

Abstract
In recent papers, there has been a lively exchange concerning theories for enzyme catalysis, especially the role of protein dynamics/pre-chemistry conformational changes in the catalytic cycle of enzymes. Of particular interest is the notion that substrate-induced conformational changes that assemble the polymerase active site prior to chemistry are required for DNA synthesis and impact fidelity (i.e., substrate specificity). High-resolution crystal structures of DNA polymerase β representing intermediates of substrate complexes prior to the chemical step are available. These structures indicate that conformational adjustments in both the protein and substrates must occur to achieve the requisite geometry of the reactive participants for catalysis. We discuss computational and kinetic methods to examine possible conformational change pathways that lead from the observed crystal structure intermediates to the final structures poised for chemistry. The results, as well as kinetic data from site-directed mutagenesis studies, are consistent with models requiring pre-chemistry conformational adjustments in order to achieve high fidelity DNA synthesis. Thus, substrate-induced conformational changes that assemble the polymerase active site prior to chemistry contribute to DNA synthesis even when they do not represent actual rate-determining steps for chemistry.
Keywords: Enzyme catalysis, Intrinsic protein dynamics, Pre-chemistry conformational adjustments, Nucleotidyl transfer, DNA polymerase β, Catalytic cycle chemical step
1 Introduction
Recent papers have provided a lively exchange concerning theories for enzyme catalysis, specifically the role of intrinsic dynamics in the catalytic cycles as deduced from molecular simulations (e.g., [1–8]). The issues involve the functional significance of these dynamics, the relation of conformational to chemical transitions, and the utility of theories that emerge from atomistic simulations concerning motions and associated energy landscapes. Various papers have pointed out issues of semantics in these discussions concerning what is dynamics: correlated conformational changes that occur throughout the enzyme’s pathway and act to facilitate the chemical reaction on one hand, versus rearrangements that have a direct link to the transition state in the chemical reaction on the other [6]. It has also been generally concluded in many of the above cited reviews that reliable molecular simulations have an important role in elucidating structural changes during enzyme catalytic cycles, including achieving reactive conformations, even if they do not support a catalytic role for dynamics in driving the chemical reaction per se [4].
DNA polymerases are multi-domain enzymes with a polymerase domain and accessory domains. The polymerase domain is responsible for the nucleotidyl transferase reaction and is usually composed of three subdomains. DNA polymerase (pol) β is the simplest eukaryotic DNA polymerase and has been well characterized structurally [9]. A key structural observation that has been made for several DNA polymerases is that a subdomain closes around the nascent base pair upon binding of the incoming correct nucleoside triphosphate, to trap it between the growing DNA duplex and the polymerase [10, 11]. The transition from the open polymerase/DNA binary complex to the closed ternary polymerase/DNA/dNTP complex is accompanied by conformational adjustments in key protein side chains, DNA nucleotides, and the incoming nucleotide. In addition, the binding of two metal cofactors, Mg2+, is required for chemistry and is known to influence local conformations and subdomain motions [12, 13].
Although it is generally believed that chemistry is the rate-limiting step during DNA synthesis for DNA polymerases, the lack of a straightforward assay to assess whether polymerase and/or substrate conformational changes limit DNA synthesis in specific cases (e.g., altered enzymes or substrates) dictates that activity measurements should be interpreted cautiously. In the specific case of pol β, the role of the assembly of enzyme side chains, substrates, and metals necessary to achieve optimal geometry for chemistry has been recently discussed. Warshel and Prasad argue that conformational adjustments preceding the pol β chemical step are inconsequential to understanding catalysis or fidelity [14]. We present an alternate viewpoint: such adjustments have a role in catalysis since they are a pre-requisite for DNA synthesis. In this perspective, we elaborate on this point and review challenges to integrating results from kinetic, structural and computational approaches toward understanding polymerase catalysis.
2 Induced-fit and pre-chemistry conformational changes
As already recently argued in the literature [3–7], an understanding of enzyme-catalyzed reactions requires knowledge of many steps, conformational and chemical, with the later being just one part of the big picture [5]. There is increasing support for pre-chemistry conformational changes as an important component in the overall catalytic cycle for pol β. Many of the pre-chemistry atomic adjustments have been revealed through X-ray crystallography [12, 15], NMR spectroscopy [13], and fluorescence spectroscopy [16] and have also been studied using computational approaches (e.g., [17–21]). The combined picture of the energy landscape is needed to link atomistic details with results from various experimental methods and pursue enzyme design applications.
The induced-fit hypothesis [22] argues that the alignment of catalytic groups be optimized for high activity and that substrate binding provides energy to overcome the unfavorable conformation. As noted above, crystallographic structures of DNA polymerases in various liganded states indicate that subdomain re-positioning is thought to be an essential step to ready the system for the chemically competent state. Originally, these subdomain motions were thought to be rigid, but computer simulations revealed an unexpected complexity in subtle side-chain motions that evolve the system into the state ready for chemistry [23, 24]. This work also suggested that it is these side-chain motions that are rate limiting in the conformational closing pathway rather than the overall subdomain closing motion. Such comparative studies for matched (correct) and mismatched (incorrect) incoming nucleotides (e.g., [17, 25–28]) thus led to a better understanding of the role of specific enzyme residues as well as ions in organizing the active site. Our suggestion of pre-chemistry conformational changes in this connection [23] is a specific application of the induced-fit hypothesis. For this discussion, the terms dynamics or pre-chemistry conformational changes refer to substrate or enzyme atomic rearrangements that have catalytic consequences, that is, events that impact phosphoryl transfer chemistry.
There have been many studies exploring alternative or conjunctive mechanisms to induced-fit, namely conformational selection [29–33]. Though the difference is subtle and refers to whether the enzyme has the intrinsic capacity to undergo the requisite conformational changes (which are merely favored when substrate binds via a shift in equilibrium in the conformational energy landscape) or whether the substrate actually triggers these changes, dynamics studies suggest that a hybrid mechanism is operative for polymerases of the X-family [34]. In fact, follow-up work suggested that while subdomain motions appear intrinsic to the DNA polymerase, the subtle side-chain motions have higher activation energy barriers and their dynamics and favored states are largely determined by the binding of the substrate [35].
Figure 1 shows this behavior of the potential mean force for the pol β closing pathway prior to chemistry, as computed using the nudged elastic band method [29] for three order parameters (subdomain motion, Phe272 ring flipping, and rotation of Arg258) in three cases where the templating (coding) base is guanine: 1) without the correct dCTP substrate (top), 2) with correct dCTP (middle), and 3) with incorrect dATP (bottom). We see that while the subdomain motion is essentially a barrier-free motion in agreement with prior studies [24], substrate binding modulates the distribution of equilibrium enzyme conformations of key side-chain conformations as needed for the chemical reaction. These data are in agreement with single-molecule FRET studies of the Klenow fragment of Pol I showing a high degree of conformational flexibility in the unliganded state and occupancy of distinct conformational states other than the open and closed conformations in the presence of an incorrect substrate [36]. These results are also in line with similar single-molecule studies of HIV-1 reverse transcriptase that suggest conformational heterogeneity of the polymerase/DNA complex depending on the binding register of the DNA substrate [37]. The collective data thus indicate the applicability of the conformational selection model: substrate binding to a specific polymerase conformation shifts the energy landscape to favor one state over another rather than triggering a transition per se. The studies are applicable to many other enzymes [38–40] and further underscore the range and relevance of enzyme dynamics for understanding their complete mechanism.
Fig. 1.

Quantitative characterization of the open/closed transitions in pol β/DNA complex without dNTP and in the presence of correct (dCTP) and incorrect (dATP) incoming nucleotide (top to bottom) [35]. Shown are two-dimensional free energy surfaces as a function of the ΔDrmsd progress variable for three order parameters: α-Helix N rmsd with respect to the closed crystal state, and dihedral angles of key active-site residues, Asp192 and Arg258. ΔDrmsd is the difference in rmsd value of the instantaneous structure from the reference open and closed states: negative values correspond to the more open-like state while positive values correspond to closed-like state. Near the open and closed states, the values of the order parameters range as follows: α-helix N rmsd, from 6 to 1.5 Å; Asp192 flip, from 90° to 180°; and Arg258 rotation, from 100° to 260°. These order parameters characterize the closing transition of pol β and were previously identified as transition states along the pathway [24]. Here, the free energy profiles were obtained by performing umbrella sampling simulations on optimized NEB paths. In umbrella sampling simulations, weak harmonic restraints were placed along the ΔDrmsd progress variable. The data were unbiased using WHAM to obtain free energy surfaces
3 Influence of pre-chemistry conformational changes on enzyme efficiency and fidelity
Although protein and substrate conformational adjustments are required to optimize the active-site architecture for efficient chemistry, the role of these adjustments in substrate specificity (fidelity) is controversial (see [41] for discussion). In general, DNA polymerase fidelity is the ratio of insertion efficiencies for right and wrong incoming nucleotides. High fidelity DNA polymerases have high insertion efficiencies for the correct nucleotide, whereas the insertion efficiency for incorrect nucleotides is much lower and generally similar for all DNA polymerases irrespective of their fidelity [42].
Site-directed mutagenesis of DNA polymerases has demonstrated that some of the altered enzymes have decreased fidelity. Accordingly, correct and/or incorrect nucleotide insertion efficiencies must be differentially affected with the mutant enzyme, suggesting that the altered side chain plays a unique role(s) for correct and incorrect insertion. For example, Arg283 of wild-type pol β interacts with the template strand in the closed substrate bound enzyme, but does not interact with DNA in the absence of a nucleotide (i.e., open enzyme form) (Fig. 2) [43]. Replacing this arginine side chain with alanine would be expected to destabilize the closed form of the ternary complex. The kinetic result is a loss of correct, but not incorrect, insertion efficiency leading to a dramatically lower fidelity [42, 44]. Thus, Arg283 appears to play an essential role in pre-chemistry events for correct, but not incorrect, nucleotide insertion.
Fig. 2.
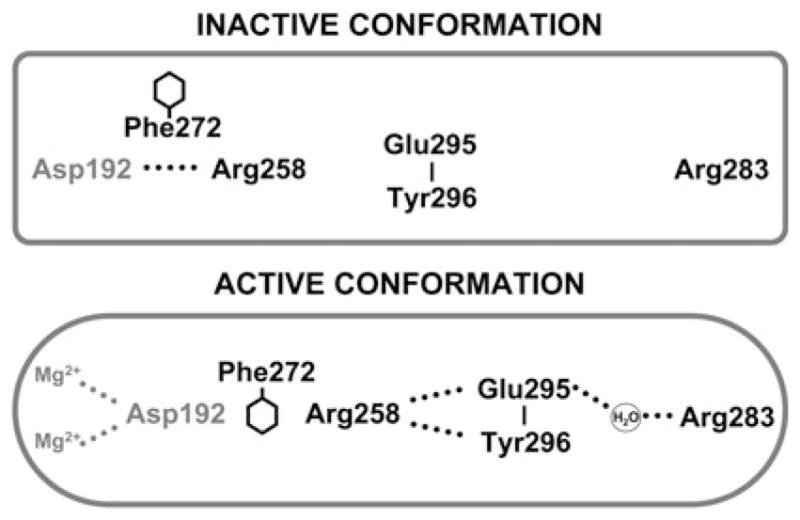
Altered side-chain interactions accompanying subdomain closure of pol β. In the open inactive conformation, Arg283 does not interact with other key residues, but in the closed active conformation, it interacts with the templating (coding) base, the upstream template nucleotide (not illustrated), and with Glu295 (indirectly). Thus, the position of the subdomain can be structurally transmitted to the active site through a series of interactions involving Arg283 permitting Asp192 to coordinate both active-site Mg2+ ions. This is also accompanied by altered interactions of Glu295/Tyr296 with Arg258 in the open (inactive) and closed (active) forms. Phe272 is postulated to transiently interfere with interactions between Asp192 and Arg258 permitting an interaction with Glu295/Tyr296. Residues of the subdomain that closes are indicated in boldface text. This figure was reproduced from Ref. [13] with permission
Likewise, Glu295 has been implicated in providing key contacts as the polymerase transitions from an open to closed complex (Fig. 2) [13]. It has also been shown that the enzyme is catalytically compromised upon lysine substitution [45, 46]. A crystal structure of a binary DNA complex of the E295K mutant indicates an intermediate conformation between open and closed forms [47]. Further computational work [47] supports a very high-energy barrier for the closing conformational pathway of this enzyme due to electrostatic and steric factors (Fig. 3). In fact, the high-energy barrier comes not from subdomain closing but from the rotation of Arg258 that must release Asp192 to coordinate active-site metals. Indeed, NMR studies are consistent with the idea that the E295K mutant is unable to achieve a closed ‘activated’ complex in the presence of a correct nucleotide/Mg2+ [13]. Thus, this example underscores the need to understand conformational as well as chemical events: mutants may exist with conformational transitions that are not only important but possibly rate limiting.
Fig. 3.
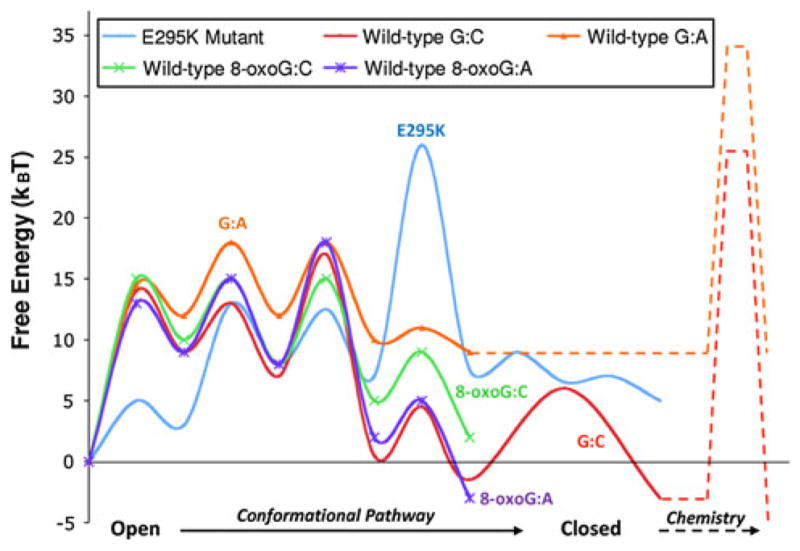
Estimated conformational and chemical pathways and/or barrier for four pol β systems as collected from several works: wild-type system with correct and incorrect incoming bases [17, 19, 20]; wild-type system with the 8-oxoG DNA lesion [59]; and for E295K mutant [47]. All conformational barrier calculations employ transition path sampling, and the chemistry estimates are obtained from various groups using QM/MM and EVB techniques
Although high-resolution structures of DNA polymerases from several families in various liganded states have provided valuable insights into the structure and mechanism of DNA polymerases, correlating these structures with observed kinetic behavior is not straightforward. These structures reveal that the protein and substrates must undergo structural changes to attain the geometry necessary for chemistry. Appropriately, these conformational changes should align catalytic groups for efficient correct nucleotide insertion and misalign catalytic atoms for inefficient incorrect nucleotide misinsertion. Indeed, structures of pol β with active-site mismatches reveal that subdomain closure misaligns key catalytic atoms [19, 48].
For nucleotide binding to a polymerase/DNA complex, a simple representation of a two-step nucleotide-binding model proposed by Johnson and Benkovic [49] is illustrated in Fig. 4. Historically, indirect kinetic evidence suggested that a non-chemical step limited the overall rate (i.e., k2 and k−2 were slow relative to k3, Fig. 4); in other words, a conformational change believed to be the open (ES) to closed (*ES) polymerase transition was rate limiting for correct nucleotide insertion. More recently, the rate of the open to closed enzyme transition [50–53] or template conformational adjustments [16, 54] has been experimentally determined to be faster than chemistry. This indicated that the chemical step was the slowest step for the forward reaction. Tsai and Johnson point out, however, the efficiency (kcat/Km) of correct nucleotide insertion is independent of the chemical step (k3) [41, 52] even when it is slow. For correct insertion, efficiency is approximately K1k2 where K1 = k1/k−1. Critically, catalytic efficiency is linked to the reversal of the polymerase conformational change (k−2; where k−2,incorrect ≫ k−2,correct; i.e., K2 = k2/k−2 = [*ES]/[ES] ≪ 1). Thus, the concentration of *ES accumulates for correct insertion, but not for incorrect insertion (i.e., [ES] is high). The significance of this simple result is that chemistry, even when the slowest step, cannot be directly measured in all situations since the measured rate is k3[*ES] and [*ES] will depend on the magnitude of several rate constants including k−2. The conformational change forming *ES insulates the correct nucleotide from rapidly dissociating from the ES complex (nucleotide binding is in rapid equilibrium; i.e., k−1 ≫ k2) by forming a complex (*ES) with a slower reverse rate constant (k−2). In contrast, when k−2 is rapid, as in the case for an incorrect nucleotide, *ES does not accumulate and the wrong nucleotide can quickly dissociate from the ES complex. In this situation, the flux through the catalytic cycle is significantly reduced due to the low concentration of *ES. Since the magnitude of k−2 is not known in most instances, the observed reaction rate cannot be directly related to chemistry or conformational changes.
Fig. 4.
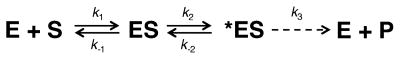
Two-step substrate binding model. Enzyme (E) binds a substrate/ligand (S) to form an initial collision complex (ES). This complex undergoes a conformational change to form a productive complex (*ES) that can undergo chemistry to form products (P). For many DNA polymerases, the ES and *ES states would represent the open DNA binary and closed dNTP ternary complexes, respectively, observed crystallographically. Note that when chemistry (k3) is rate limiting, the observed velocity will depend on the concentration of *ES. Observed changes in the rate of product formation can be due to changes in k3 and/or changes in the concentration of *ES. Nucleotide binding to DNA polymerases is believed to utilize a two-step binding mechanism [41, 49, 52]. Accordingly, attempting to interpret or assign energy barriers to activity measurements is problematic, since the distribution of intermediate enzyme species is not known
This simple analysis applied to DNA polymerase only accounts for nucleotide binding and does not address other conformational adjustments in the substrates (i.e., DNA translocation) that also must occur. Additionally, pyrophosphorolysis (reverse of step 3 in Fig. 4, not shown) and post-chemistry conformational changes would alter the distribution of enzyme species thereby confounding the interpretation of experimentally determined rates. Kinetic characterization of the sensitivity of the mitochondrial DNA polymerase γ to nucleoside analogs has demonstrated that the reverse reaction and post-chemistry conformational changes can influence nucleotide insertion [55]. From this discussion, it is apparent that interpretations of activity measurements based solely on steady-state or transient-state kinetic approaches can be error prone, since they rely on the precise mechanism and magnitude of the intrinsic rate constants. A kinetic mechanism that defines the rate constants for each pertinent step should be consistent with results from both of these kinetic approaches and provide a deeper understanding of the distribution of intermediate enzyme complexes during catalytic cycling. The ambiguity in defining the distribution of enzyme complexes during catalytic cycling makes correlating computationally determined energy barriers to specific enzymatic steps a challenge. This challenge also extends to correlating high-resolution crystallographic structures to specific enzymatic intermediate species that have been postulated through kinetic characterization and the inferred events that must occur during transition between these species.
The observation that conformational changes are required for catalytic activation supports the concept of an induced-fit mechanism proposed by Koshland [22] and related mechanisms [29–33]. It has been pointed out that induced fit can alter enzyme specificity even when critical conformational changes are not rate limiting [56], such as when the transition states for correct and incorrect incorporation are unique. Although an induced-fit model reduces enzyme specificity relative to a situation where a unique transition state exists, the reduced specificity represents an acceptable compromise for an enzyme such as a DNA polymerase, which must select a different/new substrate (DNA and dNTP) with each insertion event.
4 Concluding remarks
The complex internal motions and simulation of the entire configurational landscape are essential to understanding function as well as how enzymes achieve regulation and function with different substrates and through various protein associations. The commentary by Mulholland et al. [57] also underscores the importance of molecular simulation to make connections with experiment, to predict behavior of mutant systems and alternative substrate, and to connect the conformational to chemical pathways, as we have done for various polymerases in many different contexts of substrates, mutations, and other conditions. These combined pathways and conformations are utilized by DNA polymerases to enhance fidelity. Clearly, works that adopt these broader views and attempt to connect the many aspects of enzyme activity and function help contribute to the deciphering of complex enzyme machineries, of which polymerases are notable in both their versatility and fidelity. From an overview of catalytic efficiencies for divergent DNA polymerases, it is clear that high fidelity DNA polymerases are optimized for correct nucleotide insertion. These high fidelity enzymes exhibit subdomain, enzyme side chain, and substrate adjustments that contribute to optimizing the electrostatics and geometry of the active site [10]. Thus, substrate-induced conformational changes that assemble the polymerase active site prior to chemistry are required for DNA synthesis.
Crystallographic structures [12, 44, 47], spectroscopic NMR studies [13], and kinetic evidence [44, 46] highlight critical residues believed to play an important role in these pre-chemistry events for pol β. Although these pre-chemistry events are difficult to experimentally measure, computational approaches can provide models that bridge crystallographic structures and kinetic properties.
Computational methods are subject to the well-known approximations and imperfections of force fields and sampling [58], but perfection is not a pre-requisite for utility. Ultimately, the success of such predictions and interpretations can be tested experimentally. Work that attempts to merge a range of experimental knowledge and computational insights is valuable. Many computational studies by a variety of approaches have added further insights to the behavior of mutant enzymes by describing altered energetic and dynamic scenarios for alternate enzyme/substrate conformations, their effect on the overall enzymatic reaction, and associated feasible reaction pathways. Such cumulative data provide a framework to interpret a wide range of behavior observed with different polymerase systems across several DNA polymerase families and stimulate the design of further experiments. A view that focuses on the chemical barrier [14] may overlook important steps required to achieve a catalytically competent state and, accordingly, cannot always reliably predict the behavior of altered enzymes.
Acknowledgments
Research described in this Article was supported in part by Philip Morris USA Inc. and Morris International and by NSF award MCB-0316771, and NIH award R01 ES012692 to T.S., and Research Project Numbers Z01-ES050158 and Z01-ES050161 to S.H.W. in the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences and was in association with NIH award 1U19CA105010.
Footnotes
Editor’s Note: This paper and papers by Mulholland AJ, Roitberg AE, Tunñón I (doi:10.1007/s00214-012-1286-8) and Ram Prasad B, Kamerlin SCL, Florián J, Warshel A (doi:10.1007/s00214-012-1288-6) document and discuss contrasting outlooks on the questions of pre-chemistry and catalysis in DNA polymerase. All authors were initially provided with one another’s manuscripts, at which point opportunities to make revisions were offered, and finally Mulholland, Roitberg, and Tunñón were given the ‘last word’ on the revised manuscripts in their role as commentators. The editors of TCA hope that this discussion will illuminate key issues affecting ongoing work in this area.
Contributor Information
Tamar Schlick, Email: schlick@nyu.edu, Department of Chemistry, New York University, 100 Washington Square East, Silver Building, New York, NY 10003, USA. Courant Institute of Mathematical Sciences, New York, University, 251 Mercer Street, New York, NY 10012, USA.
Karunesh Arora, Departments of Chemistry and Biophysics, University of Michigan, 930 N. University Avenue, Ann Arbor, MI 48109, USA.
William A. Beard, Laboratory of Structural Biology, National Institute of Environmental Sciences, National Institutes of Health, P.O. Box 12233, Research Triangle Park, NC 27709, USA
Samuel H. Wilson, Laboratory of Structural Biology, National Institute of Environmental Sciences, National Institutes of Health, P.O. Box 12233, Research Triangle Park, NC 27709, USA
References
- 1.Garcia-Viloca M, Gao J, Karplus M, Truhlar DG. How enzymes work: analysis by modern rate theory and computer simulations. Science. 2004;303:186. doi: 10.1126/science.1088172. [DOI] [PubMed] [Google Scholar]
- 2.Kamerlin SCL, Warshel A. At the dawn of the 21st century: is dynamics the missing link for understanding enzyme catalysis? Proteins. 2010;78:1339. doi: 10.1002/prot.22654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karplus M. Role of conformation transitions in adenylate kinase. Proc Natl Acad Sci USA. 2010;107:E71. doi: 10.1073/pnas.1002180107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGeagh JD, Ranaghan KE, Mulholland AJ. Protein dynamics and enzyme catalysis: insights from simulations. Biochim Biophys Acta. 2011;1814:1077. doi: 10.1016/j.bbapap.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Nagel ZD, Klinman JP. Tunneling and dynamics in enzymatic hydride transfer. Chem Rev. 2006;106:3095. doi: 10.1021/cr050301x. [DOI] [PubMed] [Google Scholar]
- 6.Nashine VC, Hammes-Schiffer S, Benkovic SJ. Coupled motions in enzyme catalysis. Curr Opin Chem Biol. 2010;14:644. doi: 10.1016/j.cbpa.2010.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Villali J, Kern D. Choreographing an enzyme’s dance. Curr Opin Chem Biol. 2010;14:636. doi: 10.1016/j.cbpa.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hammes GG, Benkovic SJ, Hammes-Schiffer S. Flexibility, diversity, and cooperativity: pillars of enzyme catalysis. Biochemistry. 2011;50:10422. doi: 10.1021/bi201486f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beard WA, Wilson SH. Structure and mechanism of DNA polymerase β. Chem Rev. 2006;106:361. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- 10.Beard WA, Wilson SH. Structural insights into the origins of DNA polymerase fidelity. Structure (Camb) 2003;11:489. doi: 10.1016/s0969-2126(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 11.Joyce CM, Benkovic SJ. DNA polymerase fidelity: kinetics, structure, and checkpoints. Biochemistry. 2004;43:14317. doi: 10.1021/bi048422z. [DOI] [PubMed] [Google Scholar]
- 12.Batra VK, Beard WA, Shock DD, Krahn JM, Pedersen LC, Wilson SH. Magnesium induced assembly of a complete DNA polymerase catalytic complex. Structure (Camb) 2006;14:757. doi: 10.1016/j.str.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirby TW, DeRose EF, Cavanaugh NA, Beard WA, Shock DD, Mueller GA, Wilson SH, London RE. Metal-induced DNA translocation leads to DNA polymerase conformational activation. Nucl Acids Res. 2012;40:2974. doi: 10.1093/nar/gkr1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prasad BR, Warshel A. Prechemistry versus preorganization in DNA replication fidelity. Proteins. 2011;79:2900. doi: 10.1002/prot.23128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sawaya MR, Prasad P, Wilson SH, Kraut J, Pelletier H. Crystal structures of human DNA polymerase β complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry. 1997;36:11205. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- 16.Zhong X, Patel SS, Tsai M-D. DNA polymerase β. 5. Dissecting the functional roles of the two metal ions with Cr(III)dTTP. J Am Chem Soc. 1998;120:235. [Google Scholar]
- 17.Radhakrishnan R, Schlick T. Fidelity discrimination in DNA polymerase β: differing closing profiles for a mismatched (G:A) versus matched (G:C) base pair. J Am Chem Soc. 2005;127:13245. doi: 10.1021/ja052623o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin P, Pedersen LC, Batra VK, Beard WA, Wilson SH, Pedersen LG. Energy analysis of chemistry for correct insertion by DNA polymerase β. Proc Natl Acad Sci USA. 2006;103:13294. doi: 10.1073/pnas.0606006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin P, Batra VK, Pedersen LC, Beard WA, Wilson SH, Pedersen LG. Incorrect nucleotide insertion at the active site of a G:A mismatch catalyzed by DNA polymerase β. Proc Natl Acad Sci USA. 2008;105:5670. doi: 10.1073/pnas.0801257105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Radhakrishnan R, Schlick T. Correct and incorrect nucleotide incorporation pathways in DNA polymerase β. Biochem Biophys Res Commun. 2006;350:521. doi: 10.1016/j.bbrc.2006.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Schlick T. Quantum mechanics/molecular mechanics investigation of the chemical reaction in Dpo4 reveals water-dependent pathways and requirements for active site reorganization. J Am Chem Soc. 2008;130:13240. doi: 10.1021/ja802215c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koshland DE., Jr Application of a theory of enzyme specificity to protein synthesis. Proc Natl Acad Sci USA. 1958;44:98. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Radhakrishnan R, Arora K, Wang Y, Beard WA, Wilson SH, Schlick T. Regulation of DNA repair fidelity by molecular checkpoints: “Gates” in DNA polymerase β’s substrate selection. Biochemistry. 2006;45:15142. doi: 10.1021/bi061353z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radhakrishnan R, Schlick T. Orchestration of cooperative events in DNA synthesis and repair mechanism unraveled by transition path sampling of DNA polymerase β’s closing. Proc Natl Acad Sci USA. 2004;101:5970. doi: 10.1073/pnas.0308585101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang L, Arora K, Beard WA, Wilson SH, Schlick T. Critical role of magnesium ions in DNA polymerase β’s closing and active site assembly. J Am Chem Soc. 2004;126:8441. doi: 10.1021/ja049412o. [DOI] [PubMed] [Google Scholar]
- 26.Yang L, Beard W, Wilson S, Roux B, Broyde S, Schlick T. Local deformations revealed by dynamics simulations of DNA polymerase β with DNA mismatches at the primer terminus. J Mol Biol. 2002;321:459. doi: 10.1016/s0022-2836(02)00617-4. [DOI] [PubMed] [Google Scholar]
- 27.Yang L, Beard WA, Wilson SH, Broyde S, Schlick T. Polymerase β simulations suggest that Arg258 rotation is a slow step rather than large subdomain motions per se. J Mol Biol. 2002;317:679. doi: 10.1006/jmbi.2002.5450. [DOI] [PubMed] [Google Scholar]
- 28.Yang L, Beard WA, Wilson SH, Broyde S, Schlick T. Highly organized but pliant active site of DNA polymerase β: compensatory mechanisms in mutant enzymes revealed by dynamics simulations and energy analyses. Biophysical J. 2004;86:3392. doi: 10.1529/biophysj.103.036012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arora K, Brooks CL., III Large-scale allosteric conformational transitions of adenylate kinase appear to involve a population-shift mechanism. Proc Natl Acad Sci USA. 2007;104:18496. doi: 10.1073/pnas.0706443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bucher D, Grant BJ, McCammon JA. Induced fit or conformational selection? The role of the semi-closed state in the maltose binding protein. Biochemistry. 2011;50:10530. doi: 10.1021/bi201481a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hammes GG, Chang Y-C, Oas TG. Conformational selection or induced fit: a flux description of reaction mechanism. Proc Natl Acad Sci USA. 2009;106:13737. doi: 10.1073/pnas.0907195106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma B, Nussinov R. Enzyme dynamics point to stepwise conformational selection in catalysis. Curr Opin Chem Biol. 2010;14:652. doi: 10.1016/j.cbpa.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wlodarski T, Zagrovic B. Conformational selection and induced fit mechanism underlie specificity in noncovalent interactions with ubiquitin. Proc Natl Acad Sci USA. 2009;106:19346. doi: 10.1073/pnas.0906966106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foley MC, Arora K, Schlick T. Intrinsic motions of DNA polymerases underlie their remarkable specificity and selectivity and suggest a hybrid substrate binding mechanism. In: Schlick T, editor. Innovations in bimolecular modeling and simulations. Vol. 2. Royal Society of Chemistry; London, UK: 2012. pp. 81–110. [Google Scholar]
- 35.Arora K, Brooks CL, III, Schlick T. Computational support for hybrid induced fit and conformational selection mechanisms. 2012. In preparation. [Google Scholar]
- 36.Santoso Y, Joyce CM, Potapova O, Le Reste L, Hohlbein J, Torella JP, Grindley NDF, Kapanidis AN. Conformational transitions in DNA polymerase I revealed by single-molecule FRET. Proc Natl Acad Sci USA. 2010;107:715. doi: 10.1073/pnas.0910909107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rothwell PJ, Berger S, Kensch O, Felekyan S, Antonik M, Wöhrl BM, Restle T, Goody RS, Seidel CAM. Multiparameter single-molecule fluorescence spectroscopy reveals heterogeneity of HIV-1 reverse transcriptase: primer/template complexes. Proc Natl Acad Sci USA. 2003;100:1655. doi: 10.1073/pnas.0434003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arora K, Brooks CL., III Functionally important conformations of the Met20 loop in dihydrofolate reductase are populated by rapid thermal fluctuations. J Am Chem Soc. 2009;131:5642. doi: 10.1021/ja9000135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhabha G, Lee J, Ekiert DC, Gam J, Wilson IA, Dyson HJ, Benkovic SJ, Wright PE. A dynamic knockout reveals that conformational fluctuations influence the chemical step of enzyme catalysis. Science. 2011;332:234. doi: 10.1126/science.1198542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- 41.Johnson KA. Role of induced fit in enzyme specificity: a molecular forward/reverse switch. J Biol Chem. 2008;283:26297. doi: 10.1074/jbc.R800034200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beard WA, Shock DD, Vande Berg BJ, Wilson SH. Efficiency of correct nucleotide insertion governs DNA polymerase fidelity. J Biol Chem. 2002;277:47393. doi: 10.1074/jbc.M210036200. [DOI] [PubMed] [Google Scholar]
- 43.Pelletier H, Sawaya MR, Kumar A, Wilson SH, Kraut J. Structures of ternary complexes of rat DNA polymerase β, a DNA template-primer, and ddCTP. Science. 1994;264:1891. [PubMed] [Google Scholar]
- 44.Beard WA, Osheroff WP, Prasad R, Sawaya MR, Jaju M, Wood TG, Kraut J, Kunkel TA, Wilson SH. Enzyme-DNA interactions required for efficient nucleotide incorporation and discrimination in human DNA polymerase β. J Biol Chem. 1996;271:12141. doi: 10.1074/jbc.271.21.12141. [DOI] [PubMed] [Google Scholar]
- 45.Iwanaga A, Ouchida M, Miyazaki K, Hori K, Mukai T. Functional mutation of DNA polymerase β found in human gastric cancer—inability of the base excision repair in vitro. Mutat Res. 1999;435:121. doi: 10.1016/s0921-8777(99)00036-1. [DOI] [PubMed] [Google Scholar]
- 46.Lang T, Dalal S, Chikova A, DiMaio D, Sweasy JB. The E295K DNA polymerase beta gastric cancer-associated variant interferes with base excision repair and induces cellular transformation. Mol Cell Biol. 2007;27:5587. doi: 10.1128/MCB.01883-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y, Gridley CL, Jaeger J, Sweasy JB, Schlick T. Unfavorable electrostatic and steric interactions in DNA polymerase β E295K mutant interfere with the enzyme’s pathway. J Am Chem Soc. 2012;134:9999. doi: 10.1021/ja300361r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Batra VK, Beard WA, Shock DD, Pedersen LC, Wilson SH. Structures of DNA polymerase β with active site mismatches suggest a transient abasic site intermediate during mis-incorporation. Mol Cell. 2008;30:315. doi: 10.1016/j.molcel.2008.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bryant FR, Johnson KA, Benkovic SJ. Elementary steps in the DNA polymerase I pathway. Biochemistry. 1983;22:3537. doi: 10.1021/bi00284a001. [DOI] [PubMed] [Google Scholar]
- 50.Joyce CM, Potapova O, DeLucia AM, Huang X, Basu VP, Grindley NDF. Fingers-closing and other rapid conformational changes in DNA polymerase I (Klenow fragment) and their role in nucleotide selectivity. Biochemistry. 2008;47:6103. doi: 10.1021/bi7021848. [DOI] [PubMed] [Google Scholar]
- 51.Rothwell PJ, Mitaksov V, Waksman G. Motions of the fingers subdomain of Klentaq1 are fast and not rate limiting: implications for the molecular basis of fidelity in DNA polymerases. Mol Cell. 2005;19:345. doi: 10.1016/j.molcel.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 52.Tsai Y-C, Johnson KA. A new paradigm for DNA polymerase specificity. Biochemistry. 2006;45:9675. doi: 10.1021/bi060993z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kirmizialtin S, Nguyen V, Johnson KA, Elber R. How conformational dynamics of DNA polymerase select correct substrates: experiments and simulations. Structure (Camb) 2012;20:618. doi: 10.1016/j.str.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang H, Cao W, Zakharova E, Konigsberg W, De La Cruz EM. Fluorescence of 2-aminopurine reveals rapid conformational changes in the RB69 DNA polymerase-primer/template complexes upon binding and incorporation of matched deoxy-nucleoside triphosphates. Nucl Acids Res. 2007;35:6052. doi: 10.1093/nar/gkm587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hanes JW, Johnson KA. A novel mechanism of selectivity against AZT by the human mitochondrial DNA polymerase. Nucl Acids Res. 2007;35:6973. doi: 10.1093/nar/gkm695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Post CB, Ray WJ., Jr Reexamination of induced fit as a determinant of substrate specificity in enzymatic reactions. Biochemistry. 1995;34:15881. doi: 10.1021/bi00049a001. [DOI] [PubMed] [Google Scholar]
- 57.Mulholland A, Roitberg AE, Tuñón I. Enzyme dynamics and catalysis in the mechanism of DNA polymerase. Theor Chem Acc. 2012;131:1286. [Google Scholar]
- 58.Schlick T, Collepardo-Guevara R, Halvorsen LA, Jung S, Xiao X. Biomolecular modeling and simulation: a field coming of age. Q Rev Biophys. 2011;44:191. doi: 10.1017/S0033583510000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Y, Reddy S, Beard WA, Wilson SH, Schlick T. Differing conformational pathways before and after chemistry for insertion of dATP versus dCTP opposite 8-oxoG in DNA polymerase β. Biophys J. 2007;92:3063. doi: 10.1529/biophysj.106.092106. [DOI] [PMC free article] [PubMed] [Google Scholar]