

Abstract
Background
Kindlin-3 is a novel integrin activator in hematopoietic cells and its deficiency leads to immune problems and severe bleeding, known as LAD-III. Our current understanding of Kindlin-3 function primarily relies on analysis of animal models or cell lines.
Objectives
To understand the functions of Kindlin-3 in human primary blood cells.
Patients/Methods
Here we analyze primary and immortalized hematopoietic cells obtained from a new LAD-III patient with immune problems, bleeding, a history of anemia and abnormally shaped red blood cells.
Results
Patient’s WBC and platelets showed defect in agonist induced integrin activation and botrocetin induced platelet agglutination. Primary leukocytes from this patient exhibited abnormal activation of beta1 integrin. Integrin activation defects were responsible for observed deficiency of botrocetin induced platelet response. Analysis of patient’s genomic DNA revealed a novel mutation in kindlin-3 gene. The mutation abolished Kindlin-3 expression in primary WBC and platelets due to abnormal splicing. Kindlin-3 is expressed in erythrocytes and its deficiency proposed to lead to abnormal shape of RBC. Immortalized patient’s WBCs expressed a truncated form of Kindlin-3 which was not sufficient to support integrin activation. Expression of Kindlin-3 cDNA in immortalized patient’s WBCs rescued integrin activation defects while overexpression of the truncated form did not.
Conclusions
Kindlin-3 deficiency impairs integrin function, including activation of beta 1 integrin.
Abnormalities in GPIb-IX function in kindlin-3 deficient platelets are secondary to integrin defects.
Region of Kindlin-3 encoded by Exon 11 is crucial for its ability to activate integrins in humans.
Keywords: Integrins, Kindlins, Leukocyte Adhesion Deficiency, Platelets, Red Blood Cells, White Blood Cells
INTRODUCTION
Integrin-mediated binding of extracellular matrix ligands is essential for the development and functioning of multi-cellular organisms. Ligand binding activity of integrins is regulated by intracellular stimuli, and this process, known as integrin inside-out signaling, is particularly important for blood cells. Under normal conditions, leukocytes and platelets circulate in the presence of excessive amounts of their adhesive ligands; however, the inactive conformation of integrins on their surface precludes ligand binding. Activating cues trigger intracellular signaling cascades, inducing a transformation of integrins from resting to active conformation, which allows ligand binding. In platelets, this process leads to adhesion and aggregation responses, essential for hemostasis. In leukocytes, integrin activation enables adhesion to extracellular matrix components and endothelial cells, thus mediating leukocyte recruitment to inflammatory sites. A lack of leukocyte-specific β2 integrin in mice leads to immune deficiency, whereas the absence of platelet integrin β3 causes defective platelet aggregation and thrombus formation, resulting in bleeding and hemorrhages (1). In humans, spontaneous mutations in leukocyte integrin β2 cause an impairment of leukocyte adhesion and immune deficiency, known as LAD I (leukocyte adhesion deficiency). Mutations in platelet integrins αIIb or β3 cause defective platelet adhesion and aggregation, leading to the bleeding disorder known as Glanzmann’s thrombasthenia.
A rare recessive syndrome, LAD III, features a combined dysfunction of β1, β2, and β3 integrin families on platelets and leukocytes, and it is caused by the deficiency in intracellular integrin activator Kindlin-3 (2–3). This disease is characterized by excessive bleeding (often more severe than in Glanzmann’s thrombasthenia patients) and abnormal immune responses. Despite the considerable number of reported cases (a total of 22, to our knowledge), there are still a number of unanswered questions and disagreements, often due to the limited analysis of primary patient cells.
Although all reported mutations in LAD III patients were found to abolish Kindlin-3 expression completely, the phenotypic features vary substantially. Certain symptoms, such as bleeding and recurrent infections, are common; others, such as osteopetrosis, have been described in some, but not all, patients (4). There is also a discrepancy in the literature regarding the functioning of leukocyte integrin VLA (α4β1) in LAD III patients and the role of Kindlin-3 in its activation (2, 5–6). Interestingly, the consequences of Kindlin-3 deficiency in mice result in a more severe phenotype as compared to humans. Kindlin-3 null mice die during the first week of life due to severe anemia and hemorrhage (7–8). At the same time, many human patients survive through childhood, and only one case of mild anemia in two siblings was reported (4). Importantly, severe anemia in Kindlin-3 null mice coincided with an abnormal shape of red blood cells, suggesting a role of Kindlin-3 in erythrocytes. Thus, while the causative role of Kindlin-3 in patients with immune deficiency and bleeding is now well-documented, many issues regarding Kindlin-3 function in human blood cells remain unclear.
Here we describe a patient with Kindlin-3 deficiency due to a novel mutation in the splicing site of Kindlin-3. Importantly, the phenotypic characteristics of this patient deviate substantially from the conventional LAD III described in the literature. While bleeding and immune problems are milder than in many described cases (2, 9), this patient has a history of severe anemia and abnormally shaped red blood cells.
METHODS
Blood cell isolation and cell lines
Blood samples were obtained after IRB approved informed consent. Blood was drawn into acid-citrate-dextrose (ACD; 85 mM tri-sodium citrate, 65 mM citric acid, and 110 mM D-glucose [pH 4.6]) solution containing PGE1 (1 µg/ml of ACD). Twenty-four hours later, platelet-rich plasma (PRP) was separated by centrifugation at 100g for 10 minutes at 22°C. Platelets were purified from the PRP by gel filtration, and WBC were separated by Ficoll gradient and washed four times in PBS to remove any remaining platelets. For RBC analysis, the bottom layer was isolated and the RBC were washed five times with PBS. To deplete granulocytes, top layers of RBC were removed and the remaining RBC were passed through nylon nets. RBC purity was then assessed microscopically.
EBV-transformed cell lines were established by Cleveland Clinic’s Media Core Facility following standard protocols.
Antibodies and other reagents
Two rabbit polyclonal antibodies against separate Kindlin-3 regions were previously described (10): the first to the c-terminal peptide ELDEDLFLQLTGGHEAF, the second to the peptide, corresponding to an area between N-terminal F2 and PH domains, GEVGEPAGTDPGLD (gift from Dr. E. Plow). Monoclonal antibody to Kindlin-3 was raised to the full length protein (gift from Dr. E. Plow). Clone 3d6 was selected for further use. FITC-labeled PAC-1 and the isotype-matched non-immune antibodies were from BD Bioscience (San Jose, CA). Monoclonal antibody to Kindlin-2 and FITC-labeled HUTS-4 antibody were from Chemicon (Temecula, CA). Fc IgG-fused VCAM-1Fc was from R&D systems (Minneapolis, MN). The secondary fluorescein isothiocyanate (FITC)–conjugated, anti-human Fc IgG antibody was from Jackson ImmunoResearch Laboratories (West Grove, PA). FITC-conjugated antibodies for β1 and β3 integrin subunits were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). FITC-conjugated Fibrinogen was from Invitrogen (Carlsbad, CA). Recombinant CS1 fibronectin fragment was a gift from Dr. M. Ginsberg. Human α-thrombin was from Enzyme Research Laboratories (South Bend, IN). Adenosine diphosphate (ADP) was from Chrono-log (Havertown, PA). Convulxin was from Axxora LLC (San Diego, CA).
Platelets aggregation and agglutination assays
Platelet aggregation and agglutination were determined using a Chrono-log Model 560VS aggregometer with AGGRRO/LINK version 5.1.9 software. Platelets were isolated by gel filtration and used at 2 × 108 platelets/mL. Thrombin (0.5 U/ml), collagen (5 µg/ml), PMA (200 nM), or adenosine diphosphate (ADP; 5 µM) were used as agonists. For platelet agglutination Ristocetin (1mg/ml) or Botrocetin (2ug/ml) were used. Platelet-poor plasma was used to set the baseline. To test platelet agglutination in the absence of Ca2+, EDTA and Botrocetin were added at the final concentration of 5mM, 2.4 µg/ml and 10 µg/ml, respectively.
Flow cytometry
Analysis of αIIbβ3 activation was described previously (11). Platelets were stimulated with agonists for 5 minutes in Tyrode’s buffer containing 1 mM Ca2+ and 1 mM Mg2+ followed by addition of FITC-conjugated PAC-1 or isotype matched non-immune antibody and incubation for additional 20 minutes at room temperature.
For analysis of soluble fibrinogen binding, washed WBC (0.5 × 106) were incubated with FITC FG for 30 minutes at room temperature. To analyze the activation of leukocyte integrins, cells were incubated with VCAM-1Fc fusion protein (12) or HUTS-4 antibody, at room temperature in PBS containing 1 mM Ca2+ and 1 mM Mg2+. PMA or fMLP were added during this phase. Cells were washed and incubated with FITC-conjugated anti-human or anti-mouse antibodies IgG (Jackson Immunoresearch, West Grove, PA) for 30 minutes on ice and washed again twice. FACS Canto II flow cytometer (Becton Dickinson, Mountain View, CA), and FACSDiva and FlowJo software (Tree Star, Ashland, OR) were used for analysis.
Adhesion and spreading assays
The WBC adhesion assay was performed in 96 well plates coated with the indicated substrate for one hour at 37°C, and then blocked with a 0.5% BSA solution overnight at 4° C. 1.3 × 104 cells/well were plated and allowed to adhere for one hour. The cells were then washed with PBS, fixed and stained with Hoechst reagent (Invitrogen). Digital fluorescent images were acquired at low magnification, and the cells were counted using ImageJ software.
For the Kindlin-3 reconstitution experiments, EBV-transformed patient and control cell lines were transfected with GFP Kindlin-3 or GFP alone using the Amaxa nucleofector, according to the manufacturer’s instructions. Briefly, 5 × 106 cells were mixed with 100 µL of the solution C and 5 ug DNA, and nucleofected with program Z-001. GFP expressing cells were imaged and counted using ImageJ software in adhesion assay. Transfection efficiency and expression level were determined by flow cytometry. The number of adhered GFP expressing cells was normalized to the percentage of GFP expressing cell in the total population.
For analysis of spreading, gel-filtered platelets or washed WBC were plated on cover slips coated with FG or CS1 fibronectin fragment, respectively. At the indicated times, cells were fixed with 2% formaldehyde, permeabilized with 0.2% Triton X-100, and stained with Alexa-Fluor 568 -conjugated Phalloidin. Digital fluorescent images of randomly selected fields were acquired and analyzed using the threshold function of Image-J software (NIH).
Immunoprecipitation and Western blot analysis
For Western analysis, platelet lysates were separated by SDS PAGE and immunoblotted using polyclonal anti-Kindlin-3 antibodies. The same membrane was re-blotted for β3 integrin subunit as a loading control.
For immunoprecipitation analysis, WBC or RBC were lysed in a buffer containing 1% Triton, 150 mM NaCl, 50 mM Tris pH 7.4, and a protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). The lysates were then clarified by centrifugation at 1.6 × 104 g for 10 minutes at 4° C. 400 µg of lysate was used for WBC IP analysis. For RBC analysis, lysate from 25 µL of packed cells was used for each sample. RBC lysates were pre-cleared by one hour incubation with plain agarose beads, followed by centrifugation. WBC or RBC supernatants were incubated with Kindlin-3 antibody for two hours, and protein A/G agarose beads for one additional hour. The beads were then sedimented, washed three times, and immunoprecipitated proteins were eluted and separated by SDS PAGE on 7.5% gels and analyzed by immunoblotting.
Sequencing analysis
For Kindlin-3 mRNA sequencing, total mRNA was isolated from the patient’s, parental and control WBC and RT PCR was then performed. For genomic sequencing, genomic DNA was isolated from the patient’s and parental WBC and analyzed in all exons and intron-exon boundaries of Kindlin-3. Analysis of α globin gene was performed by CompGene (Milwaukee, WI).
Scanning electron microscopy
See supplemental methods.
Statistical analysis
Values are expressed as mean plus or minus SEM. P-values for data were calculated with student t-test, P values < 0.05 were considered statistically significant.
RESULTS
Case report
The subject is a seven year old female, born to a consanguineous couple of Sudanese background. Her parents and two siblings are alive and well, whereas a third sibling died at the age of 19 days from pseudomonas meningitis. The patient suffered from a congenital CMV infection followed by intermittent CMV viremia, which was successfully treated with ganciclovir. She also presented with multiple bacterial infections, including pneumonia, and recurrent otitis media. She developed hematomas in the knee and arm joints, and the latter was complicated with a Pseudomonas super-infection. Laboratory tests demonstrated high WBC counts, ranging between 17 and 70,000 per µL (Supplemental Table 1).
Additionally, the patient showed signs of haemostatic deficiency, such as frequent gum bleedings and easy bruising. Platelet counts were low at birth, but became and remained normal after the first few days (Supplemental Table 1). A standard clinical whole blood platelet aggregation test showed a lack of response to arachidonic acid, collagen, ADP, and Ristocetin, whereas other platelet functions, such as ATP release, in response to thrombin and ADP, were normal (Supplemental Table 2).
Although no heavy bleeding incidents were recorded, the patient had a history of severe hypochromic anemia. The level of hemoglobin (Hb) was at the lower limit for the first 20 days, but then dropped down to a range between 5.4 and 8.5 g/dL thereafter (Supplemental Table 1). A drop to 2.6 g/dL was recorded after a two-day episode of gum bleeding. RBC indices persistently showed low Hb content per cell (mean corpuscular hemoglobin, MCH) and a high variation in size (RBC distribution width, RDW). The results of blood smear tests showed the presence of abnormal RBC, including microcytes, hypochromia, and dacryocytes (tear drop RBC) from the age of one day to seven years. Hb analysis at six months showed normal results: HbA 94.4%, HbA2 3%, and HbF 2.6%. Iron panel showed low levels of serum iron, ferritin, and iron saturation (Supplemental Table 3).
Platelet deficiency
To clarify the mechanism of the haemostatic deficiency, we further characterized the platelet functional abnormality. Aggregation responses of patient’s platelets to collagen, thrombin, and PMA were detectable but reduced, compared to parental platelets used as controls (Figure 1A). After five minutes of stimulation, the responses of patient’s platelets were ~35, 20 and 40% of her parents’ responses to collagen, thrombin, and PMA, respectively. The patient’s platelets exhibited delayed spreading on a fibrinogen-coated surface when stimulated by PMA, as compared to the parent (Figure 1B, C). After one hour of incubation, ~60% of PMA-treated parental platelets, but none of the patient’s platelets, displayed a fully flat morphology on fibrinogen (Figure 1B–D). However, some of the patient’s platelets extended filopodial protrusions, or exhibited partial spreading in response to PMA (Figure 1B, D). Surprisingly, the patient’s unstimulated platelets expressed more filopodia than control ones, indicating an elevated level of basal actin polymerization.
Figure 1. Deficiency of the patient’s platelet function.


A. Agonist stimulated aggregation of platelets from the patient and parents was measured using an optical aggregometer. Platelets were isolated from PRP by gel filtration and their concentration in Tyrode’s buffer was adjusted to 2 × 108 platelets/mL. Thrombin (0.5 U/mL), collagen (5 µg/mL), or PMA (200 nM), were used as agonists. Data are representative of two experiments. B, C. Spreading of the patient’s and parental platelets on fibrinogen. Gel purified platelets were platted on a fibrinogen coated surface in Tyrode’s buffer with or without PMA (200nM) for 20 min. Representative DIC images (B) and fluorescent images of phalloidin stained actin (C) are shown. Scale bars represent 10 µm. D. The extent of spreading from eight random fields/sample was evaluated by shape (i) and by surface area (ii). Platelets with elevated central parts and completely flat platelets and were counted as partially spread and fully spread respectively (i); ***P<0.0005. Data are representative from two experiments.
Since integrin-mediated platelet functions, aggregation, and spreading appeared abnormal in the patient’s cells, we analyzed the expression and function of the main platelet integrin αIIbβ3. Results of FACS analysis showed comparable expression levels of β3 integrin subunit in the patient’s and control samples (Figure 2A). However, agonist-induced activation of αIIbβ3 on the patient’s platelets was diminished in response to three different agonists, including PMA, thrombin, and convulxin, whereas the platelet responses of both parents were normal (Figure 2B). Together, the data suggested a deficiency in integrin inside-out activation affecting multiple activation pathways.
Figure 2. Deficiency of the patient’s integrin activation.

Gel purified platelets were analyzed by flow cytometry using FITC conjugated β3 integrin antibodies to measure: (A) total β3 integrin expression or (B) FITC conjugated PAC-1 antibodies to measure activated αIIbβ3 integrin. Data labels are MFI. Data are representative from two experiments. C. VWF mediated agglutination of platelet rich plasma from the patient and parents was tested in response to Ristocetin (1µg/ml), Botrocetin (2mg/ml), or Botrocetin (10mg/ml) in the presence of EDTA. Data are representative for two experiments.
The standardized clinical test (Supplemental Table 3) showed the lack of the patient’s platelets response to Ristocetin. As a similar abnormality was previously recorded for similar patients (13), we assessed the function of GP1bIX complex, a receptor for VWF. The expression of GP1bIX complex components was similar (not shown). To induce VWF-GP1bIX mediated platelet agglutination, we used two well-characterized reagents, Ristocetin and Botrocetin, known to expose differential binding sites for GP1bIX upon their interaction with VWF. In lab settings, Botrocetin-induced platelet agglutination was reduced in the patient’s platelets as compared to control ones, whereas response to Ristocetin was comparable (Figure 2C). To determine whether reduced VWF-mediated agglutination of the patient’s platelets was secondary to abnormal integrin function, we repeated the assay under conditions which disabled integrin function. In the presence of EDTA, a Ca2+ chelator, a five-fold higher dose of Botrocetin was required to induce platelet agglutination, suggesting that agglutination induced by the lower dose was Ca2+ dependent. Furthermore, in the presence of EDTA, responses of the patient’s and control platelets to Botrocetin were comparable. The results indicate that in the presence of Ca2+, integrin activation contributes to platelet responses to Botrocetin, and that GP1bIX function seems to be intact in the patient’s platelets.
Leukocyte deficiency
As mentioned earlier, the patient’s history of multiple and recurrent infections strongly suggests deficient immune function. As we found that platelet integrin activation was impaired, we investigated the function of leukocyte integrins. We analyzed leukocyte adhesion to fibronectin, which is a ligand for α4β1 and α5β1 integrins, and α4β1-specific FN fragment, CS1 (Figure 3A, B). The patient’s cells showed a reduced adhesion as compared to control cells. Treatment with integrin activators PMA or fMLP increased the adhesion to both substrates; however, adhesion of the patient’s cells remained low. Thus, the patient’s leukocytes are characterized by a deficiency in integrin β1-mediated adhesion with and without stimulation.
Figure 3. Deficiency of the patient’s WBC function.


WBC were isolated from control or patient blood samples by Ficoll separation. A, B. WBC were platted on surface coated with FN fragment, CS1, (A) a specific ligand for α4β1 integrin, or on full length FN (B), a ligand for α5β1 and α4β1 integrins. Cell adhesion assay was performed in the presence of PMA (200nM), fMLP (100nM), or without stimulation as described in the Material and Methods section. Data are a mean of triplicate wells. C, D. WBC were platted on CS1 coated coverslip in the presence of PMA (200nM), fixed and fluorescently labeled with phalloidin shown in red nuclear stain in blue. Fluorescent images of control and patient cells are shown, scale bar represents 10 µm (C). The extent of spreading was evaluated by measuring cell surface area from 25–30 cells from two experiments (D); ***P<0.0005, NS indicates not significant. E. Integrin activation in response to PMA or fMLP was determined by flow cytometry using soluble ligands (Fibrinogen and VCAM) or conformation specific antibody for β1 integrin, HUTS-4. Data are representative of two experiments.
To further support the key role of Kindlin-3 in α4β1 function, we tested PMA-induced spreading of the patient’s and control leukocytes on CS1. In contrast to control cells, the patient’s leukocytes failed to spread (Figure 3C, D). However, β1 integrin expression levels in control and the patient’s cells were similar (data not shown), suggesting that leukocyte integrin activation is impaired.
To test integrin activation directly and investigate the function of individual integrin subclasses, we analyzed the binding of soluble integrin ligands fibrinogen (for αVβ3 and αMβ2), VCAM-1 (for α4β1), and conformation specific antibody HUTS-4 (for β1 integrins) (Figure 3E). In contrast to control cells, the patient’s cells showed no agonist-induced increase in the binding of fibrinogen, VCAM or HUTS-4 upon stimulations with PMA or fMLP. These data indicate that the patient’s leukocytes are deficient in the activation of all three classes of integrins: β1, β2, and β3.
Red blood cell abnormalities
As previously mentioned, the patient’s blood tests showed severe microcytic hypochromic anemia (Supplemental Table 1) with abnormal RBC morphology. When processing blood samples from the patient’s parents and non-related control subjects, using Ficoll separation, we noticed a high degree of hemolysis in the patient’s blood (Supplemental Figure 1). To characterize the morphological abnormalities in RBC, we used phase contrast and scanning electron microscopy. Images showed the presence of abnormally-shaped RBC, including dacrocytes (tear drop-shaped) and elliptocytes (Figure 4).
Figure 4. Red blood cell abnormalities in LAD III patient.

Representative phase contrast (A) and optical (B) images of the patient’s and control RBC. Arrows indicate abnormally shaped RBC. Scale bar represents 10 µm. C. Tear drop- shaped patient’s RBCs shown by scanning electron microscopy. Scale bar represents 1 µm.
The most frequent cause of abnormal RBC shape in children from African and Arabic origins is a reduced expression of α or β globin genes (α or β thalassemia, respectively). As mentioned earlier, our patient’s ratio of HbA2 to HbA was normal, thus ruling out β thalassemia, characterized by elevated HbA2 to HbA ratio (14). A possibility of α thalassemia, however, needed to be addressed as a potential cause of RBC abnormalities. An analysis of the patient’s α globin genes did not identify any of the seven most common deletions: -α 3.7; -α 4.2; SEA; FIL; THAI; -α 20.5; and MED (data not shown). Since these deletions cover more than 95% of the causes of α thalassemia cases in the relevant areas (15), the results indicate that thalassemia is highly unlikely in this patient.
Lack of Kindlin-3 expression in the patient’s platelets, leukocytes, and red blood cells
A defect in integrin activation in the patient’s platelets and leukocytes suggested a possibility of Kindlin-3 deficiency. Therefore, we analyzed Kindlin-3 expression levels in the patient’s cells. As opposed to normal platelets, no Kindlin-3 expression was detected in the patient’s platelets by Western blot analysis (Figure 5A, i). Increased loading of patient’s platelet lysate still did not allow for the detection of Kindlin-3 (Figure 5A, ii). As the patient’s platelets showed a heterogeneous response to PMA stimulation, it is possible that Kindlin-3 could be selectively expressed in a small subpopulation of the patient’s platelets. Accordingly, adherent platelets were stained for Kindlin-3. Whereas platelets from the control subject as well as those from the patient’s mother and father all showed clear positive staining, no signal was detected in the patient’s platelets (Figure 5A, iii). While some of the patient’s platelets partially spread in response to PMA as described earlier, there was no correlation between the amount of stimulation-induced cytoskeletal rearrangements and staining intensity. Thus, the partial response of the patient’s platelets to PMA was not due to selective Kindlin-3 expression.
Figure 5. Lack of Kindlin-3 protein expression in the patient’s cells.

A. Comparison of Kindlin-3 expression in the patient’s (P), parental (M), and control (C) platelets was analyzed by western blotting using a Kindlin-3 specific antibody. β3 integrin subunit was used as a loading control (i). Decreasing amounts of the control platelet lysate were loaded for comparison of Kindlin-3 expression to the patient’s platelets (ii). Fluorescent images of the patient’s and parental platelets stained with Kindlin-3 specific antibody and Phalloidin, scale bar represents 10 µm (iii). B. Kindlin-3 expression in control (C) and the patient’s (P) WBC was analyzed by immunoprecipitation and Western blotting. Non-specific IgG was used as a control. C. Kindlin-3 expression in control (C), parental (F and M), and the patient’s RBC (P) was analyzed by immunoprecipitation and Western blotting. Non-specific IgG was used as a control. Data are representative for three different Kindlin-3–specific antibodies.
We analyzed Kindlin-3 expression in the patient’s and control WBC by Western blotting. Similarly to platelets, we could not detect Kindlin-3 in the patient’s leukocytes, even upon immunoprecipitation with a Kindlin-3 antibody (Figure 5B). As expected, Kindlin-3 was expressed in control WBC. Thus a lack of Kindlin-3 in platelets and leukocytes is consistent with haemostatic and immune deficiency in our patient. We also analyzed expression of Kindlin-2 to address its potential compensatory up-regulation in patient leukocytes. We could not detect any expression of Kindlin-2 in either patient, parental or control leukocytes by using Kindlin-2 specific antibody (Supplementary Figure 5).
Since, in addition to conventional LAD III symptoms, our patient displayed abnormal RBC morphology, we asked whether this abnormality could also result from a Kindlin-3 deficiency. To determine whether Kindlin-3 is expressed in RBC, we analyzed highly purified RBC from the patient, the parents, and an unrelated control subject by immunoprecipitation. To ensure specificity, two different Kindlin-3 antibodies were used to precipitate and probe for Kindlin-3. The results showed that Kindlin-3 could be immunoprecipitated from control and parental RBC, but not from the patient’s RBC (Figure 5C). Thus, normal RBC express Kindlin-3, and therefore Kindlin-3 deficiency in the patient’s RBC could contribute to their abnormality.
Mutational analysis
To determine the cause for a Kindlin-3 deficiency in the patient, we performed reverse transcriptase-PCR analysis of mRNA from the patient’s WBC. Sequence analysis revealed two mutant Kindlin-3 cDNA products: one with a deletion of six bases encoding the first two amino acids of exon 11 (438 and 439), and a second missing the entire sequence of exon 11 (438–515) that encodes for 78 amino acids (Supplemental Figure 2). In contrast to control cells, no WT Kindlin-3 cDNA product was detected in the patient’s cells under the same conditions (data not shown) implying that a lack of normally spliced mRNA resulted in a Kindlin-3 protein deficiency.
We then performed a sequencing of the genomic DNA from the patient, the parents, and a sibling for exon 11 and exon-intron boundaries of Kindlin-3 gene. The analysis had identified a G to A base substitute within an AG dinucleotide preceding the start of exon 11 (Figure 6). The mutation was homozygous in the patient, whereas the parents and a sibling were heterozygous. Presence of the AG dinucleotide at the end of introns was reported as essential for the U2 spliceosome-mediated splicing (reviewed in (16)). Therefore, we predict that the identified mutation destroys the slicing acceptor site for the exon 11, resulting in abnormally spliced mRNA products as described above. Genomic database search showed no polymorphism at this position; no other mutations were identified.
Figure 6. Mutational analysis. Comparison of genomic Kindlin-3 sequences at the intron10-exon11 boundary from the database, the patient, and the parents.

The non coding DNA strain is shown. The beginning of the 11th exon is indicated by an arrow, the position of C to T substitution is highlighted in a rectangle. The mutation is homozygous in the patient and heterozygous in both parents.
Reconstitution of Kindlin-3 in patient-derived cell lines
To confirm the causative nature of Kindlin-3 mutation in the leukocyte deficiency, we established EBV-induced lymphoblastoid cell lines from the patient, the parents, and a non-related healthy control. As expected, Kindlin-3 was expressed in control and parent-derived cell lines. There was no detectable expression of full length Kindlin-3 in the patient’s cells, however we observed a band of a slightly lower molecular weight that reacted with several non-related Kindlin-3 specific antibodies. This band was also present in parental, but not control, cells (Figure 7A, Supplemental Figure 4). Based on molecular weight, reactivity with Kindlin-3 specific antibodies, and expression pattern, this band is likely to represent a product of the aberrantly spliced mRNA. Preliminary experiments showed that patient-derived cells were deficient in PMA-induced adhesion to FN, compared to control and parental cells (data not shown). However, we could not conclude whether the aberrantly spliced product was functional, as its expression in the patient’s cells was substantially lower than the expression of WT Kindlin-3 in control or parental cells. Because EBV-transformed cells were able to express the aberrantly spliced Kindlin-3 product, we could not exclude the possibility that some stimuli might induce an expression of this protein in the patient’s primary cells. Thus, to assess whether the mutated protein is functional, we generated GFP-fused WT or mutant Kindlin-3. We then expressed these proteins to comparable levels in the patient-derived cells, and compared their ability to rescue defective cell adhesion to FN. WT Kindlin-3 restored the ability of cells to adhere in response to PMA stimulation, whereas the alternatively spliced protein or GFP alone did not (Figure 7B, Supplemental Figure 3). Therefore, the data shows that the expression of WT Kindlin-3 in the patient’s cells can rescue cell adhesion, and that the aberrantly spliced Kindlin-3 product is not functional.
Figure 7. Adhesion rescue in patient’s cells, expressing WT Kindlin-3.
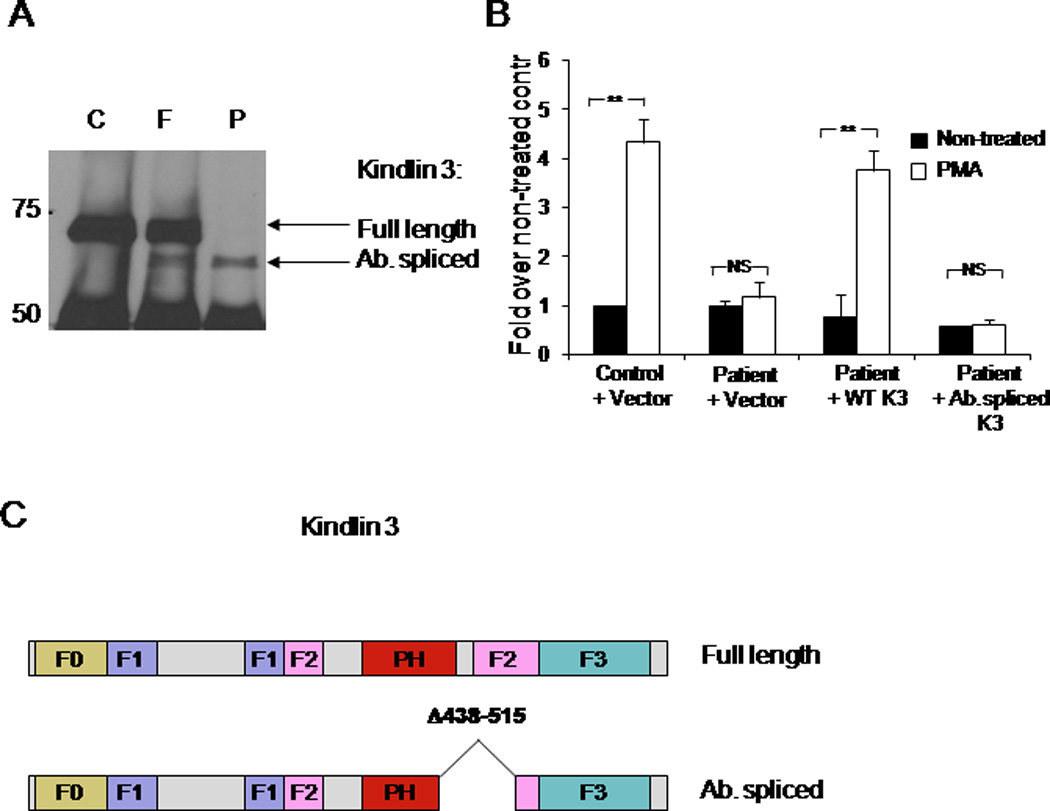
A. Endogenous expression of Kindlin-3 in EBV-transformed WBC from control subject (C), mother (M), and the patient (P) was analyzed by immunoprecipitation and Western blotting using polyclonal Kindlin-3 specific antibody. Bands corresponding to the full length and the aberrantly spliced Kindlin-3 proteins are indicated by arrows. B. Adhesion of transfected lymphoblastoid cells to FN is shown. GFP conjugates of wild type Kindlin-3 Kindlin-3 (Kindlin-3 wt), Kindlin-3 with deletion of exon 11 (amino acids 438–515, Kindlin-3 ab), or GFP alone (vector) were transiently transfected into the patient’s or control cell lines and expressed at a similar level (not shown). The cells were analyzed by adhesion assay with or without PMA stimulation. Data are expressed as fold increase over the adhesion of non-stimulated cells from control subject expressing GFP alone (mean of three experiments). **P<0.005, NS indicates not significant. C. Schematic representation of the mutated form of Kindlin-3 protein product from the patient- derived lymphoblastoid cell lines.
DISCUSSION
In this study we described a novel case of LAD III deficiency caused by a novel mutation in Kindlin-3 gene. Mutations in this gene, abolishing Kindlin-3 protein expression, were recently established as a cause of a human disease known as LAD I-variant or LAD III. However, despite the complete lack of Kindlin-3 expression in all patients described to date, clinical features of these patients vary quite dramatically. Whereas immune and hemostatic deficiencies are common, the severity of symptoms differs substantially from relatively mild (4), to life-threatening conditions leading to death at early age (17). It is possible that additional mutations in genes related to integrin signaling such us CalDAG GEF (17) might lead to more serious complications.
In this study we describe a patient with a new Kindlin-3 mutation in the splicing acceptor site of exon 11, resulting in a loss of detectable Kindlin-3 protein expression in primary platelets, leukocytes, and erythrocytes. Presumably, the abnormality of mRNA splicing leads to a compromised translation or a rapid degradation of the mutant protein product. This patient presented with relatively mild bleeding problems compared to other LADIII patients. In contrast to two cases described by us previously (2), agonist induced aggregation and spreading on fibrinogen were not completely abolished in the patient’s platelets. There was a possibility that the expression of the aberrantly spliced form of Kindlin-3 is in part responsible for this relatively mild phenotype. Indeed, in EBV-immortalized cells, but not in primary cells, we were able to detect the presence of the short form of Kindlin-3. However, when expressed in the patient’s cells, this truncated form could not rescue the integrin activation deficiency, demonstrating a lack of function. These data indicate that exon 11, encoding for parts of PH and FERM2 domains, is essential for Kindlin-3 function in humans. Therefore, even if truncated Kindlin-3 might be expressed in this patient under certain conditions, it is not able to support integrin activation. Thus, the phenotypic differences between LADIII patients with different mutations in Kindlin-3 are not due to the presence of mutated forms of the protein. We also ruled out the possibility of functional compensation by Kindlin-2. In sum, the plausible scenario is that the lack of Kindlin-3 alone in humans might result in relatively mild abnormalities, however, mutations or polymorphisms in genes related to integrin signaling pathway (such us GALDEG (17)) might exacerbate the complications of this disease. We cannot exclude the possibility that other unknown effectors are able to compensate for the lack of Kindlin-3, emphasizing the complexity and robustness of the signaling network regulating adhesiveness of hematopoietic cells. For the patient described in this report, re-expression of WT Kindlin-3 in patient cells completely rescued the functional deficiency, thus ruling out the possibility of other functional causative mutations.
While previous studies of LAD III cases agree that a lack of Kindlin-3 expression caused an inability to activate platelet integrin αIIbβ3 (8) and leukocyte integrins α2β1, αLβ2, αMβ2, αVβ3, and αVβ5 (9), there is a discrepancy regarding the role of Kindlin-3 in the regulation of α4β1 integrin (VLA-4). A study reported that adhesion of leukocytes from two LAD III patients to VLA-4 was similar to that of the control subject (5), whereas two other studies using three patients from two different families showed that leukocyte adhesion to VLA-4 was dramatically impaired (6), (2). The reason for this discrepancy is likely to be a different methodology used by these groups to measure leukocyte adhesiveness. While adhesion of leukocyte to surfaces coated with integrin ligands is regulated by integrin activation, it may also depend on integrin clustering. Therefore, to address this issue we used soluble ligand (VCAM-1) binding assay as a direct measure of α4 β1 integrin activation. In this assay, in contrast to control cells, the patient’s leukocytes were not able to bind soluble VCAM-1 when stimulated with PMA or the chemokine fMLP. Thus, Kindlin-3 in primary human leukocytes is required for agonist-stimulated activation of α4β1 integrin. Moreover, we show that LAD III patient’s leukocytes fail to spread on the α4β1 integrin substrate, CS1 fragment of fibronectin. Therefore, in primary human leukocytes Kindlin-3 regulates the activation of several integrins, including α4β1.
The distinguishing and unique feature of this patient is abnormal RBC shape and anemia. At the same time, there was no evidence of additional mutations in the hemoglobin genes. Importantly, we show for the first time that Kindlin-3 is, indeed, expressed in mature human RBC from control subjects, but not from the patient, suggesting a possibility of Kindlin-3 function in erythrocytes
Indeed, in mice, the lack of Kindlin-3 alone was sufficient to cause severe anemia and abnormal RBC shape (7). To date, the consequences of Kindlin-3 deficiency in mice are reported to be similar to that in humans, including bleeding, immune problems and osteopetrosis (3). The only exception seems to be an abnormal erythrocyte shape, which was not previously observed in human Kindlin-3 deficient patients. Analysis of previous studies in humans and mice suggests that the abnormal shape of erythrocytes might be masked in many patients, but not in the patient described in this report. This creates a possibility for unique compensatory changes in Kindlin-3 deficient subjects. Detailed analysis of these patients and their genetic background will be helpful to address this issue.
As for anemia, this complication can be solely a consequence of excessive bleeding. However, low hemoglobin levels in this particular patient, combined with the lack of severe bleeding, indicate that other causes might be involved. RBC shape abnormality was observed in this patient from an early age on a regular basis, which is not typical for children with anemia resulting solely from bleeding or iron deficiency (4, 18). It is possible, however, that the iron deficiency in this patient partially contributed to the phenotype, thus unmasking the role of Kindlin-3 in human RBC.
Therefore, the consequences of Kindlin-3 deficiency in humans might differ substantially from the results of gene ablation in animal experimental models, emphasizing the importance of studies in human subjects. Also, results observed using immortalized human cell lines may be somewhat misleading due to the differential gene expression stimulated by EBV transformation. Detailed analysis of freshly isolated primary cells from peripheral blood of human patients with monogenic deficiencies provides a unique opportunity to address the role of a given protein in human physiology. To conclude, our study describes a novel mutation in the Kindlin-3 gene, expands our knowledge of the consequences of Kindlin-3 deficiency in humans and supports potential integrin-independent function of Kindlin-3 in regulation of erythrocyte shape.
Supplementary Material
Acknowledgments
The authors thank Judith A. Drazba and Mei Yin for help with imaging; Carmel Burns for help with cell immortalization, Dr. Meller, N. for helpful discussions, Kamila Bledzka and Yongzhong Zhao for generation of Kindlin-3 antibodies. This study was supported by HL073311 and HL071625 US National Institutes of Health grants. J.M is supported by HL104920-02 NRSA postdoctoral fellowship of Health/National Heart, Lung, and Blood Institute.
Footnotes
Authorship
Contribution: J.M performed the research, analyzed the data and wrote the manuscript; N.L.M, S.P, B.A.K and Y.M. performed the research and analyzed the data; A.P. generated critical tools and performed research, L.V. and I.B.R. provided critical insight into interpretation of the data; N.M. and E.A.P performed the research, analyzed data and provided input into interpretation of the data; M.S.E. collected and analyzed the data, J.C. recruited the subject, collected the samples, provided critical insight into interpretation of the data and edited the manuscript; T.V.B. designed the study, supervised and performed the research, analyzed the data and wrote the manuscript.
Conflict of interest disclosure: The authors declare no competing financial interests.
References
- 1.Wang H, Lim D, Rudd CE. Immunopathologies linked to integrin signalling. Semin Immunopathol. 2010 Jun;32:173–182. doi: 10.1007/s00281-010-0202-3. [DOI] [PubMed] [Google Scholar]
- 2.Malinin NL, Zhang L, Choi J, Ciocea A, Razorenova O, Ma YQ, Podrez EA, Tosi M, Lennon DP, Caplan AI, Shurin SB, Plow EF, Byzova TV. A point mutation in KINDLIN3 ablates activation of three integrin subfamilies in humans. Nat Med. 2009 Mar;15:313–318. doi: 10.1038/nm.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malinin NL, Plow EF, Byzova TV. Kindlins in FERM adhesion. Blood. 2010 May 20;115:4011–4017. doi: 10.1182/blood-2009-10-239269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jurk K, Schulz AS, Kehrel BE, Räpple D, Schulze H, Möbest D, Friedrich WW, Omran H, Deak E, Henschler R, Scheele JS, Zieger B. Novel integrin-dependent platelet malfunction in siblings with leukocyte adhesion deficiency-III (LAD-III) caused by a point mutation in FERMT3. Thromb Haemost. 2010 May;103:1053–1064. doi: 10.1160/TH09-10-0689. [DOI] [PubMed] [Google Scholar]
- 5.Manevich-Mendelson E, Feigelson SW, Pasvolsky R, Aker M, Grabovsky V, Shulman Z, Kilic SS, Rosenthal-Allieri MA, Ben-Dor S, Mory A, Bernard A, Moser M, Etzioni A, Alon R. Loss of Kindlin-3 in LAD-III eliminates LFA-1 but not VLA-4 adhesiveness developed under shear flow conditions. Blood. 2009 Sep 10;114:2344–2353. doi: 10.1182/blood-2009-04-218636. [DOI] [PubMed] [Google Scholar]
- 6.McDowall A, Inwald D, Leitinger B, Jones A, Liesner R, Klein N, Hogg N. A novel form of integrin dysfunction involving beta1, beta2, and beta3 integrins. J Clin Invest. 2003 Jan;111:51–60. doi: 10.1172/JCI14076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krüger M, Moser M, Ussar S, Thievessen I, Luber CA, Forner F, Schmidt S, Zanivan S, Fässler R, Mann M. SILAC mouse for quantitative proteomics uncovers kindling-3 as an essential factor for red blood cell function. Cell. 2008 Jul 25;134:353–364. doi: 10.1016/j.cell.2008.05.033. [DOI] [PubMed] [Google Scholar]
- 8.Moser M, Nieswandt B, Ussar S, Pozgajova M, Fassler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. 2008 Mar;14:325–330. doi: 10.1038/nm1722. [DOI] [PubMed] [Google Scholar]
- 9.Svensson L, Howarth K, McDowall A, Patzak I, Evans R, Ussar S, Moser M, Metin A, Fried M, Tomlinson I, Hogg N. Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat Med. 2009 Mar;15:306–312. doi: 10.1038/nm.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bialkowska K, Ma YQ, Bledzka K, Sossey-Alaoui K, Izem L, Zhang X, Malinin N, Qin J, Byzova T, Plow EF. The integrin co-activator Kindlin-3 is expressed and functional in a non-hematopoietic cell, the endothelial cell. J Biol Chem. 2010 Jun 11;285:18640–18649. doi: 10.1074/jbc.M109.085746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shattil SJ, Hoxie JA, Cunningham M, Brass LF. Changes in the platelet membrane glycoprotein IIb.IIIa complex during platelet activation. J Biol Chem. 1985 Sep 15;260:11107–11114. [PubMed] [Google Scholar]
- 12.Rose DM, Cardarelli PM, Cobb RR, Ginsberg MH. Soluble VCAM-1 binding to alpha4 integrins is cell-type specific and activation dependent and is disrupted during apoptosis in T cells. Blood. 2000 Jan 15;95:602–609. [PubMed] [Google Scholar]
- 13.Shurin SB, Tosi M, Fox J, Zhang L. Congenitally defective platelet and leukocyte adhesion receptor function. Blood. 1996;88:96a. [Google Scholar]
- 14.Mosca A, Paleari R, Galanello R, Sollaino C, Perseu L, Demartis FR, Passarello C, Giambona A, Maggio A. New analytical tools and epidemiological data for the identification of HbA2 borderline subjects in the screening for beta-thalassemia. Bioelectrochemistry. 2008 Aug;73:137–140. doi: 10.1016/j.bioelechem.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 15.Bergstrome JA, Poon A. Evaluation of a single-tube multiplex polymerase chain reaction screen for detection of common alpha-thalassemia genotypes in a clinical laboratory. Am J Clin Pathol. 2002 Jul;118:18–24. doi: 10.1309/3VK2-UCJ1-5GBJ-QV8Q. [DOI] [PubMed] [Google Scholar]
- 16.Alder MN, Rogozin IB, Iyer LM, Glazko GV, Cooper MD, Pancer Z. Diversity and function of adaptive immune receptors in a jawless vertebrate. Science. 2005 Dec 23;310:1970–1973. doi: 10.1126/science.1119420. [DOI] [PubMed] [Google Scholar]
- 17.Alon R, Etzioni A. LAD-III, a novel group of leukocyte integrin activation deficiencies. Trends Immunol. 2003 Oct;24:561–566. doi: 10.1016/j.it.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 18.Janus J, Moerschel SK. Evaluation of anemia in children. Am Fam Physician. 2010 Jun 15;81:1462–1471. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.