-induced activation of macrophage K+ channels requires activation of caspase-1, a well-known downstream target of LeTx (14). To address this question, we used BMDMs from C57BL/6 mice, which have a mutant nalp1 gene that renders these cells unable to form active caspase-1, thus making these macrophages resistant to LeTx-induced IL-1β release (27). Similarly to macrophages derived from Nalp1-competent BALB/c mice, LeTx-induced activation of both Kir and Kv channels was also observed in C57BL/6 BMDMs, indicating that activation of macrophage K+ channels is independent of caspase-1 activation (Fig. 5). As was shown earlier (28), macrophages isolated from C57BL/6 mice did not release IL-1β in response to LeTx stimulation (data not shown). These observations also demonstrate that LeTx-induced activation of Kir and Kv channels is observed in primary BMDMs as well as in macrophages of alveolar and peritoneal origin.
FIGURE 5.
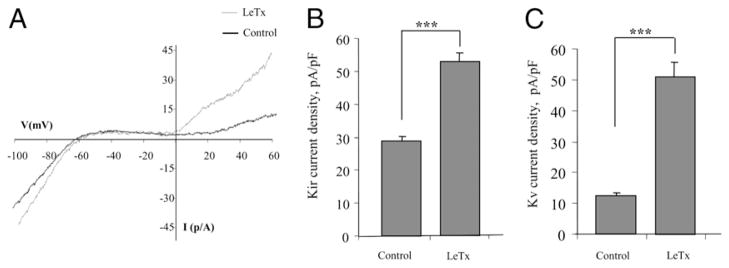
LeTx-induced activation of macrophage K+ channels does not require caspase-1 activation. A, Overlay of typical current traces recorded from C57BL/6 BMDMs treated with LeTx (LF and PA, 100 ng/ml), as compared with an untreated control cell. Mean peak Kir (B) and Kv (C) current densities recorded at −95 mV and +60 mV, respectively, for the cell populations described above with 5–20 cells per condition in three to five independent experiments and are expressed as the mean ± SEM. The LeTx induction of increased Kir and Kv current densities were significant compared with the untreated control cells. ***p < 0.001.
LeTx-induced macrophage production of IL-1β is related to K+ channel expression and function
Two experimental strategies were used to test further the requirement for K+ channel function in LeTx-induced macrophage production of IL-1β: 1) blocking K+ channels with Ba2+ or 4-AP to inhibit Kir or Kv channels, respectively; and 2) transducing macrophages with either a DN Kir2.1 or WT Kir2.1 construct.
First, we tested the effect of LeTx on primary peritoneal macrophage production of four proinflammatory cytokines (IL-1β, IL-1α, TNF-α, and IL-6) using the Bio-plex assay (Fig. 6A). A significant increase in LeTx-induced cytokine production was observed for IL-1β and IL-1α but not for TNF-α and IL-6. The effect on IL-1α was unexpected because although multiple earlier studies reported that LeTx induces release of IL-1β, no LeTx-induced release of IL-1α was observed in earlier studies (5). The source of this discrepancy is not clear. Most importantly, the LeTx-induced increase in both IL-1β and IL-1α release was significantly inhibited by blocking Kir channels with Ba2+. These cytokine response data indicate that the LeTx effect on macrophages is cytokine-selective and suggest involvement of caspase-1 activation in cytokine overproduction, because caspase-1 is involved in IL-1β and IL-1α responses (29) but not production of TNF-α or IL-6.
FIGURE 6.
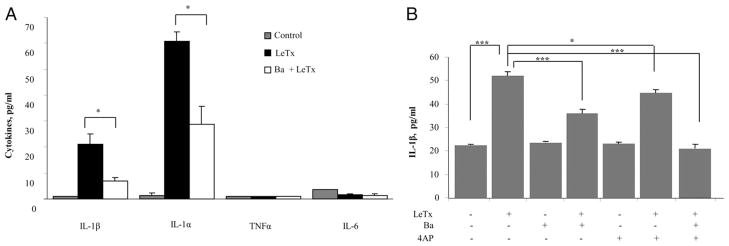
K+ channel dependence of LeTx-induced cytokine production. A, Cytokine Bio-plex assay in peritoneal macrophages was performed to measure IL-1β, IL-1α, TNF-α, and IL-6 cytokines. Cells were preincubated for 1 h with or without 300 μM Ba2+ and treated with LeTx (LF and PA, 100 ng/ml) or Ba2+ (300 μM) + LeTx for 3 h. Cells not treated with LeTx or LeTx + Ba2+ were used as untreated control. Each data set is shown as mean ± SEM of three different experiments with p values between LeTx and Ba2+ + LeTx. B, IL-1β release in MH-S cells was measured in culture supernatants from cells pretreated for 1 h with 300 μM Ba2+, 3 mM 4-AP, or both and then exposed to 4 h of LeTx (LF and PA, 100 ng/ml) in the presence or in the absence of the blockers. Each value represents the mean ± SEM of data from three experiments done with duplicate macrophage cell cultures on 3 separate days. *p < 0.05, ***p < 0.01.
To evaluate further the role of macrophage K+ channels in LeTx-induced release of IL-1β, we measured IL-1β release in MH-S macrophages in the presence and in the absence of Ba2+ or 4-AP to block Kir or Kv channels, respectively (Fig. 6B). Our results show that blocking either Kir or Kv channels significantly inhibits LeTx-induced IL-1β release with the combined Ba2+ and 4-AP treatment completely abrogating the effect. These experiments demonstrate that both Kir and Kv channels contribute to K+ efflux that is necessary for the IL-1β release (Fig. 6B).
As expected, cells overexpressing WT Kir2.1 channels had significantly larger Kir current densities compared with control cells expressing only the endogenous Kir channels or cells transduced with an empty vector. Exposing Kir2.1 WT-expressing cells to LeTx induced further increase in the current. In contrast, cells expressing dnKir2.1 (DN) had significantly less current density than control cells (Fig. 7A, 7B, DN versus Empty). In agreement with the results observed using Ba2+ pretreatment to block Kir (Fig. 6), the reduced Kir activity in macrophages transduced with the dnKir2.1 construct was associated with a loss of LeTx-induced secretion of IL-1β (Fig. 7C, DN versus Empty). Conversely, overexpression of WT Kir2.1 channels resulted in a marked increase in IL-1β release by LeTx-treated macrophages (Fig. 7C).
FIGURE 7.

LeTx-induced IL-1β production by macrophages depends on the level of Kir channel function. A, Representative Kir current recordings from alveolar macrophages (MH-S) under four experimental conditions: control untreated (−), transduced with an empty vector (the control and Empty traces overlap), transduced with a vector containing WT Kir, or transduced with a vector containing DN Kir. B, Mean peak current densities measured at −95 mV for the four conditions described above and, in addition, in cells transduced with WT Kir and then treated with LeTx. Each data set is expressed as mean ± SEM (n = 6 cells per condition in three independent experiments). C, Levels of IL-1β produced by alveolar macrophages (MH-S) under the four conditions of Kir expression described above: control-nontransduced, transduced with empty vector, transduced with a vector containing WT Kir, and transduced with a vector containing DN Kir. The results represent the mean ± SEM of data from three experiments done with duplicate macrophage cell cultures on 3 separate days. **p < 0.05, ***p < 0.01.
Discussion
The goal of this study was to identify the mechanism of LeTx-induced K+ efflux and its relationship to toxin-induced macrophage production of IL-1β. Our observations show that treatment of alveolar and peritoneal macrophages with LeTx results in an LF-metalloprotease activity-dependent activation of two types of K+ channels, Kir and Kv, and link this LF-induced enhancement of K+ activity to macrophage production of IL-1β. These studies suggest that both Kir and Kv channels represent potential targets for therapeutic interventions to treat LeTx-induced proinflammatory cytokine responses.
Consideration of the system
These studies were performed mainly on cells derived from BALB/c mice, a mouse strain that is highly susceptible to the lethal toxin and that is used, therefore, in multiple studies as a murine model for LeTx-induced toxic effects (e.g., Refs. 30–33). The susceptibility of BALB/c mice to LeTx has been attributed earlier to the rapid lysis of macrophages and release of proinflammatory cytokines (30, 34), which in turn depends on activation of the inflammatory proteins Nalp1b and caspase-1 (27). Indeed, consistent with the earlier studies (30), we observed that exposing either alveolar or peritoneal macrophages derived or freshly isolated from BALB/c mice to low doses of LeTx resulted in a significant increase in IL-1β release. Cells derived from BALB/c mice, therefore, appear to be the most appropriate model to study the mechanism of LeTx-induced release of proinflammatory cytokines from macrophages.
Furthermore, we focused on the low concentration range (up to 100 ng/ml) because both our observations and earlier studies from other investigators showed that LeTx-induced release of IL-1β in several types of macrophages plateaus at 100 ng/ml LeTx (5). Also, a similar concentration range of the PA (~100 ng/ml) was reported in blood samples of anthrax-infected mice during the early manifestation of the illness (35). We suggest, therefore, that these low concentrations of LeTx may correspond to the early stage of the disease development and play a critical role in triggering further proinflammatory responses.
The role of K+ channels in IL-1β release
Multiple studies have shown that initiation of K+ efflux from the cytosol to the extracellular space stimulates caspase-1–dependent cleavage of pro–IL-1β to mature IL-1β and facilitates IL-1β release in response to proinflammatory stimuli (e.g., Refs. 10, 11, 36). Specifically, macrophage release of IL-1β induced by LPS is increased by exposing macrophages to K+ ionophores, such as nigericin, gramicidin, and valinomycin (10, 11, 36). Conversely, exposing LPS-treated cells to a high extracellular K+ concentration to eliminate creation of a K+ gradient suppresses LPS-induced IL-1β secretion (11). The mechanism by which K+ efflux is thought to facilitate IL-1β maturation is by activation of caspase-1, the prototypic IL-1β–converting enzyme (10, 11, 37). Furthermore, Wickliffe et al. (14) have reported that K+ efflux is required for LeTx-induced activation of caspase-1 and formation of the multiprotein inflammasome complex. However, prior to the data we report in this study, there was only a limited amount of information about the specific pathways that contribute to K+ efflux induced by inflammatory stimuli. Indeed, it is expected that opening of any type of K+ channels or even any type of non-selective ion channels that are permeable to K+ might facilitate maturation and release of IL-1β. It was not clear, however, whether K+ efflux is mediated by K+ ion channels or by a non-specific disruption of the plasma membrane. It was also not known whether K+ efflux pathways are activated by the lethal toxin or whether K+ escapes through channels that are constitutively open. To our knowledge, our data provide the first direct evidence that exposure to LeTx activates two specific types of macrophage K+ channels, a Kir channel belonging to the Kir family of inward rectifies and a Kv channel belonging to the family of voltage-gated K+ channels. Furthermore, because LeTx-induced increase in K+ currents was not accompanied by any changes in the reversal potentials and was blocked by the known Kir and Kv blockers, respectively, we can exclude the possibility that LeTx-induced K+ efflux under these experimental conditions can be partially mediated by nonspecific membrane leaks or nonselective cation channels. In all of these experiments, cells were stimulated by LeTx alone in the absence of any additional stimuli, suggesting that LeTx alone is sufficient to induce activation of the channels. We also show that the mechanism by which LeTx activates macrophage K+ channels at least in part requires the proteolytic activity of LF, because an LFE687C mutant that has no metalloprotease activity (38, 39) does not activate either Kir or Kv. It remains to be determined whether LF directly cleaves and therefore modifies the channel protein or whether the LF effect on Kir and Kv activity is indirect.
The next major question is whether LeTx-induced activation of macrophage K+ channels might be an early or a late event in LeTx-induced cytotoxicity and IL-1β release. Indeed, although earlier studies suggested that K+ efflux is required before caspase-1 activation (14), other studies suggested that K+ efflux may be a late event occurring downstream of caspase-1 activation (40, 41). Our current observations showing that LeTx-induced activation of both Kir and Kv channels is also observed in macrophages isolated from C57BL/6 mice that are deficient in the ability to activate caspase-1 clearly indicate that activation of the channels is independent of caspase-1 activation.
Finally, our observations show that no increase in IL-1β release was observed in cells that overexpress WT Kir2.1 if they were not stimulated with LeTx. Thus, even though an increase in K+ current in cells overexpressing WT Kir2.1 was even higher than that in control cells treated with LeTx, an increase in Kir current alone in the absence of the toxin was not sufficient to stimulate IL-1β release. These observations are fully consistent with the previous studies showing that treating macrophages with K+ ionophore nigericin also did not induce IL-1β release in the absence of LeTx (42). We suggest, therefore, that an increase in the activity of macrophage K+ channels is necessary but not sufficient to induce IL-1β release.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health Grants HL073965 (to I.L.), HL083298 (to I.L.), 5T32HL082547 (to J.W.C.), AI53526 (to J.L.C.), AI57153 (to J.L.C.), AI56575 (to J.L.C.), and AI077949 (to J.L.C.), and by the Canadian Institutes for Health Research (MOP 44365), the Leducq Foundation (European-North American Atrial Fibrillation Research Alliance, 07/CVD/03) (to S.N.), and the James A. and Marion C. Grant Fund for Immunology Research (to J.L.C.).
We thank Dr. Stephen Leppla for reagents, methods, and advice related to toxin protein preparation and Dr. Ruxana Sadikot for helping with the IL-1β detection assay.
Abbreviations used in this article
- 4-AP
4-aminopyridine
- BMDM
bone marrow-derived macrophage
- DN
dominant-negative
- Kir
inwardly rectifying K+
- Kv
voltage-gated K+
- LeTx
lethal toxin
- LF
lethal factor
- mLeTx
mutant lethal factor protein LFE687C combined with protective Ag
- PA
protective Ag
- WT
wild-type
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Cohen S, Mendelson I, Altboum Z, Kobiler D, Elhanany E, Bino T, Leitner M, Inbar I, Rosenberg H, Gozes Y, et al. Attenuated non-toxinogenic and nonencapsulated recombinant Bacillus anthracis spore vaccines protect against anthrax. Infect Immun. 2000;68:4549–4558. doi: 10.1128/iai.68.8.4549-4558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brossier F, Mock M. Toxins of. Bacillus anthracis Toxicon. 2001;39:1747–1755. doi: 10.1016/s0041-0101(01)00161-1. [DOI] [PubMed] [Google Scholar]
- 3.Chopra AP, Boone SA, Liang X, Duesbery NS. Anthrax lethal factor proteolysis and inactivation of MAPK kinase. J Biol Chem. 2003;278:9402–9406. doi: 10.1074/jbc.M211262200. [DOI] [PubMed] [Google Scholar]
- 4.Leppla SH. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc Natl Acad Sci USA. 1982;79:3162–3166. doi: 10.1073/pnas.79.10.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cordoba-Rodriguez R, Fang H, Lankford CSR, Frucht DM. Anthrax lethal toxin rapidly activates caspase-1/ICE and induces extracellular release of interleukin (IL)-1beta and IL-18. J Biol Chem. 2004;279:20563–20566. doi: 10.1074/jbc.C300539200. [DOI] [PubMed] [Google Scholar]
- 6.Kang TJ, Basu S, Zhang L, Thomas KE, Vogel SN, Baillie L, Cross AS. Bacillus anthracis spores and lethal toxin induce IL-1beta via functionally distinct signaling pathways. Eur J Immunol. 2008;38:1574–1584. doi: 10.1002/eji.200838141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loving CL, Osorio M, Kim YG, Nuñez G, Hughes MA, Merkel TJ. Nod1/Nod2-mediated recognition plays a critical role in induction of adaptive immunity to anthrax after aerosol exposure. Infect Immun. 2009;77:4529–4537. doi: 10.1128/IAI.00563-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Popov SG, Popova TG, Grene E, Klotz F, Cardwell J, Bradburne C, Jama Y, Maland M, Wells J, Nalca A, et al. Systemic cytokine response in murine anthrax. Cell Microbiol. 2004;6:225–233. doi: 10.1046/j.1462-5822.2003.00358.x. [DOI] [PubMed] [Google Scholar]
- 9.Thornberry NA. Interleukin-1 beta converting enzyme. Methods Enzymol. 1994;244:615–631. doi: 10.1016/0076-6879(94)44045-x. [DOI] [PubMed] [Google Scholar]
- 10.Perregaux D, Gabel CA. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem. 1994;269:15195–15203. [PubMed] [Google Scholar]
- 11.Walev I, Reske K, Palmer M, Valeva A, Bhakdi S. Potassium-inhibited processing of IL-1 beta in human monocytes. EMBO J. 1995;14:1607–1614. doi: 10.1002/j.1460-2075.1995.tb07149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheneval D, Ramage P, Kastelic T, Szelestenyi T, Niggli H, Hemmig R, Bachmann M, MacKenzie A. Increased mature interleukin-1beta (IL-1beta) secretion from THP-1 cells induced by nigericin is a result of activation of p45 IL-1beta-converting enzyme processing. J Biol Chem. 1998;273:17846–17851. doi: 10.1074/jbc.273.28.17846. [DOI] [PubMed] [Google Scholar]
- 13.Hanna PC, Kochi S, Collier RJ. Biochemical and physiological changes induced by anthrax lethal toxin in J774 macrophage-like cells. Mol Biol Cell. 1992;3:1269–1277. doi: 10.1091/mbc.3.11.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wickliffe KE, Leppla SH, Moayeri M. Anthrax lethal toxin-induced inflammasome formation and caspase-1 activation are late events dependent on ion fluxes and the proteasome. Cell Microbiol. 2008;10:332–343. doi: 10.1111/j.1462-5822.2007.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKinney LC, Gallin EK. G-protein activators induce a potassium conductance in murine macrophages. J Membr Biol. 1992;130:265–276. doi: 10.1007/BF00240483. [DOI] [PubMed] [Google Scholar]
- 16.DeCoursey TE, Kim SY, Silver MR, Quandt FN. Ion channel expression in PMA-differentiated human THP-1 macrophages. J Membr Biol. 1996;152:141–157. doi: 10.1007/s002329900093. [DOI] [PubMed] [Google Scholar]
- 17.Mackenzie AB, Chirakkal H, North RA. Kv1.3 potassium channels in human alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2003;285:L862–L868. doi: 10.1152/ajplung.00095.2003. [DOI] [PubMed] [Google Scholar]
- 18.Vicente R, Escalada A, Coma M, Fuster G, Sánchez-Tilló E, López-Iglesias C, Soler C, Solsona C, Celada A, Felipe A. Differential voltage-dependent K+ channel responses during proliferation and activation in macrophages. J Biol Chem. 2003;278:46307–46320. doi: 10.1074/jbc.M304388200. [DOI] [PubMed] [Google Scholar]
- 19.Mbawuike IN, Herscowitz HB. MH-S, a murine alveolar macrophage cell line: morphological, cytochemical, and functional characteristics. J Leukoc Biol. 1989;46:119–127. doi: 10.1002/jlb.46.2.119. [DOI] [PubMed] [Google Scholar]
- 20.Celada A, Gray PW, Rinderknecht E, Schreiber RD. Evidence for a gamma-interferon receptor that regulates macrophage tumoricidal activity. J Exp Med. 1984;160:55–74. doi: 10.1084/jem.160.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fang Y, Schram G, Romanenko VG, Shi C, Conti L, Vandenberg CA, Davies PF, Nattel S, Levitan I. Functional expression of Kir2.x in human aortic endothelial cells: the dominant role of Kir2.2. Am J Physiol Cell Physiol. 2005;289:C1134–C1144. doi: 10.1152/ajpcell.00077.2005. [DOI] [PubMed] [Google Scholar]
- 22.Petrache I, Choi ME, Otterbein LE, Chin BY, Mantell LL, Horowitz S, Choi AM. Mitogen-activated protein kinase pathway mediates hyperoxia-induced apoptosis in cultured macrophage cells. Am J Physiol. 1999;277:L589–L595. doi: 10.1152/ajplung.1999.277.3.L589. [DOI] [PubMed] [Google Scholar]
- 23.Farberman MM, Demello DE, Hoffmann JW, Ryerse JS. Morphologic changes in alveolar macrophages in response to UVEC-activated pulmonary Type II epithelial cells. Tissue Cell. 2005;37:213–222. doi: 10.1016/j.tice.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 24.Kim SO, Jing Q, Hoebe K, Beutler B, Duesbery NS, Han J. Sensitizing anthrax lethal toxin-resistant macrophages to lethal toxin-induced killing by tumor necrosis factor-alpha. J Biol Chem. 2003;278:7413–7421. doi: 10.1074/jbc.M209279200. [DOI] [PubMed] [Google Scholar]
- 25.Moayeri M, Haines D, Young HA, Leppla SH. Bacillus anthracis lethal toxin induces TNF-alpha-independent hypoxia-mediated toxicity in mice. J Clin Invest. 2003;112:670–682. doi: 10.1172/JCI17991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klimpel KR, Arora N, Leppla SH. Anthrax toxin lethal factor contains a zinc metalloprotease consensus sequence which is required for lethal toxin activity. Mol Microbiol. 1994;13:1093–1100. doi: 10.1111/j.1365-2958.1994.tb00500.x. [DOI] [PubMed] [Google Scholar]
- 27.Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet. 2006;38:240–244. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 28.Moayeri M, Crown D, Dorward DW, Gardner D, Ward JM, Li Y, Cui X, Eichacker P, Leppla SH. The heart is an early target of anthrax lethal toxin in mice: a protective role for neuronal nitric oxide synthase (nNOS) PLoS Pathog. 2009;5:e1000456. doi: 10.1371/journal.ppat.1000456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- 30.Hanna PC, Acosta D, Collier RJ. On the role of macrophages in anthrax. Proc Natl Acad Sci USA. 1993;90:10198–10201. doi: 10.1073/pnas.90.21.10198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muehlbauer SM, Evering TH, Bonuccelli G, Squires RC, Ashton AW, Porcelli SA, Lisanti MP, Brojatsch J. Anthrax lethal toxin kills macrophages in a strain-specific manner by apoptosis or caspase-1-mediated necrosis. Cell Cycle. 2007;6:758–766. doi: 10.4161/cc.6.6.3991. [DOI] [PubMed] [Google Scholar]
- 32.Rivera J, Cordero RJB, Nakouzi AS, Frases S, Nicola A, Casadevall A. Bacillus anthracis produces membrane-derived vesicles containing biologically active toxins. Proc Natl Acad Sci USA. 2010;107:19002–19007. doi: 10.1073/pnas.1008843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muehlbauer SM, Lima HJ, Jr, Goldman DL, Jacobson LS, Rivera J, Goldberg MF, Palladino MA, Casadevall A, Brojatsch J. Proteasome inhibitors prevent caspase-1-mediated disease in rodents challenged with anthrax lethal toxin. Am J Pathol. 2010;177:735–743. doi: 10.2353/ajpath.2010.090828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanna PC, Kruskal BA, Ezekowitz RA, Bloom BR, Collier RJ. Role of macrophage oxidative burst in the action of anthrax lethal toxin. Mol Med. 1994;1:7–18. [PMC free article] [PubMed] [Google Scholar]
- 35.Tang S, Moayeri M, Chen Z, Harma H, Zhao J, Hu H, Purcell RH, Leppla SH, Hewlett IK. Detection of anthrax toxin by an ultra-sensitive immunoassay using europium nanoparticles. Clin Vaccine Immunol. 2009;16:408–413. doi: 10.1128/CVI.00412-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perregaux D, Barberia J, Lanzetti AJ, Geoghegan KF, Carty TJ, Gabel CA. IL-1 beta maturation: evidence that mature cytokine formation can be induced specifically by nigericin. J Immunol. 1992;149:1294–1303. [PubMed] [Google Scholar]
- 37.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 38.Park S, Leppla SH. Optimized production and purification of Bacillus anthracis lethal factor. Protein Expr Purif. 2000;18:293–302. doi: 10.1006/prep.2000.1208. [DOI] [PubMed] [Google Scholar]
- 39.Xu Q, Zeng M. Detoxified lethal toxin as a potential mucosal vaccine against anthrax. Clin Vaccine Immunol. 2008;15:612–616. doi: 10.1128/CVI.00402-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alileche A, Squires RC, Muehlbauer SM, Lisanti MP, Brojatsch J. Mitochondrial impairment is a critical event in anthrax lethal toxin-induced cytolysis of murine macrophages. Cell Cycle. 2006;5:100–106. doi: 10.4161/cc.5.1.2283. [DOI] [PubMed] [Google Scholar]
- 41.Averette KM, Pratt MR, Yang Y, Bassilian S, Whitelegge JP, Loo JA, Muir TW, Bradley KA. Anthrax lethal toxin induced lysosomal membrane permeabilization and cytosolic cathepsin release is Nlrp1b/Nalp1b-dependent. PLoS ONE. 2009;4:e7913. doi: 10.1371/journal.pone.0007913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.