

Abstract
Purpose
Interleukin-10 (IL-10) is an anti-inflammatory molecule that has achieved interest as a therapeutic for neuropathic pain. In this work, the potential of plasmid DNA-encoding IL-10 (pDNA-IL-10) slowly released from biodegradable microparticles to provide long-term pain relief in an animal model of neuropathic pain was investigated.
Methods
PLGA microparticles encapsulating pDNA-IL-10 were developed and assessed both in vitro and in vivo.
Results
In vitro, pDNA containing microparticles activated macrophages, enhanced the production of nitric oxide, and increased the production of IL-10 protein relative to levels achieved with unencapsulated pDNA-IL-10. In vivo, intrathecally administered microparticles embedded in meningeal tissue, induced phagocytic cell recruitment to the cerebrospinal fluid, and relieved neuropathic pain for greater than 74 days following a single intrathecal administration, a feat not achieved with unencapsulated pDNA. Therapeutic effects of microparticle-delivered pDNA-IL-10 were blocked in the presence of IL-10-neutralizing antibody, and elevated levels of plasmid-derived IL-10 were detected in tissues for a prolonged time period post-injection (>28 days), demonstrating that therapeutic effects are dependent on IL-10 protein production.
Conclusions
These studies demonstrate that microparticle encapsulation significantly enhances the potency of intrathecally administered pDNA, which may be extended to treat other disorders that require intrathecal gene therapy.
Keywords: interleukin-10, microparticle, pDNA, PLGA, neuropathic pain
INTRODUCTION
Approximately 2–7% of the population in Europe and the U.S. suffer from neuropathic pain, a condition caused by damage and/or inflammation to nerves following traumatic injury, viral infection or chemotherapy (1). Non-neuronal cells, glia within the spinal cord parenchyma, become activated in response to inflammation or trauma and play a major role in the development and maintenance of neuropathic pain (2) by producing proinflammatory cytokines (3) which act on neurons in pain pathways to sensitize and enhance pain processing (4). The anti-inflammatory cytokine interleukin-10 (IL-10) has emerged as a novel drug candidate to treat this condition, as IL-10 has been shown to suppress the production and function of many proinflammatory cytokines released by activated glia (5,6). While intrathecal administration of IL-10 protein has been shown to provide relief from pain in animal models, effects are short-lived and closely parallel the half-life of IL-10 in the cerebrospinal fluid (CSF) (7).
To provide prolonged pain relief, gene therapy vectors encoding for the production of IL-10 have been utilized (viral vectors result in 10–14 days of relief (8), non-viral vectors result in greater than 60 days of relief (9)). Histological analysis of tissue collected after IL-10 gene delivery demonstrates that this therapeutic approach reduces glial activation (10). In order for non-viral vectors to provide long-term relief in these treatments, however, two high-dose intrathecal administrations are required, and the duration of time in between injections is critical to the success of the therapeutic. Successful long-term pain relief is ultimately achieved when the first administered dosage of plasmid DNA (pDNA) is 100μg, the second dosage is 25 μg and the two doses are given no sooner than 5 hr and no later than 3 d apart (11,12).
The cellular milieu present following nerve injury and a subsequent injection of pDNA is thought to play a key role in the success of the therapeutic. Within 6 hr to 5 d following nerve injury, macrophages infiltrate into the CSF and the surrounding meningeal tissue (13). Similar to this, an additional wave of macrophages is recruited to the CSF within 6 hr after an injection of unencapsulated pDNA (11). The majority of cells recruited are ED1 positive (blood-derived macrophages or monocytes (14)) and/or ED2 positive (mature tissue or resident macrophages (15)). Importantly, both of these macrophage populations are highly phagocytic (16). Thus, the first dose of pDNA creates a cellular environment enriched in cells which can phagocytose pDNA provided in the second injection and subsequently express the gene of interest. A fraction of transfected macrophages likely undergo natural apoptosis or natural clearance to the lymph nodes, consequently removing the delivered gene and source of therapeutic protein from the targeted delivery site over a relatively short time-scale (17). However, a significant population of the recruited macrophages may differentiate into ED2 positive resident macrophages (18) or microglial cells (19), which may persist for extended periods of time (20), potentially providing a longer-term source of IL-10 protein and pain relief. Recent work with pDNA-IL-10 delivery indicates that the number and state of these macrophages present in the CSF at the time of gene delivery is a key factor in the overall success of a pDNA-IL-10-mediated therapy for neuropathic pain (11). For pDNA-IL-10 delivery to facilitate long-term pain resolution, it has recently been shown that phagocytic immune cells must first be activated and recruited to the CSF for subsequent exposure to the therapeutic pDNA, (21) thereby indicating that enhanced phagocytic cell activation and recruitment will consequently enhance the efficacy of pDNA-mediated therapy for neuropathic pain.
While the therapeutic window of greater than 60 days is highly promising for the treatment of neuropathic pain, the success of non-viral gene therapy requires multiple high-dose (100 μg /25 μg) injections. This poses serious economical and clinical constraints, as larger doses will likely be necessary in humans, and pain patients are unlikely to adhere to a multi-day spinal injection schedule. Synthetic biodegradable polymer microparticles, designed to slowly release the gene of interest as they degrade, may offer solutions to the problems associated with intrathecally administering a dose of unencapsulated pDNA. Poly (lactic-co-glycolic-acid) (PLGA) is perhaps the most widely studied synthetic degradable polymer used to prepare microparticles and has been approved by the U.S. FDA for the slow release of peptides and proteins (22). Encapsulating pDNA within PLGA improves the structural integrity of pDNA and its activity in vivo, likely by protecting it from degradative enzymes (23). In addition, PLGA has been shown to have adjuvant-like properties, activating cells of the innate immune system. For example, when immature dendritic cells are cultured in the presence of PLGA, expression of MHC-II molecules is increased, as is the expression of co-stimulatory factors (CD80 and CD83) (24). In addition, cytokine (IL-12, IL-1beta, IL-6, TNF-alpha, IL-8 and IL-10) secretion is upregulated (25), and T-cell proliferation is enhanced (26). This adjuvant effect of PLGA is particularly advantageous for vaccine delivery applications, as antigen presentation is enhanced upon PLGA microparticle-mediated delivery, a factor which leads to a stronger specific immune response and a better vaccine. Similarly, PLGA microparticles have been shown to increase cytokine production (27) and the production of reactive oxygen intermediates by macrophages (28). Macrophages activated by PLGA microparticles are more phagocytic, and gene expression levels are higher when pDNA is present within the PLGA microparticle (29). Due to the abundance of macrophages in the CSF and in surrounding meningeal tissue during neuropathic pain (11,30), activating and enhancing the phagocytic capacity of this cell population presents an opportunity for improved intrathecal gene delivery when an appropriate balance between enhanced phagocytosis and reduced inflammation due to the presence of IL-10 is attained.
In the current work, the possibility that the adjuvancy of PLGA combined with the continued release of pDNA may lead to the development of a more efficient, single low-dose injection of pDNA-IL-10 for treating neuropathic pain was examined. Studies were designed to compare the potency of a total dose of 8.8 μg of pDNA-IL-10 when intrathecally administered either as an unencapsulated dosage or via PLGA microparticles. In addition to determining the extent to which neuropathic pain was relieved in animal models, experiments were designed to identify differences in the level of macrophage activation by assessing nitric oxide production, IL-10 gene expression and protein production in culture. In vivo, experiments were designed to elucidate differences in the pattern of cellular recruitment to the CSF, which may be a key factor involved in the success of the pDNA-IL-10 therapeutic.
MATERIALS AND METHODS
Amplification and Purification of pDNA
The plasmid construct encoding for rat interleukin-10 (pDNA-IL-10F129S) has been previously described in detail (9), and pDNA-Control was identical except for a polyA sequence in the place of the IL-10 encoding region. Plasmids were amplified and verified as previously described (9) and purified using an endotoxin-free plasmid Giga purification kit (Qiagen). The endotoxin content of the pDNA-IL-10, as assessed by the photometric Limulus amebocyte lysate (LAL) assay (BioWhittaker Inc.), was 0.0021±0.00034 ng LPS/μg DNA, a level that is generally considered negligible (31).
Microparticle Preparation and Characterization
Microparticles were prepared using a modified double emulsion/solvent evaporation protocol (32). Briefly, a 50:50 PLGA copolymer (MW 75,000, Lactel Absorbable Polymers) was dissolved in ethyl acetate (Sigma). Vehicle alone (phosphate-buffered saline (PBS) + 3% (w/v) sucrose (Sigma)) or pDNA in vehicle were emulsified in the PLGA solution followed by a second emulsion in a 5% (w/v) polyvinyl alcohol, 28% calcium chloride, 3% sucrose (Sigma) and 7% (v/v) ethyl acetate solution. After 4 hr of hardening in a wash solution, the resulting microparticles were collected, lyophilized and stored at 4°C. Scanning electron microscopy (SEM) was used to examine microparticle morphology. The diameters of >1000 microparticles present in 10 different images were measured with NIH ImageJ software, and binned particle diameters were used to generate a normalized frequency distribution. The zeta potential of the microparticles was measured with a Nicomp 380 ZLS Zeta Potential Analyzer, and the endotoxin levels of the resultant microparticles were tested by the LAL assay, using serial dilution as a control for inhibition. For immunohistochemical analysis, the aqueous phase used prior to the first emulsification consisted of rhodamine-dextran at a concentration of 10 mg/ml in PBS.
Total pDNA encapsulation was assessed by extracting pDNA from microparticles via sodium hydroxide dissolution (32), measuring the absorbance at 260 nm and comparing obtained values to DNA standards at known concentrations. Final pDNA loadings were 8.78±0.65 μgpDNA/mgPLGA for PLGA-pDNA-IL-10 microparticles and 8.78±0.07 μgpDNA/mgPLGA for PLGA-pDNA-Control microparticles. Aqueous extraction of pDNA was conducted by dissolving microparticles in chloroform and allowing the pDNA to migrate into aqueous buffer (32). The extracted pDNAwas subsequently concentrated by precipitation with ethanol and re-suspended in PBS+3% sucrose vehicle. The structural integrity of the aqueous extracted pDNAwas compared against unencapsulated pDNA (which was similarly exposed to the aqueous extraction process) by loading 2 μg of total pDNA into the wells of a 1.0% agarose gel containing ethidium bromide, running the gel at 75 V for 2 hr, and imaging the gel with UV trans-illumination at 305 nm. Biological activity of aqueous extracted pDNA was assessed by lipofectamine-mediated transfection into human embryonic kindey-293 cells according to manufacturer protocols (Invitrogen), and IL-10 protein concentrations in cell culture supernatants collected 24 hr after transfection with aqueous extracted and unencapsulated pDNAwere assessed by ELISA (R&D Systems). In vitro release profiling was conducted by incubating microparticles in PBS over time in a water bath at 37°C, and pDNA contents in the supernatant were quantified by a PicoGreen assay (33).
NO Production, IL-10 Gene Expression and IL-10 Protein Production by Macrophages Exposed to Unencapsulated or Microparticle-Delivered pDNA-IL-10 in Culture
The stimulatory effects of pDNA and/or PLGA microparticles were assessed using primary peritoneal macrophages isolated by lavage. Activation of the cell population was assessed by the measurement of nitric oxide (NO). IL-10 mRNA detection in cell pellets and IL-10 protein detection in the supernatants were used as indicators of the extent of transfection and gene expression. For NO, IL-10 mRNA and IL-10 protein production assays, cells were allowed to adhere to 96-well tissue culture plates for 2 h at 37°C, and the medium was then replaced with 100 μl of medium containing (1) 5 mg/ml vehicle microparticles (PLGA-Vehicle), (2) 5 mg/ml PLGA-Vehicle suspended in 44 μg/ml pDNA-Control, (3) 5 mg/ml of microparticles encapsulating pDNA-Control (PLGA-pDNA-Control), (4) 44 μg/ml pDNA-IL-10, (5) 5 mg/ml PLGA-Vehicle suspended in 44 μg/ml pDNA-IL-10 and (6) 5 mg/ml PLGA-pDNA-IL-10 microparticles. A separate lipopolysaccharide (LPS, Sigma L7770) condition (100 μl/well at 0.1 μg/ml) was also included as a comparison control. The supernatants from sample wells were then collected separately for analysis of nitrite (a breakdown product of nitric oxide; Griess reagent system; Promega) and IL-10 production (ELISA; R&D Systems). For RT-PCR, the adhered cells were detached from the tissue culture dishes with the application of trypsin for 5 min and transferred to a separate tube where cell culture medium was added to inactivate the trypsin. The samples were then centrifuged (1,000g for 10 min at 4°C), and the pellets were resuspended in ice-cold PBS and stored at −80°C until further analysis (see method for RT-PCR).
Behavioral Testing in CCI Animal Models of Neuropathic Pain Treated with Intrathecally Administered Unencapsulated or Microparticle-Delivered pDNA-IL-10
Viral-free adult male Sprague-Dawley rats (350–450 g; Harlan Labs) were used in all experiments, all procedures were approved by the University of Colorado Institutional Animal Care and Use Committee, and all research adhered to the Principles of Laboratory Animal Care (NIH #85-23). Chronic constriction injury (CCI) was created at mid-thigh level of the left hind leg as previously described (34). Briefly, under isoflurane anesthesia, the left sciatic nerve was exposed via blunt dissection, and 4 chromic gut sutures were tied loosely around the nerve. Sham-operated rats had the sciatic nerve identically isolated but not ligated. Unless otherwise specified, CCI-operated rats were used in all animal experiments.
Intrathecal injections and CSF collections were conducted as previously described (7). All intrathecal administrations occurred 10 d post-CCI surgery, and injection volumes were 20 μl. Injections consisted of vehicle, pDNA-IL-10 (8.8 μg in 20 μl), PLGA-Vehicle, PLGA-pDNA-IL-10 and PLGA-pDNA-Control microparticles (1 mg in 20 μl) or pDNA-IL-10+ PLGA Vehicle (1 mg of microparticles in 8.8 μg of pDNA-IL-10 in 20 μl). For antibody administration, lyophilized bulk ion exchange purified sheep anti-rat interleukin-10 IgG neutralizing antibody (Anti-IL-10-IgG) as well as non-specific IgG control (Control-IgG) antibody (National Institute for Biological Standards and Control; Potters Bar, UK) were diluted with PBS to a dose of 0.2 μg in10 μl. Testing for mechanical allodynia was conducted as previously described (10), and behavioral measures were performed blind with respect to treatment group.
Tissue Collection, Microparticle Distribution and IHC Protocol
Following transcardial perfusions with 0.9% saline, intact spinal columns were collected and transferred to 4% paraformal-dehyde (Sigma) for 24 hr. Spinal columns were decalcified in a 10% EDTA (Sigma)+0.1% sodium azide (Sigma) solution for 7 d, equilibrated in 30% sucrose +0.1% sodium azide solution for 2 d, and cryo-sectioned at a 30 μm thickness. Microparticle distributions were examined under a 546 nm reflector cube using a Zeiss Pascal LSM microscope with a 40× Plan NeoFluor (1.3) oil immersion objective. Microparticle counts at marked distances were recorded with a hand tally counter (averaged from 6 tissue sections from 5 animals). IHC analysis of spinal cord tissues was conducted according to standard procedures for the antigens CD11b/c (1:200 dilution, BD Pharmingen 554859) and MHCII (1:200 dilution AbD Serotec MCA46R) using a fluorescence-conjugated secondary antibody (Invitrogen A11029), the nuclear stain DAPI, and confocal fluorescence microscopy.
RT-PCR
RNA was isolated from lumbar cord tissue via a standard method of phenol:chloroform extraction (35). Total RNA was reverse transcribed into cDNA using the Super-Script II First Strand Synthesis System for RT-PCR (Invitrogen) according to manufacturer’s instructions and stored at −80°C until RT-PCR was performed. cDNA from peritoneal macrophage and CSF samples was synthesized using a Cells-Direct III cDNA synthesis kit (Invitrogen). The samples were centrifuged (1,000g for 10 min at 4°C), the pellet washed with ice cold PBS and centrifuged again (1,000g for 10 min at room temperature). Cells were re-suspended and processed according to synthesis kit instructions. Primers were designed to measure the housekeeping gene glyceraldehyde-6-phosphate dehydrogenase (GAPDH) and plasmid-derived IL-10, which utilized a unique sequence that forms the 3′ UTR of the plasmid and is not found in the rat genome. PCR amplification of cDNA was performed in duplicate using the Quantitect SYBR Green PCR Kit (Qiagen) according to manufacturer’s instructions and monitored using the MyiQ Single-Color Real-Time PCR Detection System (Bio-Rad). The threshold cycle (CT; number of cycles to reach threshold of detection) was determined for each reaction, and mRNA was quantified using the comparative CT(ΔCT) method (36). The PCR primer specifications are shown in Table I.
Table I.
PCR Primer Specifications
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| GAPDH | CTTCCAGGAGCGAGATCCC | TTCAGGTGAGCCCCAGCCTT |
| Plasmid IL-10 | ACGCTGTCATCGATTTCT | CCGCTAGCTCAATTTTTCA |
Identification of Cell Types Present in the CSF by Immunocytochemistry Following Intrathecally Administered Unencapsulated or Microparticle-Delivered pDNA-IL-10
After intrathecal injections identical to the conditions used for behavioral testing, 20 μl of the CSF samples were plated onto glass coverslips (Fisher Scientific, 12 mm circular No.1) in the wells of a 24-well plate immediately following collection. Cells within CSF were allowed to adhere to the coverslips for 40 min in an incubator (37°C, 5% CO2). Following incubation, immunohistochemical staining for cell phenotype markers was conducted according to standard procedures for the antigens ED1 (AbD Serotec MCA341R) and ED2 (AbD Serotec MCA342R) at 1:200 dilutions using fluorescence-conjugated secondary antibody (Invitrogen A11029) and the nuclear stain DAPI. Samples were analyzed for cell density and phenotype via NIH ImageJ manual counts of fluorescence microscopy captured images.
Data Analysis
Statistical comparisons utilized two-sample t-tests assuming unequal variances on a 95% confidence interval with two-tailed p-values as the output unless otherwise indicated. Data from the von Frey test were analyzed as the interpolated 50% threshold (absolute threshold) in log base 10 of stimulus intensity (monofilament stiffness in milligrams × 10).
RESULTS
PLGA Microparticle Characterization
The microparticles utilized in this work exhibited a spherical and smooth morphology (Fig. 1a) and a zeta-potential of −28.04±2.12 mV. The microparticles exhibited a heterogeneous size distribution (Fig. 1b) with an overall median diameter of 4.67±0.26 μm, which is consistent with similar methods of microparticle manufacturing (32), and the pDNA encapsulation efficiency for the particles was 55.1%. Agarose gel electrophoresis of aqueous extracted pDNA from microparticles compared to unencapsulated pDNA indicated that a significant amount of the relaxed and supercoiled pDNA structural integrity was preserved after encapsulation, although a slight detection of linearized pDNA and slight alterations in the migration of multimeric pDNA species were observed after encapsulation (Fig. 1c). By comparing resultant IL-10 protein expression levels in the supernatants of human embryonic kidney-293 cells 24 hr after lipofectamine-mediated transfection with dose-matched microparticle extracted or unencapsulated pDNA (data not shown), it was determined that the microparticle extracted pDNA-IL-10 exhibited a 96.8% biological activity retention for the resultant production of IL-10, thereby indicating the microparticle formation process did not substantially reduce the activity of the encapsulated pDNA. In vitro pDNA release analysis demonstrated that 30% of the pDNA was released after 3 days, and steady release was achieved for greater than 75 days (Fig. 1d). This two-phase release profile is a common characteristic of macromolecule release from emulsion-based PLGA microparticles, where the enhanced phase of initial pDNA release is due to an increased pDNA content on or near the surface of the microparticles, which is followed by a sustained release and diffusion of pDNA from the microparticle interior (37). Endotoxin levels from microparticles with and without encapsulated pDNA were below the limits of detection for the LAL assay up to a microparticle concentration of 10 mg/ml (1 mg of microparticles/well).
Fig. 1.

Characterization of microparticles. a Representative image of microparticles under scanning electron microscopy. b Normalized frequency distribution of binned microparticle diameters. c Agarose gel electrophoresis of aqueous extracted pDNA from microparticles, lane 1 contains size markers, lane 2 contains unencapsulated pDNA and lane 3 contains aqueous extracted pDNA. (d) In vitro cumulative pDNA release from microparticles over time.
NO Production, IL-10 Gene Expression and IL-10 Protein Production in Response to Unencapsulated or Microparticle-Delivered pDNA in Primary Macrophage Culture
As classically activated macrophage populations are known to exhibit an enhanced production of nitric oxide (NO) upon stimulation (38), and as the production of NO correlates with a higher level of phagocytic activity (39), the potential of microparticles to stimulate macrophages in a non-endotoxi-mediated manner was assessed in vitro. Cellular morphology and total cell numbers were not affected by any of the conditions at the 24 and 72 hr time points (data not shown). Unencapsulated pDNA-IL-10 did not significantly increase the production of NO from the macrophages relative to the medium-only control, while all of the PLGA conditions exhibited significantly elevated NO production both at 24 and 72 hr relative to the medium-only control and unencapsulated pDNA-IL-10 conditions (Fig. 2a). Lipopolysaccharide (LPS) has also been shown to stimulate peritoneal macrophages (40), and the magnitude of NO response for the PLGA conditions was comparable to that observed from LPS stimulation.
Fig. 2.

In vitro macrophage responses to unencapsulated and dose-matched pDNA and microparticles 24 or 72 hr after incubation. a Nitric oxide production in cell culture supernatants, b plasmid-derived IL-10 mRNA expression relative to GAPDH mRNA expression in collected macrophage cell pellets and (c) IL-10 production in cell culture supernatants as detected by ELISA. LPS (0.1 μg/ml) background responses are indicated by dotted lines. * indicates p value <0.05 compared to time-matched medium only control. # indicates p value <0.05 compared to time-matched pDNA-IL-10F129S. & indicates p value <0.05 compared to time-matched PLGA-Vehicle. PLGA-Vehicle represents microparticles prepared with PBS +3% sucrose as encapsulant.
The ability of pDNA encapsulated and/or released from PLGA microparticles to transfect macrophages during the middle (24 hr) and near the end (72 hr) of the initial pDNA release phase was assessed by measuring plasmid-derived IL-10 mRNA expression levels. The levels for plasmid-derived gene expression in the medium-only and PLGA-Vehicle controls represented primer background levels for basal IL-10. The PLGA-Vehicle and PLGA-pDNA-Control conditions did not exhibit significantly elevated (p>0.05) plasmid-derived mRNA expression relative to the medium-only control (Fig. 2b). Unencapsulated pDNA-IL-10 measurably and/or significantly (p<0.05) enhanced plasmid-derived and total IL-10 mRNA expression at the 24 and 72 hr time points relative to the controls. When delivered alongside of PLGA particles, pDNA-IL-10 exhibited a similar magnitude of plasmid-derived IL-10 gene expression as the unencapsulated condition within 72 h of incubation. When pDNA-IL-10 was encapsulated within microparticles, IL-10 gene expression levels were substantially elevated above the controls and the unencapsulated pDNA-IL-10 and the PLGA vehicle + pDNA-IL-10 conditions, suggesting that encapsulation within the particles improved the transfection efficiency.
To further assess the effects of PLGA-mediated macrophage stimulation on protein expression from pDNA-IL-10 exposure, the IL-10 protein content in medium supernatants at the 24 and 72 hr time points was also quantified by ELISA. The PLGA-Vehicle and pDNA-Control conditions were not significantly elevated from the culture medium at any of the time points (Fig. 2c). Unencapsulated pDNA-IL-10 with or without PLGA-Vehicle co-administrations also did not elevate resultant IL-10 protein levels substantially. PLGA-pDNA-IL-10 did not lead to measurable IL-10 protein at 24 hr, but exhibited a significantly elevated (p<0.05) level of secreted IL-10 protein within 72 hr relative to the medium-only control and unencapsulated pDNA-IL-10 conditions. Therefore, even though the effect was delayed with protein expression, both the IL-10 gene expression and IL-10 protein expression data ultimately indicated that the macrophages were more efficiently transfected by pDNA that was physically encapsulated within and released by microparticles. The activation of macrophages by PLGA for an increased phagocytic potential likely facilitated the enhanced IL-10 expression within 72 hr from pDNA-IL-10-containing microparticles relative to unencapsulated pDNA-IL-10.
Microparticle Localization in Tissue Sections
Spinal columns were collected 3 d after the administration of PLGA-pDNA-IL-10 microparticles to determine that the number of microparticles embedded in the meningeal tissue surrounding the spinal cord decreased in the rostral direction from the injection site (no microparticles were observed in brain sections), and increased in the caudal direction from the injection site, which parallels the direction of CSF bulk flow (Fig. 3a). Microparticles were also observed in collected CSF samples with a high degree of variability (p>0.05 relative to baseline controls). In addition to the 3 d time point, PLGA-pDNA-IL-10 microparticles were also observed in the meningeal tissue surrounding the spinal cord at 1, 14, 21, 28 and 60 d after administration, where the rostral-to-caudal trend was stable over time. In both meningeal tissue and CSF samples, no measurable difference in the size distribution of microparticles detected in the rostral vs. the caudal direction was observed. Microparticles that were observed in the CSF and in meningeal tissue collections were both internal and external to phagocytic cell membranes. PLGA microparticle penetration into the spinal cord parenchyma was not observed at any of these time points (representative image at 3 d is shown in Fig. 3b and c). The use of antibodies for phagocytic cell types indicated that meningeal layer-embedded microparticles were embedded within a matrix of cells staining positively for major histocompatibility complex-II (MHCII), a marker of antigen-presenting cells, including macrophages, and OX-42 (CD11b/c), a classical marker for macrophages and microglia (representative images of OX-42 and MHCII staining at 3 d are shown in Fig. 3b and c). Although microparticles that did not associate with any cell types were observed on the outer periphery of the spinal cord meningeal tissue sections, when embedded within the meningeal tissue microparticles only closely associated with regions that were positive for MHCII and OX-42 (MHCII and OX-42 positive macrophages were also observed in meningeal tissue regions without microparticles present).
Fig. 3.
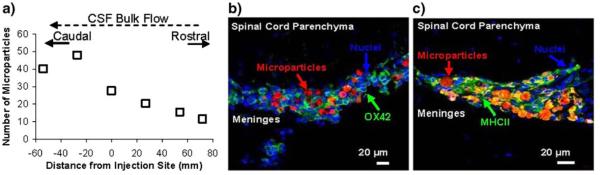
In vivo microparticle localization 72 hr post-injection. a In vivo microparticle distributions assessed by total microparticle counts in spinal cord cross-sections at known distances from the injection site (N=6 rats). b OX-42 immunostaining and (c) and MHCII immunostaining of spinal cord cross-sections; PLGA-pDNA-IL-10F129S microparticles (labeled with rhodamine-dextran) are shown in red, cell nuclei stained by DAPI are shown in blue and antibody stained cells are shown in green (yellow regions are attributed to a significant detection of both red and green fluorphores due the abundance of microparticles present within an abundantly positive MHCII population).
Reversal of Neuropathic Pain by pDNA-IL-10-Releasing PLGA Microparticles
The long-term ability of a single intrathecal administration of PLGA-pDNA-IL-10 to control neuropathic pain was examined. Animals that have received a chronic constriction injury (CCI) exhibit mechanical allodynia, while sham-operated animals do not exhibit mechanical allodynia and represent a baseline control for the reversal of neuropathic pain in this model. PLGA-pDNA-IL-10 fully reversed the effects of CCI-induced mechanical allodynia (i.e. return to the non-allodynic levels of the sham-operated controls) within 3 d of administration and lasted fully for 74 d (repeated measures ANOVA (F(3,19)=656, p<0.001); Fig. 4a). No measurable therapeutic effects were observed 90 d after administration. A single injection of unencapsulated pDNA-IL-10, PLGA-Vehicle, and PLGA-pDNA-Control did not exhibit therapeutic effects, and PLGA-Vehicle administration did not alter the mechanical sensitivity to touch of sham-operated animals (Fig. 4a). To verify that IL-10 plays a role in the mediation of the therapeutic effects from PLGA-pDNA-IL-10 administrations, a neutralizing antibody for IL-10 (anti-IL-10-IgG) was administered during ongoing therapy efficacy. A transient return of mechanical allodynia, due to the transient residence of the antibody, was observed 24 hr after anti-IL-10-IgG administration, but not with control IgG (p<0.05), and a restoration of therapeutic efficacy was observed within 48 hr of anti-IL-10-IgG administration (Fig. 4b), likely due to the short (1–2 hr) half-life of IgG in the CSF (41) as well as ongoing IL-10 gene expression. The blockade of the behavioral efficacy after the administration of anti-IL-10-IgG validates that the PLGA-pDNA-IL-10 therapeutic effects require functional IL-10. To verify that physical encapsulation of pDNA-IL-10 was necessary, the behavioral efficacy of unencapsulated pDNA-IL-10 co-administered with 1 mg of PLGA-Vehicle for the reversal of CCI-induced mechanical allodynia was also assessed. One mg of PLGA-Vehicle suspended in a solution of unencapsulated pDNA-IL-10 (20 μl total administration) did not exhibit significant therapeutic effects for the reversal of CCI-induced mechanical allodynia (i.e. did not therapeutically reverse the CCI-operated animals to the non-allodynic levels represented by the sham-operated animals, Fig. 4c), which indicated that physical encapsulation of pDNA within the PLGA microparticles was a necessary requirement for therapeutic efficacy in the microparticle-mediated single administration paradigm.
Fig. 4.

Assessments of mechanical allodynia. a Single intrathecal microparticle administrations (N=6 rats/group). b Blockade of IL-10 protein by intrathecal administration of anti-IL-10 IgG and control IgG 31 d after the administration of PLGA-pDNA-IL-10F129S microparticles (N=4 rats/group). * indicates p value <0.05 compared to time-matched control IgG administration. c Co-administration of pDNA-IL-10F129S and PLGA-Vehicle microparticles to CCI-operated animals compared to intrathecal administrations of Vehicle (PBS +3% Sucrose) to CCI- and Sham-operated animals (N=6 rats/group).
Gene Expression In Vivo Following Delivery of pDNA-IL-10-Releasing PLGA Microparticles
To further assess the role of IL-10-mediated signaling in early and ongoing therapeutic efficacy from PLGA-pDNA-IL-10, a gene expression study was conducted on tissue samples collected prior to achieving behavioral efficacy (1 d), initially on achieving behavioral efficacy (3 d), at mid-behavioral efficacy (28 d) and at post-behavioral efficacy (93d). As unencapsulated pDNA-IL-10 and PLGA-Vehicle did not lead to behavioral efficacy, to minimize the unnecessary use of animals, sample collections for these conditions were not conducted over time. As a control for background, however, samples were collected at the 28 d time point from both Sham- and CCI-operated PLGA-pDNA-Control-injected animals and at the 93 d time point for Sham- and CCI-operated PLGA-Vehicle-injected animals, both of which coincided with the cessation of behavioral testing for these conditions, again to minimize animal use. As IL-10 mRNA expression levels in these conditions did not exhibit statistical differences from one another (p>0.05), the mRNA levels detected in these tissues were averaged together as an assessment of background expression and are represented by the dotted lines in Fig. 5.
Fig. 5.
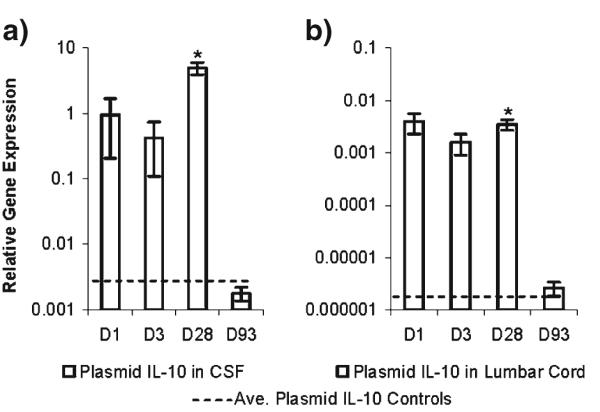
RT-PCR analysis for plasmid-derived IL-10 mRNA relative to GAPDH mRNA expression at the indicated time points after administration of PLGA-pDNA-IL-10F129S microparticles. (a) CSF cells and (b) Lumbar cord tissue (N=6 rats/group). There were no statistical differences between baseline controls consisting of Sham- and CCI-operated animals 28 d post-administration of PLGA-pDNA-Control and Sham- and CCI-operated animals 93 d post-administration of PLGA-Vehicle; dotted lines represent the average values of these baseline controls for each tissue region. * indicates p value <0.05 compared to tissue-matched baseline controls (one-tailed t-tests).
mRNA expression was assessed in cells collected from the CSF, as recent work with high-dose dual administrations of unencapsulated pDNA-IL-10 has highlighted the importance of CSF cell populations in the mediation of therapeutic effects (11). mRNA expression was also analyzed from spinal cord tissues collected at the injection site with the meningeal layer intact (lumbar cord), as it has been shown that cells within meningeal tissues surrounding the lumbar spinal cord can take up large amounts of intrathecally administered pDNA as well (9). Plasmid-derived IL-10 mRNA expression was present at elevated levels in cellular samples from the CSF and lumbar cord tissue at 1, 3 and 28 d after administration (Fig. 5a and b). The magnitude of IL-10 mRNA expression in CSF cells during mid-efficacy (28 d) was substantially elevated relative to the early (1 d and 3 d) time points (Fig. 5a). Moreover, the magnitude of IL-10 mRNA expression levels in CSF cells at all of the collected time points was more than two orders of magnitude above the expression levels in the lumbar cord tissue samples (Fig. 5a vs. b). Samples collected from PLGA-pDNA-IL-10-injected animals at the cessation of behavioral efficacy (93 d) in both regions were not statistically different from the background controls (p>0.05), indicating that a loss in behavioral efficacy correlated with a loss of transgene expression.
Recruitment of Phagocytic Immune Cells Following Injection of pDNA-IL-10-Releasing PLGA Microparticles
Experiments were conducted to determine whether phagocytic cellular infiltration to the CSF was also a component of the successful single PLGA-pDNA-IL-10 administration paradigm. Infiltration studies focused on dose-matched single administrations of pDNA-IL-10, PLGA-Vehicle and PLGA-pDNA-IL-10 relative to time-matched vehicle injections. To minimize the unnecessary use of animals, cellular recruitment assessments were conducted at the expected peak cell infiltration time point (24 hr) and at a time point that would be likely to result in partial attenuation (72 hr) (11), to determine whether therapeutically effective PLGA-pDNA-IL-10 administrations promoted increased cellular infiltration to the CSF.
At the 24 hr time point, pDNA-IL-10 administration increased the number of cells in the CSF relative to a time-matched vehicle injection by 119%. PLGA-Vehicle and PLGA-pDNA-IL-10 administrations, however, substantially increased the number of cells in the CSF relative to a vehicle injection by 268% and 307%, respectively (Fig. 6a). The cellular infiltration level for both of the PLGA conditions was also significantly elevated relative to the cellular increases as a result of unencapsulated pDNA (p<0.05), but the cellular increases resulting from PLGA administrations were not significantly different from one another. At the 72 hr time point, the number of cells in the CSF after a pDNA-IL-10 administration had attenuated to a level that was no longer significant relative to a time-matched vehicle administration (16% increase, p>0.05). The number of cells in the CSF as a result of PLGA-Vehicle and PLGA-pDNA-IL-10 administrations had also significantly decreased relative to their levels at the 24 hr time point, but were still at levels that were significantly elevated relative to both time-matched vehicle (35% and 21% above vehicle respectively) and pDNA-IL-10 administrations (p<0.05). In other words, PLGA micro-particle administrations significantly enhanced the recruitment of cells to the CSF at both of the time points tested, irrespective of whether or not pDNA was present within the microparticles.
Fig. 6.

In vivo CSF cell responses to microparticles. a CSF macrophage infiltration in response to pDNA and microparticles assessed by total cell nuclei counts in the CSF represented as a percentage above time-matched cell nuclei counts for vehicle-only administrations (N=6 rats/group). b In vivo phenotypic maturation of CSF cells in response to pDNA and microparticles assessed by immunostaining (N=6 rats/group). * indicates p value <0.05 compared to time matched PBS +3% sucrose vehicle control. # indicates p value <0.05 for comparison of PLGA to unencapsulated pDNA-IL-10F129S. & indicates p value <0.05 for comparison of 24 hr versus 72 hr percentage phenotype for each condition.
As the total cell numbers were significantly elevated upon microparticle administration, immunohistochemistry was also conducted on the cells of the CSF collected at the 24 and 72 hr time points. For all of the administration conditions and at both of the analyzed time points, a substantial percentage of the recruited cells were ED1-positive and ED2-positive macrophages (Fig. 6b), which are both capable of phagocytosing pDNA and/or microparticles (16). At the 24 hr time point, the percentage of ED1-positive cells relative to a vehicle administration was significantly (p<0.05) greater for unencapsulated pDNA and microparticle administrations with and without pDNA. Within 72 hr, the macrophage phenotypes were not significantly different between the vehicle and pDNA administration conditions; however, the number of ED1- and ED2-positive cells were substantially elevated for both of the microparticle conditions. This sustained presence of phagocytic cells relative to the unencapsulated pDNA profile may be one factor that enhanced the potency of microparticles relative to unencapsulated pDNA. Additionally, there was a greater percentage of ED2-positive macrophages after microparticle administrations between the 24 and 72 hr time points, which, in addition to contributing to pDNA uptake and transfection, may also contribute to the resolution of neuropathic pain by creating an anti-inflammatory wound healing and tissue-regenerative environment (42).
DISCUSSION
Many efforts in microparticle-mediated gene delivery have efficient microparticle internalization as a primary goal. However, a significant non-internalized fraction of microparticles was thought to play an important role in the observed therapeutic effects. In other words, transfection was likely occurring from pDNA that was released from the microparticles prior to internalization and from internalization of the microparticles themselves. While specialized epithelial cells can internalize microparticles up to a size of 10 μm (43), microparticle diameters of less than 1–3 μm are most efficiently internalized by macrophages and antigen-presenting cells (44), and, in general, smaller particles (1 μm) are more readily internalized than larger (3 μm) ones (45). Surface properties are also known to affect phagocytic potential, where positive microparticle surface charges (46) and highly porous and irregular surfaces (47) both increase the likelihood of microparticle phagocytosis. The size distribution of the microparticles used in this work (median diameter of 4.67±0.26 μm), their smooth surfaces (Fig. 1a) and their negative surface charge (−28.04±2.12 mV) are all factors that decrease the likelihood of microparticle internalization by the phagocytic cell types observed in the system, although, due to the heterogeneity in the microparticle size distribution (Fig. 1a and b), a fraction of the microparticle population was likely internalized by a phagocytic route, in which microparticles subsequently released pDNA-IL-10 for intracellular uptake and expression. Microparticle internalization by other known cell entry routes, including fluid-phase pinocytosis and receptor-mediated endocytosis, did not likely occur in appreciable amounts, as much smaller diameters (20–200 nm) are typically required in these cases (48).
Due to these considerations, two different mechanisms of action potentially contributed to the microparticle-mediated therapeutic effects. The fraction of the microparticle population suitable in size for efficient phagocytosis by cells in the CSF or the surrounding meningeal tissue was likely internalized, where intracellularly released pDNA would be translocated to the nucleus and ultimately expressed as mRNA. Endosomal and lysosomal entrapment of the internalized microparticles and poor nuclear entry (49) likely decrease the effectiveness of the directly phagocytosed microparticle fraction, as modifications to facilitate lysosomal escape and nuclear targeting were not conducted herein. Microparticles that were not readily internalized were either cleared from the intrathecal compartment (likely by bulk flow to the bloodstream (50), or in the course of CSF flow embedded within the meningeal tissue matrix (Fig. 3) for a prolonged time period. This meningeal tissue-embedded microparticle population then released pDNA extracellularly where it could diffuse and be taken up by cells in the meningeal tissue or in the CSF by a combination of phagocytosis (51) and receptor-mediated endocytosis (52). Hence, a significant population of the non-internalized, but meningeal tissue-embedded, microparticles was also hypothesized to have played an important role in long-term therapeutic effects by an extracellular release of pDNA for subsequent phagocytosis.
Phagocytosis occurs only in a limited number of cell types, including macrophages, requires molecular attachment to the cell surface and can be significantly enhanced by factors present in the local extracellular environment (53). Components of bacterial cell walls, including the well-characterized lipopolysaccharide molecule (54), cytokines including granulocyte colony-stimulating factor and tumor necrosis factor-alpha (55), and synthetic PLGA microparticles (56) have all been shown to stimulate phagocytosis in macrophages. The most common indicators of macrophage activation and enhanced phagocytic activity include the production and secretion of tumor necrosis factor-alpha (2–8 hr following activation) and nitric oxide production (12–24 h following activation) (38).
Measurement of one indicator of activation, nitric oxide production in macrophage cultures, indicates that PLGA microparticles (both in the presence and absence of pDNA) significantly enhance nitric oxide production relative to dose-matched unencapsulated pDNA, and thereby enhanced the internalization of extracellularly released pDNA. The stimulatory effect of PLGA microparticles may be attributed to the foreign and particulate nature of the material which can initiate a protective response (57), the acidic lactic and glycolic acid degradation byproducts of PLGA (58), and the creation of a low pH microenvironment in the vicinity of the surface of the degrading microparticles (59). When pDNA was delivered within or in combination with PLGA microparticles, a further increase in nitric oxide production was observed relative to PLGA that only contained vehicle at 24 h. This is consistent with prior work demonstrating that CpG motifs within pDNA can significantly enhance phagocytic activation when they are presented to cells alongside of other stimulatory substances (52). pDNA complexation and structural variations (including degradation) can also enhance its immuno-stimulatory effects (60,61). While PLGA microparticles exhibit a stimulatory effect on phagocytic cells as observed in these and prior studies, PLGA microparticles have consistently demonstrated negligible toxicity for prolonged time periods in vivo (62) and are not likely to substantially contribute to peripheral nerve injury and/or ongoing inflammation during neuropathic pain, as supported by the in vivo data presented in this manuscript (Fig. 4a).
In cultured macrophages, both IL-10 gene expression and protein production were highest when pDNA-IL-10 was delivered to cells via PLGA microparticles, even though a fraction (~30%) of the total dose of pDNA-IL-10 in the microparticle was released in that interval (Fig. 1d). While gene expression levels were higher at both 24 h and 72 h, a delay in protein expression was observed. Given that pDNA internalization by both phagocytosis and receptor-mediated endocytosis in macrophages occurs within 1–4 hr (53,63) and pDNA transport to the nucleus and the initiation of protein expression occurs over a 1–8 hr interval (depending on the rate of lysosomal release) (64), each of these processes may contribute to the observed delay in protein production. It is possible that the dosage of pDNA released over the first 24 hr interval was insufficient for the generation of large amounts of secreted protein. Although it was not specifically explored in this work, prior results also indicate that pDNA released from microparticles over a 1–72 hr interval may not retain a full conformational stability, as it has been shown that slight pDNA degradation occurs over the first 16–24 h of incubation due to the local PLGA environment (65), but that a significant percentage of released pDNA still retains conformational stability throughout the first 72 hrs of release (66). Nevertheless, the time-course over which IL-10 protein production was observed in vitro aligned with the time-scale over which behavioral effects were observed in vivo, where PLGA-pDNA-IL-10 administration did not facilitate therapeutic effects within 24 hr, but fully reversed neuropathic pain within 72 hr (Fig. 4a).
While nitric oxide production was not assessed in vivo, cellular recruitment to the CSF following delivery of unencapsulated pDNA or pDNA within PLGA microparticles was assessed. Similar to our findings with cultured macrophages, PLGA microparticles with or without pDNA stimulated the highest levels of cellular recruitment to the CSF. The unencapsulated dose of pDNA stimulated cellular recruitment to the CSF to a lesser extent than the PLGA conditions, but this effect was not sustained over the course of 72 h, likely due to rapid clearance of the pDNA from the CSF (t1/2 for pDNA in rat peripheral tissue is 10–20 min (67). Consistent with cell infiltration studies, a dose of unencapsulated pDNA did not facilitate therapeutic effects in vivo (Fig. 4a). Even when co-administered with PLGA microparticles to enhance the stimulatory effects of the pDNA (Fig. 4c), therapeutic effects from unencapsulated pDNA were not realized, likely due to pDNA degradation and removal from the local environment prior to significant interaction with the infiltrating phagocytic cell population. Prior work conducted by our group has also shown that polyethylenimine, a vector which is known to be effective for transfection but does not provide sustained release, cannot promote long-term therapeutic efficacy for pDNA-IL-10 in a single injection paradigm (33). All findings to date therefore indicate that single injection therapeutic efficacy with pDNA-IL-10 for neuropathic pain is only observed when pDNA-IL-10 is encapsulated by and released from PLGA microparticles over time. Future studies with this system and more advanced polymeric delivery systems for pDNA will be needed to address the relative contribution of pDNA that is released at later (30,45, 60) time points, as prior work indicates that the physical integrity of encapsulated pDNA deteriorates with extended release times in vitro (32).
Both in prior work with dual administrations of high doses of unencapsulated pDNA (9) and in the current work, therapeutic effects have been observed for an eight-to-ten week period. The administration of an antibody blocking the effects of IL-10 (Fig. 4b) negated the therapeutic effects in a transient manner, thereby indicating that ongoing IL-10 protein expression (as a result of the elevated IL-10 mRNA expression observed in Fig. 5) was responsible for therapeutic efficacy. As plasmid-derived IL-10 was detected in both the CSF collected cells and in the spinal cord tissue, the results of this work indicate that the transfected cell population included potentially resident and potentially non-resident cells. While a fraction of transfected cell population may leave the CSF, carrying the gene along with them, some transfected cells are likely to reside within the meningeal tissue to serve as a long-term source of IL-10 protein. The detection of IL-10 mRNA expression in the lumbar spinal cord tissue with its surrounding meninges (Fig. 5b) provides evidence that tissue-resident cell populations contribute to the therapy. Macrophages may also enter from the CSF and embed within the nearby meningeal tissue to differentiate into resident microglia (19,20) and consequently prolong the therapeutic effect. It is clear that macrophages were recruited as a consequence of PLGA microparticle administrations (Fig. 6a and b), but IL-10 production as a result of cellular transfection may also play a role in macrophage recruitment and differentiation (68,69).
The results of this and prior work (11,21) indicate that macrophages play an essential role in non-viral pDNA-IL-10 therapy for the resolution of neuropathic pain, but future work in the field will be necessary to specifically ascertain all of the particular macrophage cell types that may be transfected or altered in vivo as a result of pDNA-IL-10 therapy. While the mechanism by which gene delivery reverses neuropathic pain in this study has not been fully elucidated, the results presented herein are consistent with the following possibility: Microparticles embed within meningeal tissue to release pDNA-IL-10 slowly over time, which is then taken up by a combination of resident and non-resident, or circulating, cells. The non-resident cells are capable of transforming into a more resident cell population as evidenced by the fact that macrophages in the central nervous system have been shown to differentiate into microglia (19). Thus, both of these cell populations may contribute to long-term IL-10 protein production in the CSF for the ultimate resolution of neuropathic pain.
The loss of therapeutic efficacy may be due to natural apoptosis and turnover of the transfected cell population, or the loss of plasmid expression over time. The dose of pDNA released at later time points may also limit the length of time over which therapeutic effects are observed as pDNA release from microparticles in vitro is exhausted after 75 days, a time-scale close to that over which behavioral effects begin to decline. A reduction in the integrity of pDNA released at later time points might also limit the duration of therapeutic efficacy, as the low pH microclimate within the microparticle with ongoing degradation has been shown to decrease the integrity of pDNA over time (32). Indeed, elevated IL-10 mRNA levels at the end-point of therapeutic behavioral testing (93 d) were not detected (Fig. 5), indicating that ongoing plasmid expression was no longer occurring at a time-point when therapeutic effects were not observed.
Relative to the successful dual injection strategy, the total dose of pDNA necessary to achieve therapeutic effects was reduced 14-fold, and the number of injections necessary was minimized to one. The improved potency of pDNA when delivered from microparticles is likely due to two factors: (1) the stimulatory nature of PLGA microparticles and (2) the continued release of pDNA over time to phagocytic cell populations. A potential contributing factor in therapeutic mediation is that the PLGA microparticles encouraged macrophages to adopt an ED2-positive phenotype, as reflected by the sustained increase in ED2-positive cell types within the CSF over time after PLGA administrations. ED2-positive macrophages have been identified as alternatively activated cells that promote tissue regeneration and wound healing (70). These cells typically reside in meningeal tissue over a long time period and are regarded as anti-inflammatory (71). Regardless, ED2 labeling may have a broader specificity than just tissue regenerative phenotypes, and future work in the field of pDNA-IL-10 delivery will be needed to investigate the range of cytokines that may be produced by these macrophages in response to pDNA-IL-10 and PLGA administrations. The ED2 effect in and of itself was not sufficient for therapeutic effects, as the PLGA-Vehicle condition promoted an elevation in ED2-positive cells but did not reverse neuropathic pain.
The current production cost of pDNA on a laboratory scale ($100–$150/mg) represents only a fraction (<10%) of ultimate production and clinical manufacturing costs in order to generate active pharmaceutical ingredients and drug products (72). Moreover, the current cost of a single intrathecal administration is $500–$600 (73). Consequently, the substantial (>14-fold) pDNA dosage reduction and the reduced number of required intrathecal interventions both significantly reduce the overall cost of therapeutic pDNA treatments. The reduced administration requirement also represents a significant clinical advantage, as repeated and continuous intrathecal administration paradigms can lead to deleterious side-effects and patient non-compliance over time (74).
CONCLUSIONS
This work demonstrates that unmodified PLGA microparticles can significantly enhance the delivery of pDNA in an intrathecal administration paradigm. PLGA microparticles promoted cell infiltration into the CSF and, based on in vitro results, likely improved pDNA uptake and expression in the meningeal tissue surrounding the spinal cord. This enabled the successful long-term reversal of neuropathic pain in a single intrathecal administration, a feat that has not been accomplished previously with single administrations of unencapsulated non-viral pDNA. Further optimization of the PLGA delivery system, and the use of more advanced polymer delivery systems, may enable additional dosage reductions and more rapid and prolonged therapeutic efficacy for neuropathic pain and other inflammatory neuropathies.
ACKNOWLEDGEMENTS
Funding for this work was provided by NIH grant DA018156, and E. Dengler was supported by NSF Grant DGE-0549500. The authors would like to acknowledge the expert assistance of Dr. Travis Hughes and Dr. Leslie Leinwand at the University of Colorado for consultations on plasmid DNA growth and characterization. We would also like to thank Avigen Inc. (Alameda, CA) for the purification of Anti-IL-10-IgG and Control-IgG antibodies used in this work and Jenny L. Wilkerson at the University of New Mexico for her assistance with surgical procedures.
Evan M. Sloane made a substantial contribution to the in vivo components of this work.
The authors wish to dedicate this work to Dr. Evan Sloane, who lost his life to cancer prior to the publication of this work.
ABBREVIATIONS
- Anti-IL-10-IgG
anti-rat interleukin-10 IgG neutralizing antibody
- CCI
chronic constriction injury
- CSF
cerebrospinal Fluid
- CT
threshold cycle
- GAPDH
glyceraldehyde-6-phosphate dehydrogenase
- IL-10
interleukin-10
- LAL
limulus amebocyte lysate
- LPS
lipopolysaccharide
- MHCII
major histocompatibility complex-II
- NO
nitric oxide
- pDNA
plasmid DNA
- PLGA
poly (lactic-co-glycolic-acid)
REFERENCES
- 1.Miller G. The dark side of glia. Science. 2005;308:778–81. doi: 10.1126/science.308.5723.778. [DOI] [PubMed] [Google Scholar]
- 2.Suter MR, Wen YR, Decosterd I, Ji RR. Do glial cells control pain? Neuron Glia Biol. 2007;3:255–68. doi: 10.1017/S1740925X08000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watkins LR, Hutchinson MR, Ledeboer A, Wieseler-Frank J, Milligan ED, Maier SF. Glia as the “bad guys”: Implications for improving clinical pain control and the clinical utility of opioids. Brain Behav Immun. 2007;21:131–46. doi: 10.1016/j.bbi.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reeve AJ, Patel S, Fox A, Walker K, Urban L. Intrathecally administered endotoxin or cytokines produce allodynia, hyperalgesia and changes in spinal cord neuronal responses to nociceptive stimuli in the rat. Eur J Pain. 2000;4:247–57. doi: 10.1053/eujp.2000.0177. [DOI] [PubMed] [Google Scholar]
- 5.Sawada M, Suzumura A, Hosoya H, Marunouchi T, Nagatsu T. Interleukin-10 inhibits both production of cytokines and expression of cytokine receptors in microglia. J Neurochem. 1999;72:1466–71. doi: 10.1046/j.1471-4159.1999.721466.x. [DOI] [PubMed] [Google Scholar]
- 6.Pahan K, Khan M, Singh I. Interleukin-10 and interleukin-13 inhibit proinflammatory cytokine-induced ceramide production through the activation of phosphatidylinositol 3-kinase. J Neurochem. 2000;75:576–82. doi: 10.1046/j.1471-4159.2000.0750576.x. [DOI] [PubMed] [Google Scholar]
- 7.Milligan ED, Langer SJ, Sloane EM, He L, Wieseler-Frank J, O’Connor K, et al. Controlling pathological pain by adenovirally driven spinal production of the anti-inflammatory cytokine, interleukin-10. Eur J Neurosci. 2005;21:2136–48. doi: 10.1111/j.1460-9568.2005.04057.x. [DOI] [PubMed] [Google Scholar]
- 8.Milligan ED, Sloane EM, Langer SJ, Cruz PE, Chacur M, Spataro L, et al. Controlling neuropathic pain by adenoassociated virus driven production of the anti-inflammatory cytokine, interleukin-10. Mol Pain. 2005;1:1–9. doi: 10.1186/1744-8069-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milligan ED, Sloane EM, Langer SJ, Hughes TS, Jekich BM, Frank MG, et al. Repeated intrathecal injections of plasmid DNA encoding interleukin-10 produce prolonged reversal of neuropathic pain. Pain. 2006;126:294–308. doi: 10.1016/j.pain.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 10.Sloane E, Ledeboer A, Seibert W, Coats B, van Strien M, Maier K, et al. Anti-inflammatory cytokine gene therapy decreases sensory and motor dysfunction in experimental Multiple Sclerosis. Brain Behav Immun. 2009;23:92–100. doi: 10.1016/j.bbi.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sloane EM, Soderquist RG, Maier SF, Mahoney MJ, Watkins LR, Milligan ED. Long term control of neuropathic pain in a non-viral gene therapy paradigm. Gene Ther. 2009;16:470–5. doi: 10.1038/gt.2009.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ledeboer A, Jekich BM, Sloane EM, Mahoney JH, Langer SJ, Milligan ED, et al. Intrathecal interleukin-10 gene therapy attenuates paclitaxel-induced mechanical allodynia and proinflammatory cytokine expression in dorsal root ganglia in rats. Brain Behav Immun. 2007;21:686–98. doi: 10.1016/j.bbi.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomez-Nicola D, Valle-Argos B, Suardiaz M, Taylor JS, Nieto-Sampedro M. Role of IL-15 in spinal cord and sciatic nerve after chronic constriction injury: regulation of macrophage and T-cell infiltration. J Neurochem. 2008;107:1741–52. doi: 10.1111/j.1471-4159.2008.05746.x. [DOI] [PubMed] [Google Scholar]
- 14.Dijkstra CD, Dopp EA, Joling P, Kraal G. The heterogeneity of mononuclear phagocytes in lympoid organs - distinct macrophage subpopulations in the rat recognized by monoclonal antibody-ED1, antibody-ED2 and antibody-ED3. Immunology. 1985;54:589–99. [PMC free article] [PubMed] [Google Scholar]
- 15.Polfliet MMJ, Goede PH, van Kesteren-Hendrikx EML, van Rooijen N, Dijkstra CD, van den Berg TK. A method for the selective depletion of perivascular and meningeal macrophages in the central nervous system. J Neuroimmun. 2001;116:188–95. doi: 10.1016/s0165-5728(01)00282-x. [DOI] [PubMed] [Google Scholar]
- 16.Duffield JS. The inflammatory macrophage: a story of Jekyll and Hyde. Clin Sci. 2003;104:27–38. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 17.de Vos AF, van Meurs M, Brok HP, Boven LA, Hintzen RQ, van der Valk P, et al. Transfer of central nervous system autoantigens and presentation in secondary lymphoid organs. J Immunol. 2002;169:5415–23. doi: 10.4049/jimmunol.169.10.5415. [DOI] [PubMed] [Google Scholar]
- 18.Honda H, Kimura H, Rostami A. Demonstration and phenotypic characterization of resident macrophages in rat skeletal-muscle. Immunology. 1990;70:272–7. [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci. 2007;27:12396–406. doi: 10.1523/JNEUROSCI.3016-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El Khoury J, Luster AD. Mechanisms of microglia accumulation in Alzheimer’s disease: therapeutic implications. Trends Pharmacol Sci. 2008;29:626–32. doi: 10.1016/j.tips.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 21.Sloane E, Langer S, Jekich B, Mahoney J, Hughes T, Frank M, et al. Immunological priming potentiates non-viral anti-inflammatory gene therapy treatment of neuropathic pain. Gene Ther. 2009;16:1210–22. doi: 10.1038/gt.2009.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hedley ML. Formulations containing poly(lactide-co-glycolide) and plasmid DNA expression vectors. Exp Opin Biol Ther. 2003;3:903–10. doi: 10.1517/14712598.3.6.903. [DOI] [PubMed] [Google Scholar]
- 23.Remaut K, Sanders NN, De Geest BG, Braeckmans K, Demeester J, De Smedt SC. Nucleic acid delivery: Where material sciences and bio-sciences meet. Mater Sci Eng R-Rep. 2007;58:117–61. [Google Scholar]
- 24.Yoshida M, Babensee JE. Molecular aspects of microparticle phagocytosis by dendritic cells. J Biomater Sci-Polym Ed. 2006;17:893–907. doi: 10.1163/156856206777996844. [DOI] [PubMed] [Google Scholar]
- 25.Kanangat S, Nair S, Babu JS, Rouse BT. Expression of cytokine messenger-mRNA in murine splenic dendritic cells and better induction of T-cell-derived cytokines by dendritic cells than by macrophages during in-vitro costimulation assay using specific antigens. J Leukoc Biol. 1995;57:310–6. doi: 10.1002/jlb.57.2.310. [DOI] [PubMed] [Google Scholar]
- 26.Kelleher P, Knight SC. IL-12 increases CD80 expression and the stimulatory capacity of bone marrow-derived dendritic cells. Int Immunol. 1998;10:749–55. doi: 10.1093/intimm/10.6.749. [DOI] [PubMed] [Google Scholar]
- 27.Ding TT, Sun J, Zhang P. Immune evaluation of biomaterials in TNF-alpha and IL-1 beta at mRNA level. Springer; 2007. pp. 2233–2236. [DOI] [PubMed] [Google Scholar]
- 28.Prior S, Gander B, Blarer N, Merkle HP, Subira ML, Irache JM, et al. In vitro phagocytosis and monocyte-macrophage activation with poly(lactide) and poly(lactide-co-glycolide) microspheres. Eur J Pharm Sci. 2002;15:197–207. doi: 10.1016/s0928-0987(01)00218-4. [DOI] [PubMed] [Google Scholar]
- 29.Parsa S, Wang Y, Fuller J, Langer R, Pfeifer BA. A comparison between polymeric microsphere and bacterial vectors for macrophage P388D1 gene delivery. Pharm Res. 2008;25:1202–8. doi: 10.1007/s11095-008-9563-x. [DOI] [PubMed] [Google Scholar]
- 30.Mert T, Gunay I, Ocal I, Guzel AI, Inal TC, Sencar L, et al. Macrophage depletion delays progression of neuropathic pain in diabetic animals. Naunyn-Schmiedebergs Arch Pharmacol. 2009;379:445–52. doi: 10.1007/s00210-008-0387-3. [DOI] [PubMed] [Google Scholar]
- 31.Qiagen . Endofree plasmid purification handbook. Qiagen Inc; 2006. pp. 38–41. [Google Scholar]
- 32.Tinsley-Bown AM, Fretwell R, Dowsett AB, Davis SL, Farrar GH. Formulation of poly(D, L-lactic-co-glycolic acid) microparticles for rapid plasmid DNA delivery. J Control Rel. 2000;66:229–41. doi: 10.1016/s0168-3659(99)00275-8. [DOI] [PubMed] [Google Scholar]
- 33.Milligan ED, Soderquist RG, Malone SM, Mahoney JH, Hughes TS, Langer SJ, et al. Intrathecal polymer-based interleukin-10 gene delivery for neuropathic pain. Neuron Glia Biol. 2006;2:293–308. doi: 10.1017/S1740925X07000488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartmann B, Ahmadi S, Heppenstall P, Lewin G, Schott C, Borchardt T, et al. The AMPA receptor subunits GluR-A and GluR-B reciprocally modulate spinal synaptic plasticity and inflammatory pain. Neuron. 2004;44:637–50. doi: 10.1016/j.neuron.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 35.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate phenol chloroform extraction. Anal Biochem. 1987;162:156–9. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 36.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 37.Yeo Y, Park KN. Control of encapsulation efficiency and initial burst in polymeric microparticle systems. Arch Pharm Res. 2004;27:1–12. doi: 10.1007/BF02980037. [DOI] [PubMed] [Google Scholar]
- 38.Ito H, Koide N, Morikawa A, Hassan F, Islam S, Tumurkhuu G, et al. Augmentation of lipopolysaccharide-induced nitric oxide production by alpha-galactosylceramide in mouse peritoneal cells. J Endotox Res. 2005;11:213–9. doi: 10.1179/096805105X46628. [DOI] [PubMed] [Google Scholar]
- 39.Tumer C, Bilgin HM, Obay BD, Diken H, Atmaca M, Kelle M. Effect of nitric oxide on phagocytic activity of lipopolysaccharideinduced macrophages: possible role of exogenous L-arginine. Cell Biol Int. 2007;31:565–9. doi: 10.1016/j.cellbi.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 40.Lichtman SN, Wang J, Lemasters JJ. LPS receptor CD14 participates in release of TNF-alpha in RAW 264.7 and peritoneal cells but not in Kupffer cells. Am J Physiol-Gastrointest Liver Physiol. 1998;38:G39–46. doi: 10.1152/ajpgi.1998.275.1.G39. [DOI] [PubMed] [Google Scholar]
- 41.Bergman I, Burckart GJ, Pohl CR, Venkataramanan R, Barmada MA, Griffin JA, et al. Pharmacokinetics of IgG and IgM antiganglioside antibodies in rats and monkeys after intrathecal administration. J Pharmacol Exp Ther. 1998;284:111–5. [PubMed] [Google Scholar]
- 42.Gretzer C, Emanuelsson L, Liljensten E, Thomsen P. The inflammatory cell influx and cytokines changes during transition from acute inflammation to fibrous repair around implanted materials. J Biomater Sci-Polym Ed. 2006;17:669–87. doi: 10.1163/156856206777346340. [DOI] [PubMed] [Google Scholar]
- 43.Eldridge JH, Hammond CJ, Meulbroek JA, Staas JK, Gilley RM, Tice TR. Controlled vaccine release in the gut-associated lymphoid-tissues. 1. Orally-administered biodegradable microspheres target the peyers patches. Elsevier Science Bv; 1990. pp. 205–214. [Google Scholar]
- 44.Champion JA, Walker A, Mitragotri S. Role of particle size in phagocytosis of polymeric microspheres. Pharma Res. 2008;25:1815–21. doi: 10.1007/s11095-008-9562-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shakweh M, Besnard M, Nicolas V, Fattal E. Poly (lactide-co-glycolide) particles of different physicochemical properties and their uptake by Peyer’s patches in mice. Eur J Pharm Biopharm. 2005;61:1–13. doi: 10.1016/j.ejpb.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 46.Foged C, Brodin B, Frokjaer S, Sundblad A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int J Pharm. 2005;298:315–22. doi: 10.1016/j.ijpharm.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 47.Henry JA, Burugapalli K, Neuenschwander P, Pandit A. Structural variants of biodegradable polyesterurethane in vivo evoke a cellular and angiogenic response that is dictated by architecture. Acta Biomater. 2009;5:29–42. doi: 10.1016/j.actbio.2008.08.020. [DOI] [PubMed] [Google Scholar]
- 48.Zhang SL, Li J, Lykotrafitis G, Bao G, Suresh S. Size-dependent endocytosis of nanoparticles. Adv Mater. 2009;21:419–424. doi: 10.1002/adma.200801393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coonrod A, Li FQ, Horwitz M. On the mechanism of DNA transfection: efficient gene transfer without viruses. Gene Ther. 1997;4:1313–21. doi: 10.1038/sj.gt.3300536. [DOI] [PubMed] [Google Scholar]
- 50.Greitz D, Hannerz J. A proposed model of cerebrospinal fluid circulation: observations with radionuclide cisternography. Am J Neuroradiol. 1996;17:431–8. [PMC free article] [PubMed] [Google Scholar]
- 51.Stacey KJ, Sweet MJ, Hume DA. Macrophages ingest and are activated by bacterial DNA. J Immunol. 1996;157:2116–22. [PubMed] [Google Scholar]
- 52.Fukuhara Y, Naoi T, Ogawa Y, Nishikawa M, Takakura Y. Plasmid DNA uptake and subsequent cellular activation characteristics in human monocyte-derived cells in primary culture. J Pharm Sci. 2007;96:1576–84. doi: 10.1002/jps.20816. [DOI] [PubMed] [Google Scholar]
- 53.Pratten MK, Lloyd JB. Pinocytosis and phagocytosis - the effect of size of a particulate substrate on its mode of capture by rat peritoneal macrophages culture in vitro. Biochim Biophys Acta. 1986;881:307–13. doi: 10.1016/0304-4165(86)90020-6. [DOI] [PubMed] [Google Scholar]
- 54.Cooper PH, Mayer P, Baggiolini M. Stimulation of phagocytosis in bone marrow-derived mouse macrophages by bacterial lipopolysaccharide - correlation with biochemical and functional parameters. J Immunol. 1984;133:913–22. [PubMed] [Google Scholar]
- 55.Collins HL, Bancroft GJ. Cytokine enhancement of complement- dependent phagocytosis by macrophages - synergy of tumor-necrosis-factor-alpha and granulocyte-macrophage colony-stimulating factor for phagocytosis of cryptococcus-neoformans. Eur J Immunol. 1992;22:1447–54. doi: 10.1002/eji.1830220617. [DOI] [PubMed] [Google Scholar]
- 56.Hasegawa T, Hirota K, Tomoda K, Ito F, Inagawa H, Kochi C, et al. Phagocytic activity of alveolar macrophages toward polystyrene latex microspheres and PLGA microspheres loaded with anti-tuberculosis agent. Colloids Surf B-Biointerfaces. 2007;60:221–8. doi: 10.1016/j.colsurfb.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 57.Anderson JM, Shive MS. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliv Rev. 1997;28:5–24. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
- 58.Samuvel DJ, Sundararaj KP, Nareika A, Lopes-Virella MF, Huang Y. Lactate boosts TLR4 signaling and NF-kappa B pathway-mediated gene transcription in macrophages via mono-carboxylate transporters and MD-2 Up-regulation. J Immunol. 2009;182:2476–84. doi: 10.4049/jimmunol.0802059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zolnik BS, Burgess DJ. Evaluation of in vivo-in vitro release of dexamethasone from PLGA microspheres. J Control Release. 2008;127:137–45. doi: 10.1016/j.jconrel.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 60.Tousignant JD, Zhao HM, Yew NS, Cheng SH, Eastman SJ, Scheule RK. DNA sequences in cationic lipid: pDNA-mediated systemic toxicities. Hum Gene Ther. 2003;14:203–14. doi: 10.1089/10430340360535760. [DOI] [PubMed] [Google Scholar]
- 61.Yasuda K, Wagner H, Takakura Y. Role of immunostimulatory DNA and TLR9 in gene therapy. Crit Rev Ther Drug Carr Syst. 2006;23:89–109. doi: 10.1615/critrevtherdrugcarriersyst.v23.i2.10. [DOI] [PubMed] [Google Scholar]
- 62.Zielhuis SW, Nijsen JFW, Seppenwoolde JH, Bakker CJG, Krijger GC, Dullens HFJ, et al. Long-term toxicity of holmium-loaded poly(L-lactic acid) microspheres in rats. Biomaterials. 2007;28:4591–9. doi: 10.1016/j.biomaterials.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 63.Koval M, Preiter K, Adles C, Stahl PD, Steinberg TH. Size of IgG-opsonized particles determines macrophage response during internalization. Exp Cell Res. 1998;242:265–73. doi: 10.1006/excr.1998.4110. [DOI] [PubMed] [Google Scholar]
- 64.Grosse S, Tremeau-Bravard A, Aron Y, Briand P, Fajac I. Intracellular rate-limiting steps of gene transfer using glycosylated polylysines in cystic fibrosis airway epithelial cells. Gene Ther. 2002;9:1000–7. doi: 10.1038/sj.gt.3301768. [DOI] [PubMed] [Google Scholar]
- 65.Wang DQ, Robinson DR, Kwon GS, Samuel J. Encapsulation of plasmid DNA in biodegradable poly(D, L-lactic-co-glycolic acid) microspheres as a novel approach for immunogene delivery. J Control Release. 1999;57:9–18. doi: 10.1016/s0168-3659(98)00099-6. [DOI] [PubMed] [Google Scholar]
- 66.Cohen H, Levy RJ, Gao J, Fishbein I, Kousaev V, Sosnowski S, et al. Sustained delivery and expression of DNA encapsulated in polymeric nanoparticles. Gene Ther. 2000;7:1896–905. doi: 10.1038/sj.gt.3301318. [DOI] [PubMed] [Google Scholar]
- 67.Houk BE, Martin R, Hochhaus G, Hughes JA. Pharmacokinetics of plasmid DNA in the rat. Pharm Res. 2001;18:67–74. doi: 10.1023/a:1011078711008. [DOI] [PubMed] [Google Scholar]
- 68.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–61. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 69.Ma J, Chen T, Mandelin J, Ceponis A, Miller NE, Hukkanen M, et al. Regulation of macrophage activation. Cell Mol Life Sci. 2003;60:2334–46. doi: 10.1007/s00018-003-3020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 71.Polfliet MMJ, Fabriek BO, Daniels WP, Dijkstra CD, van den Berg TK. The rat macrophage scavenger receptor CD163: Expression, regulation and role in inflammatory mediator production. Immunobiology. 2006;211:419–25. doi: 10.1016/j.imbio.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 72.Pinheiro ED, Antunes OAC, Fortunak JMD. A survey of the syntheses of active pharmaceutical ingredients for antiretroviral drug combinations critical to access in emerging nations. Antivir Res. 2008;79:143–65. doi: 10.1016/j.antiviral.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 73.de Lissovoy G, Matza LS, Green H, Werner M, Edgar T. Cost-effectiveness of intrathecal baclofen therapy for the treatment of severe spasticity associated with cerebral palsy. J Child Neurol. 2007;22:49–59. doi: 10.1177/0883073807299976. [DOI] [PubMed] [Google Scholar]
- 74.Plassat R, Verbe BP, Menei P, Menegalli D, Mathe JF, Richard I. Treatment of spasticity with intrathecal baclofen administration: long-term follow-up, review of 40 patients. Spinal Cord. 2004;42:686–93. doi: 10.1038/sj.sc.3101647. [DOI] [PubMed] [Google Scholar]