

An increase in Rab1b levels induces changes in Golgi size and in gene expression. These Rab1b-dependent changes require the activity of p38 mitogen-activated protein kinase and the cAMP-responsive element binding protein consensus binding. The results show a Rab1b increase in secretory cells after stimulation and suggest that this increase is required to elicit a secretory response.
Abstract
Rab1b belongs to the Rab-GTPase family that regulates membrane trafficking and signal transduction systems able to control diverse cellular activities, including gene expression. Rab1b is essential for endoplasmic reticulum–Golgi transport. Although it is ubiquitously expressed, its mRNA levels vary among different tissues. This work aims to characterize the role of the high Rab1b levels detected in some secretory tissues. We report that, in HeLa cells, an increase in Rab1b levels induces changes in Golgi size and gene expression. Significantly, analyses applied to selected genes, KDELR3, GM130 (involved in membrane transport), and the proto-oncogene JUN, indicate that the Rab1b increase acts as a molecular switch to control the expression of these genes at the transcriptional level, resulting in changes at the protein level. These Rab1b-dependent changes require the activity of p38 mitogen-activated protein kinase and the cAMP-responsive element-binding protein consensus binding site in those target promoter regions. Moreover, our results reveal that, in a secretory thyroid cell line (FRTL5), Rab1b expression increases in response to thyroid-stimulating hormone (TSH). Additionally, changes in Rab1b expression in FRTL5 cells modify the specific TSH response. Our results show, for the first time, that changes in Rab1b levels modulate gene transcription and strongly suggest that a Rab1b increase is required to elicit a secretory response.
INTRODUCTION
In mammalian cells, more than 60 different Rab proteins (Rabs) are associated with specific organelles of the endocytic and exocytic pathways. They alternate between a GTP-bound membrane-associated form and a GDP-bound cytosolic form. GTP-bound Rabs interact with a variety of effectors and regulate all of the steps required in membrane transport, including budding, targeting, docking, and fusion of transport intermediates with their target membranes (Segev, 2010).
In addition to the role of Rabs as regulators of protein trafficking, recent reports have established links between changes in Rab expression levels and the behavior of various tumors. For example, down-regulation of the Rab32 promoter was detected in colon cancer and gastric adenocarcinomas (Mori et al., 2004; Shibata et al., 2006). On the other hand, a high level of Rab23 expression has been detected in hepatocellular carcinoma specimens (Liu et al., 2007). Also, overexpression of Rab25 has been found in ovarian and breast cancers (Cheng et al., 2004), with this increase being implicated in the progression and aggressiveness of these tumors. Moreover, Rab1a was found to be overexpressed in tongue squamous cell carcinomas (Shimada et al., 2005).
Under normal conditions, some Rabs are expressed in a tissue-specific manner, but the majority of them are ubiquitously expressed in all mammalian cells. However, the Rab expression profiles across different tissues are variable (Gurkan et al., 2005). Although a close correlation between Rabs function and signal transduction systems has been reported (Bucci and Chiariello, 2006), the importance of the variability in Rab expression levels and their impact on signal transduction pathways in normal and pathological conditions remains unclear.
Rab1 isoforms (Rab1a and Rab1b) are ubiquitous and essential for endoplasmic reticulum (ER)-to-Golgi transport, with different tissues expressing dissimilar mRNA levels of Rab1a and Rab1b. In the present study, we examined different cellular effects induced by changes of Rab1b levels. We report the first evidence that an increase in Rab1b levels induces changes in Golgi structure and gene expression. Such gene expression changes require the activation of p38 mitogen-activated protein kinase (MAPK) and the cAMP-responsive element-binding protein (CREB) binding consensus site. We also show that Rab1b levels increase when thyroid cells, FRTL5, are stimulated by thyroid-stimulating hormone (TSH), and that changes in Rab1b expression modify the specific TSH response.
RESULTS
Increased Rab1b expression induces Golgi enlargement
Rab1b expression is ubiquitous and variable in different tissues, and high mRNA levels are detected in tissues with specific secretory activity (Gurkan et al., 2005), such as prostate, lung, and thyroid that produce prostatic acid phosphatase, pulmonary surfactant-associated protein, and thyroglobulin (TG), respectively. To analyze the role of high Rab1b levels, we evaluated the long-term effect of Rab1b transfection in HeLa cells.
We have previously demonstrated that expression of the active mutant Rab1bQ67L had an effect on the overall distribution of coatomer protein 1 (COPI) and Golgi-specific brefeldin A resistance factor 1 (GBF1), as well as the Arf1 membrane association–dissociation dynamics, after 24 h of transfection (Monetta et al., 2007). However, these parameters were not modified in cells expressing green fluorescent protein (GFP)-Rab1b wild-type (Rab1bwt). In contrast, Golgi labeled with Golgi matrix protein of 130 kDa (GM130), GalT, or TGN46 exhibited a bigger size or stronger immunofluorescence signal in transfected cells (Figure 1A, asterisks) than in untransfected cells or in cells expressing low levels of Rab1bwt after 36–48 h of GFP-Rab1bwt transfection. GM130 is a Rab1b effector peripherally associated to membranes; therefore an increase in Rab1bwt could enhance GM130 recruitment. However, we considered that overexpression of Rab1bwt causes a general Golgi modification because the signal of the transmembrane Golgi proteins GalT and TGN46 was also increased. For analysis of the Golgi organization in transfected cells, correlative light electron microscopy (CLEM) assays were performed in GFP-Rab1bwt–expressing cells. As shown in Figure 1B (left), GFP-Rab1bwt–transfected cells exhibited an enlargement of the Golgi complex. In these cells, cis-, medial, and trans-Golgi cisternae were well defined, aligned, and organized with longer length than in control cells (Figure 1B, right panel). Moreover, a significant increase in the number of round shape profiles, as well as in irregular shape membrane structures, was observed at the proximity of the Golgi complex and ER exit sites (ERES)/vesicular-tubular clusters (VTCs) interface. Interestingly, a CLEM assay performed in cells expressing the GTP-restricted mutant (GFP-Rab1Q67L) showed an induction of central fragmentation of the Golgi complex and, instead of the Golgi ribbon, a large number of COPI-like round-shaped profiles were present in the juxta-nuclear area and at the ERES/VTCs interface (unpublished data). This ultrastructural information agrees with reported data showing an increase of COPI-labeled peripheral structures in Rab1Q67L–transfected cells (Monetta et al., 2007). Taken together, these data suggest that the Golgi enlargement induced by overexpression of GFP-Rab1bwt is a consequence of a function of Rab1b.
FIGURE 1:
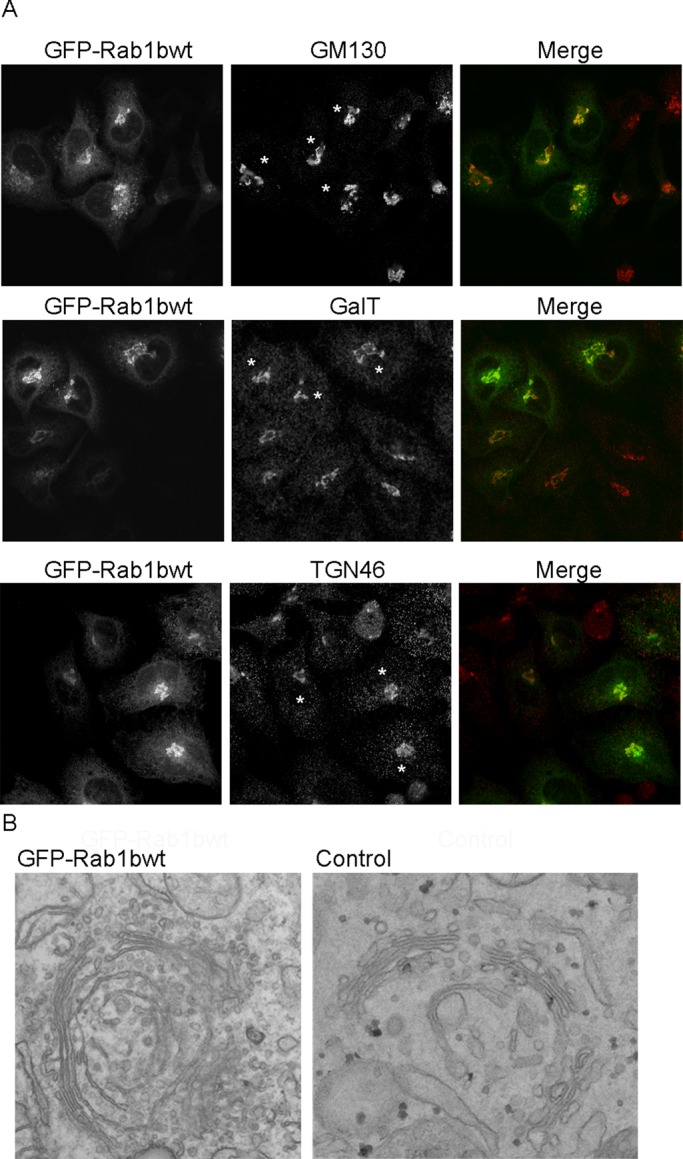
Rab1b expression induces Golgi enlargement. HeLa cells were transfected with GFP-Rab1bwt and analyzed 36–48 h after transfection. (A) Confocal immunofluorescence assay performed to detect GM130, GalT, and TGN46. Asterisks label transfected cells expressing high levels of GFP-Rab1bwt. (B) Golgi phenotype analyzed by CLEM assays in GFP-Rab1bwt–expressing cells (left) or control, untransfected cells (right).
An increase in Rab1b levels modifies gene expression of vesicle traffic-related genes
To explore how an increase in Rab1b levels induces Golgi enlargement, we performed a global gene expression microarray analysis on HeLa cells expressing Rab1bwt. Affymetrix GeneChip arrays were used with RNA samples from cells expressing GFP-Rab1bwt construct. To eliminate any nonspecific response induced by the simple fact of GFP overexpression or by a nonspecific Rab1bwt effect, we also performed microarray analyses on HeLa cells transfected with the following constructs: GFP empty vector, GFP-Rab1Q67L (GTP-Rab1b–restricted mutant), and GFP-Rab7Q67L. All samples were compared against GFP-expressing cells, and probe sets showing significantly altered expression levels greater than 1.5-fold change (p < 0.05) were considered for further analysis. By performing this first filter, we eliminated expression changes induced by overexpression of GFP. Thus a total of 1424 probe sets were up- or down-regulated in Rab1bwt-, Rab1Q67L-, and Rab7Q67L-expressing cells. All the constructs induced changes in gene expression. Although some probe sets were shared between the different samples, the majority of them were exclusively changed by only one construct, suggesting these changes are specific for each Rab construct. Venn diagrams showing the number of probe sets modified (up and down) by the indicated GFP-Rab construct and the overlap between them are displayed in Figure 2A. To select expression changes induced only by Rab1bwt, we excluded probe sets that were also modified by Rab1Q67L and Rab7Q67L. After applying this second filter we analyzed 244 (152 up and 92 down) probe sets (representing 219 genes) that were solely modified by expression of Rab1bwt, because this condition correlates with physiological situations in which Rab1b levels are increased.
It was found that Rab1bwt changed the expression of a wide functional category of genes (Table 1). Functional classification of Rab1b-modified genes indicates that probe sets were categorized in 21 functions (Tables 1 and S1 and Figure 2B). Vesicle trafficking accounted for one of the most significant sets of Rab1b-changed probe sets (34 probe sets that represent 29 genes; Table 2 and Figure 2B). Interestingly, the majority of vesicle traffic genes (24 out of 29) were up-regulated. Twelve of these vesicle traffic genes encode proteins that participate in different steps of the ER-to-Golgi transport; among them were: Sec24D and Sec31L1 (required for the budding of COPII vesicles); COPZ2 and COPG (subunits of the COPI vesicle complex); and VDP (known as p115) and GOLGA2 (known as GM130), which are Rab1 effectors required for membrane-tethering events (Allan et al., 2000; Weide et al., 2001). Moreover, Bet1, Sec22L1 (or Sec22B; Hay et al., 1997), and the syntaxin-binding protein 1 STXBP1 (or Munc18; Burgoyne and Morgan, 2007) encode proteins that participate in vesicle fusion events. Another ER-to-Golgi transport gene was KDELR3, which encodes a member of the KDEL ER protein receptor family, being required for the retention of the luminal ER proteins and also normal vesicular traffic through the Golgi (Pelham, 1991). Genes such us KDELR3, ARL1 (Munro, 2005), ARF4 (Cavenagh et al., 1996), and RAB26 (Wagner et al., 1995) were represented by multiple probe sets.
TABLE 1:
Functional classification of probe sets specifically modified under Rab1b with the numbers of genes that the probe sets represent shown in parentheses.
| Function | Number of probe sets |
|---|---|
| Vesicle trafficking | 34 (29) |
| Transcription factor | 23 (19) |
| Signal transduction | 21 (16) |
| Protein binding | 18 (17) |
| Metabolism | 14 (14) |
| Structural proteins | 15 (15) |
| Receptor | 16 (15) |
| Pathology | 12 (11) |
| Unknown | 11 (11) |
| Cell-adhesion related | 9 (7) |
| Cell cycle progress | 9 (7) |
| RNA processing | 8 (8) |
| Protein kinase | 8 (8) |
| Apoptosis related | 7 (7) |
| Immune-response related | 7 (6) |
| Hypothetical protein | 10 (10) |
| Chaperone | 6 (6) |
| Open reading frame | 5 (5) |
| Tumor | 5 (4) |
| Carrier family | 3 (3) |
| Growth factor | 3 (1) |
| Total | 244 (219) |
FIGURE 2:

Increase in Rab1b levels modifies gene expression. (A) Venn diagrams showing the number of probe sets modified (up and down) by the indicated GFP-Rab construct (with respect to GFP alone) and the overlap between them. (B) Pie chart showing number of genes, clustered by their functional classification, that were modified by Rab1b increase (the total number of genes was 219). (C) Expression analysis of mRNA levels of some interesting genes plotted as the fold change for each gene in the GFP-Rab1bwt construct cells compared with the GFP-transfected cell line, measured by either microarray (continuous line) or RT-qPCR (gray bars).
TABLE 2:
List of vesicle trafficking genes altered in Rab1b-overexpressing cells.
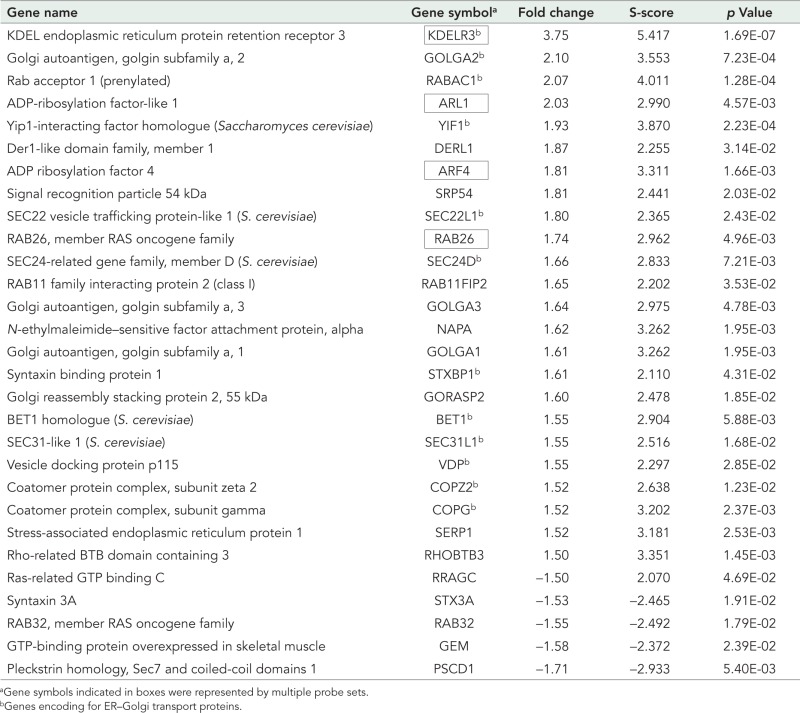 |
To validate the results obtained from microarray analyses, we assessed the expression levels of 10 of the differentially expressed genes by real-time RT-qPCR. The relative expression levels for the 10 genes had a significant correlation (Pearson's r = 0.94, p < 0.0001) with the microarray results, confirming the validity of the findings (Figure 2C). These genes were selected for three main reasons: first, KDELR3, ARF4, PRA1/RABAC1, ARL1, YIF1, and GOLGA2/GM130 encode proteins required for membrane traffic, with their mRNA fold change being among the top five up-regulated in this functional category. In addition, PRA1/RABAC1, GOLGA2/GM130, and YIF1 encode Rab1b/Ypt1p-related proteins (Martincic et al., 1997; Matern et al., 2000). Second, the c-Jun proto-oncogene (JUN), a member of the CXC chemokine superfamily, interleukin 8 (IL8), and the transcription factor, early growth response 1 (EGR1), were found to be related in functional networks (see Materials and Methods; Ingenuity Pathway Analysis 3.0 tools); also JUN and IL8 were ranked among the top five down-regulated genes (fold changes: −2.21 and −2.62 respectively; Table S1). Finally, G protein–coupled receptor 126 (GPR126) was selected as a representative member of signaling molecules.
To test whether transport activity is enhanced in Rab1b-transfected HeLa cells, we evaluated the dynamics of brefeldin A wash-out (BFA-WO) in Rab1b-transfected cells and in untransfected cells. The dynamics of Golgi reconstitution were evaluated by analyzing the phenotype of an endogenous Golgi protein (GalNAc-T2). Transfection with GFP and CFP-MannII was performed as controls. Results (Supplemental Figure S1) indicated that BFA-WO response is faster in Rab1b-overexpressing cells and strongly suggest that Rab1b overexpression confers extra transport capacity.
Rab1b increase induces changes in JUN, GM130, and KDELR3 expression
To assess whether the Rab1b effect on gene expression correlates with a concomitant modification of protein levels, we established a HeLa cell line that stably expresses the Rab1b-myc construct (Alvarez et al., 2003) in a tetracycline-inducible manner (T-Rex Rab1b cells). In these cells, addition of tetracycline induces Rab1b-myc expression in a time-dependent manner, and Rab1b levels increase from 3.4- to 5.1-fold after 12–48 h of tetracycline induction, respectively (Figure 3A). An increase in Rab1b-myc levels could be detected as early as 2 h after induction (unpublished data). Because microarray analyses were performed in cells after 48 h of transfection, we decided to analyze protein levels after 48 h of tetracycline induction. Western blotting was performed to analyze changes in GM130, KDEL-receptor (KDELR), c-Jun (because their respective mRNA levels were significantly modified 2.10- and 3.75-fold up and 2.21-fold down, respectively) and in calreticulin (because its mRNA did not show a significant change). As shown in Figure 3B, the increase in Rab1b-myc protein level was followed by an increase of GM130 and KDELR and a decrease of c-Jun protein expression. In contrast, calreticulin levels were not modified under the same conditions. Quantitative analysis of protein signals relative to the calreticulin signal indicated that GM130 and KDELR increased ∼1.4- and 3.1-fold, respectively, while the reduction in c-Jun was ∼1.6-fold relative to the control sample (without tetracycline). Interestingly, a similar analysis performed on a HeLa cell line that stably expresses a dominant-negative Rab1b mutant (Rab1N121I) indicated that induction of this mutant did not alter GM130, KDELR, or c-Jun levels (Figure 3B). Similar results were observed on cells expressing Rab1Q67L (unpublished data). In agreement with this observation, expression of the Rab1Q67L mutant did not modify the mRNA levels of GM130, KDELR, or JUN based on the microarray data. Therefore these results suggest that the observed modifications in GM130, KDELR, and JUN expression levels are not a general stress response induced by exogenous protein overexpression. Taken together, our results indicate that the increase of Rab1b levels changes gene and protein expression levels and that normal Rab1b GDP-GTP exchange seems to be required for these activities.
FIGURE 3:
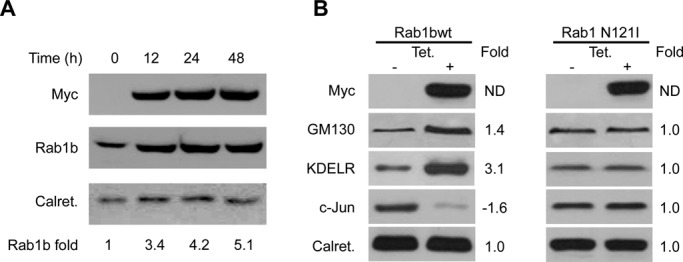
Increase in Rab1b levels modifies protein expression. Western blot analysis performed with cell extracts from stably transfected HeLa cell lines that express the indicated Rab1b-myc constructs in a tetracycline-inducible manner (T-Rex Rab1b cells). (A) Time course of Rab1b-myc (wt) induction after the indicated times of tetracycline addition. Myc antibody detected only inducible Rab1b-myc, while Rab1b antibody detect both endogenous and inducible. Rab1b fold change for each time (numbers at the bottom of the figure) was calculated as indicated in (B). (B) GM130, KDELR, and c-Jun changes induced after 48 h of tetracycline addition in T-Rex cells stably transfected with Rab1bwt or Rab1N121I. The intensity of each band relative to calreticulin (loading control) was measured, and the fold change (numbers next to each Western blot) was calculated as the ratio of the induced situation to the uninduced one (control). Relative density in control situation was set to 1.
Rab1b increase regulates JUN, GM130, and KDELR3 promoter activity
To investigate whether Rab1b regulates the expression of KDELR3, GM130, and JUN by modulating their respective promoters, we cloned the 5′-flanking regions of human GM130 (−454 to −80 base pairs) and KDELR3 (−291 to + 136 base pairs) genes containing the putative promoter sequence into the promoterless, luciferase pGL3 reporter vector (Figure 4A). The JUN promoter construct used (−1780 to +731 base pairs) has been previously reported (Wei et al., 1998). These constructs were transiently transfected into the T-Rex Rab1b cells, and luciferase activity was measured in the absence and the presence of tetracycline. Relative luciferase activity of the GM130 and KDELR3 constructs increased ∼2.2- and 18-fold, respectively, compared with the control (without tetracycline), whereas a 2.6-fold decrease in luciferase activity of the c-Jun construct was detected, compared with the control (Figure 4B). These data indicate that an increase in Rab1b levels was able to modulate activity of the KDELR3, GM130, and JUN promoters. We further explored the kinetics of Rab1b effect on promoter activity. We found that the increased response of the GM130 and KDELR3 promoters was time-dependent and was detected even after only 2 h of tetracycline addition (Figure 4C). Interestingly, the JUN promoter activity slightly increased after 2 h of Rab1b induction and then started to decrease, also in a time-dependent manner. Furthermore, the time-dependent response in the activity of the gene promoter regions assayed correlated with modifications in the expression of their respective endogenous proteins (Figure 4D). Taken together, our data indicate that an increase in Rab1b levels modifies the expression of GM130, KDELR, and c-Jun proteins, at least in part through the activation of their promoter.
FIGURE 4:

Rab1b regulates the expression of KDELR3, GM130, and c-Jun by modulating their promoter activity. (A) Schematic representation of the 5′-flanking regions of the human GM130, KDELR, and c-JUN genes cloned in a promoterless luciferase reporter vector (pGL3 basic vector). (B) Relative luciferase activities of pGL3 constructs depicted in (A) transiently transfected to the T-Rex Rab1b cells. After 12 h of transfection, the Rab1bwt expression was induced (+) or not (−) with tetracycline (for an additional 48 h). The graphs show relative luciferase activities; numbers on the right of each bar indicate the average value of relative luciferase activity from at least three experiments, and the ratio between samples for the indicated condition is shown in parentheses. (C) Time course of the relative luciferase activities of the indicated pGL3 constructs after tetracycline addition. Error bars in (B) and (C) represent SD. (D) Time course of protein induction in T-Rex Rab1b cells after the indicated times of tetracycline addition. Calreticulin was used as loading control.
Phosphorylation of the p38-MAPK is required for the Rab1b-induced response
How does Rab1b induce changes in JUN, GM130, and KDELR3 gene expression? It has been shown that a diverse array of extracellular stimuli leads to the activation (phosphorylation) of the c-Jun transcription factor mediated by the c-Jun NH2-terminal kinase (JNK) signal transduction pathway. Furthermore, endogenous signals initiated in the ER by the accumulation of unfolded proteins can also activate JNK (Urano et al., 2000). In a different cellular context, the extracellular signal–regulated protein kinase (ERK) can also regulate c-Jun mRNA levels (Lopez-Bergami et al., 2007). However, data about the potential mechanisms that control the expression of GM130 and KDELR3 are rather limited.
Considering that JNK, ERK, and p38 kinase belong to the family of MAPKs, and that a gene expression program can depend on the integration of signals provided by the temporal activation of each of the MAPK family members, we asked whether the MAPK pathway might regulate the Rab1b-dependent changes of GM130, KDELR3, and JUN transcription. Specific pharmacological inhibitors of the JNK (SP600125), MEK1/2-ERK (U0126), and p38 MAPK (SB 203580) pathways were used for this purpose. These inhibitors were added in the T-Rex Rab1b cells transiently transfected with the GM130, KDELR3, or JUN promoter constructs previously described (Figure 4A). As shown in Figure 5A, Rab1b-mediated increase in GM130 and KDELR promoter activity was specifically blocked by the p38 MAPK inhibitor but not by either the MEK1/2-ERK or JNK inhibitors. In addition, down-regulation of the JUN promoter was blocked by both the p38 MAPK and JNK inhibitors but not by the MEK1/2-ERK one. In line with these results, MAPK inhibitors also modulated GM130, KDELR, and c-Jun proteins expression levels (Figure 5B). The p38 MAPK inhibitor blocked the rise in GM130 and KDELR induced by the Rab1b increase (lanes 1–3), whereas neither the MEK1/2-ERK nor JNK (Figure 5B, lanes 4 and 5) inhibitors had this effect. Furthermore, the p38 MAPK and the JNK inhibitors (Figure 5B, lanes 3 and 5) but not the MEK1/2-ERK inhibitor (Figure 5B, lane 4) blocked the reduction in c-Jun induced by an increase in Rab1b (Figure 5B, lane 2). To test whether the phosphorylation inhibitors used in these assays were effective and specific for each MAPK, we assessed the phosphorylation state of p38, JNK, and ERK. As shown in Figure 5C (lanes 1 and 2), phosphorylation of p38, JNK, and ERK increased ∼3-fold relative to the control (noninduced) samples after induction of Rab1b, while the total MAPK levels remained unmodified (Figure 5D, lanes 1 and 2). As expected, the increase in p38, JNK, and ERK phosphorylation was blocked with the specific kinase inhibitors, and the inhibitors did not modify total MAPK levels (Figure 5, C and D). Phospho-p38 (P-p38) was analyzed in standard HeLa cells (untransfected or non–T-Rex cells) incubated in regular medium (Figure 5F, lane 1), in cells irradiated with UV (positive control of P-p38 induction, lane 2), and in the presence of tetracycline (lane 3). UV irradiation but not tetracycline addition was able to increase P-p38, indicating that in HeLa T-Rex cells p38 phosphorylation was induced by a Rab1b increase.
FIGURE 5:

Analysis of the MAPK pathways and the effect of their inhibitors on the T-Rex Rab1b cells. Inhibitors used were SB203580 (p-38), UO126 (MEK1/2), and SP600125 (JNK). (A) Effect of specific MAPK inhibitors on the relative luciferase activities of the pGL3-GM130, pGL3-KDELR, and pGL3-c-JUN constructs depicted in Figure 4A. Numbers on the right of each bar indicate the average value of relative luciferase activity from at least three experiments, and the ratio between samples for the indicated condition is shown in parentheses. Error bars represent SD. (B) Effect of specific MAPK inhibitors on the GM130, KDELR, and c-JUN changes induced by the increase in Rab1b. (C and D) Effect of Rab1b induction and MAPK inhibitors on the phosphorylation and total levels of p38, JNK, and ERK. The intensity of each band relative to calreticulin (loading control) was measured, and the fold change was calculated as the ratio of the tetracycline induced to the uninduced one. Numbers under each MAPK Western blot represent the average fold change of two independent experiments. Inhibitors of p38 and MEK1-2: 25 μM; JNK inhibitor: 50 μM. (E) Time course of the activation of the indicated phospho-MAPK in T-Rex Rab1b cells after different times of tetracycline addition. Numbers under each MAPK Western blot represent the average fold change of two independent experiments. Fold change was calculated as indicated in (C) and (D). (F) p38 phosphorylation analysis in response to tetracycline addition. P-p38 was analyzed in untransfected HeLa cells incubated in regular medium (lane 1), in cells irradiated with UV (to stimulate P-p38 induction, lane 2) and in the presence of tetracycline (lane 3).
Taken together, these data indicate that: first, an increase in Rab1b induces phosphorylation of p38, JNK, and ERK; second, phosphorylation of p38 but not phosphorylation of ERK is required for the regulation of the GM130, KDELR3, and JUN promoter activities; and finally, phosphorylation of JNK is also required for regulation of the JUN promoter. Considering that the GM130, KDELR3, and JUN promoter activity responses were detectable after 2 h (Figure 4C), and even after only 30 min of Rab1b induction (unpublished data), and that inhibition of p38 phosphorylation impedes the effects of an elevated Rab1b on GM130, KDELR3, and c-Jun changes (at both promoter activity and protein levels), we postulate that the first effect of the increase in Rab1b was the activation (phosphorylation) of p38. To test this assumption, we analyzed the dynamics of the phosphorylation of p38, JNK, and ERK after tetracycline addition. As shown in Figure 5E, enhanced phosphorylation of p38 was detectable after 30 min of Rab1b induction and reached its highest level after 75 min. Interestingly, JNK and ERK phosphorylation levels did not change, even after 2 h of Rab1b induction. These data indicate that an increase of p38 phosphorylation preceded JNK and ERK phosphorylation changes. Despite this fact, addition of a p38 kinase inhibitor was unable to block either JNK or ERK phosphorylation (Figure 5C), suggesting not only that the inhibitor was specific but also that the JNK and ERK phosphorylations were independent of p38 activation.
CREB-binding consensus site is required for the Rab1b-induced response
For extending the analysis of the GM130 and KDELR3 promoters and searching for the specific DNA-binding sites that could be responsible for the Rab1b induction effect observed at the transcription level, deletion constructs of these promoters were generated and cloned into the luciferase reporter pGL3 vector. Three new deletion constructs, named PGL3-GM130-1/2/3 and PGL3-KDELR-1/2/3, were generated from each of the cloned regions of the GM130 (PGL3-GM130) and KDLER (PGL3-KDELR) genes, respectively (Figure 6A). Relative luciferase activity of these constructs was measured in the T-Rex Rab1b cells. The activity of the promoter variant pGL3-GM130-1 increased after tetracycline addition by ∼2.5 times compared with the control without tetracycline. This activation is similar in magnitude to that seen for the PGL3-GM130 construct (Figure 6A). In contrast, the activity of promoter constructs pGL3-GM130-2 and pGL3-GM130-3 were barely up-regulated after tetracycline addition, showing only a slight increase (1.3 and 1.4 respectively) compared with control samples. Additionally, relative activities of the pGL3-KDELR-1-3 constructs indicated that Rab1b induction (by tetracycline addition) increased the promoter activity of the pGL3-KDELR-1 construct (11.9 times over the control), but did not up-regulate the activities of either pGL3-KDELR-2 or pGL3-KDELR-3.
FIGURE 6:

CREB activation and its consensus sequence are required for the effect of Rab1b. (A) Relative luciferase activities obtained with a set of reporter vectors containing the indicated 5′-flanking variants of the GM130 and KDELR promoters depicted in Figure 3A. pGL3-GM130-1/2/3 and pGL3-KDELR-1/2/3 represent different deletions, and pGL3-GM130-1M and pGL3-KDELR-1M represent mutant variants of their respective versions in which the CREB-binding consensus site was mutated. The graphs show relative luciferase activities; numbers on the right of each bar indicate the average value of relative luciferase activity from at least three experiments and the ratio between samples for the indicated condition is shown in parentheses. Error bars represent SD. (B–D) Western blot analysis of T-Rex Rab1b cells. (B) Detection of P-CREB and total CREB in the absence or presence of tetracycline (over 48 h). (C) Detection of P-CREB at the indicated times after tetracycline addition. (D) Effect of the indicated MAPK inhibitors on the CREB phosphorylation induced by an increase in Rab1b levels. Calreticulin was used as the loading control. (E) Luciferase activities obtained with the indicated pGL3 constructs (represented in Figure 4A) cotransfected with a plasmid vector encoding CREB and with a control vector (R. reiniformis phRL-TK) in the T-Rex Rab1b cells. CREB expression was able to stimulate GM130 and KDELR transcription and to inhibit c-Jun transcription. While these modifications were less pronounced than those observed by an increase in Rab1b, they were still significant (p < 0.05) and indicated that CREB can regulate their promoters. Additionally, modifications in the promoter activities in cells expressing both CREB and Rab1b had no significant differences compared with those induced by Rab1b alone.
These data indicate that regulatory promoter elements located in pGL3-GM130 between −404 to −336 (68 base pairs) and in pGL3-KDEL between −253 to −126 (127 base pairs) are required for a Rab1b-mediated effect on their promoter activities. Due to the fact that both GM130 and KDELR 5′-flanking regions required the activation of p-38 MAPK to respond to Rab1b increase, we can speculate that the same transcriptional network could be required to modulate the promoter activity of the GM130 and KDELR genes.
We conducted an in silico search for consensus binding sites for transcription factors in both 5′-flanking regions (Gene2Promoter; Genomatix [MatInspector] Munich, Germany). Only the core sequence of CREB was present in both 5′ regions at an acceptable score, suggesting the involvement of CREB in mediating the effect of Rab1b. To assess whether CREB activation occurred after Rab1b induction, we tested CREB phosphorylation in T-Rex Rab1b cells. As shown in Figure 6B, an increase of Rab1b induced CREB phosphorylation without modifying total CREB levels. Furthermore, CREB phosphorylation could be detected after 30 min of Rab1b induction (Figure 6C), as early as p38 activation, and CREB phosphorylation was inhibited by the p38 inhibitor but not by either the JNK or the ERK inhibitor (Figure 6D). Because overexpression of Creb3L1 (a CREB-like protein) can up-regulate (in HeLa cells) expression of genes required for the secretory pathway (Fox et al., 2010), we also tested the effect of CREB expression on the promoter activity of our PGL3 constructs. As shown in Figure 6E, CREB expression was able to stimulate GM130 and KDELR promoter activity and to inhibit JUN transcription. Finally, to further analyze the role of CREB, the CREB-binding consensus site was mutated in pGL3-GM130-1 and pGL3-KDELR-1, with the respective mutated versions (pGL3-GM130-1M and pGL3-KDELR-1M) being obtained (Figure 6A). The promoter activities of these mutants were basically not modified by Rab1b induction, indicating the involvement of the CREB-binding consensus site in mediating the effect of Rab1b (Figure 6A).
Rab1b levels modulate the secretory response in a thyroid cell line
It has been shown that tissues such as thyroid, prostate, and epithelial lung cells have 3- to 10-fold higher RAB1B mRNA levels than other organs (Gurkan et al., 2005). Our data imply that changes in the levels of Rab1b elicit a MAPK signaling cascade that modulates the expression of a variety of genes. We postulate that, in specialized secretory cells, the appropriate stimulus induces a rise in Rab1b that increases membrane transport and secretion of specific substrates. To test this assumption, we used a secretory thyroid cell line (FRTL5) to analyze changes in Rab1b levels and their impact on a secretory system. In these cells, TSH stimulates secretion of TG (Van Heuverswyn et al., 1984) and synthesis of the plasma membrane protein sodium iodide symporter (NIS; Kogai et al., 1997). To analyze whether TSH stimulation also modifies Rab1b and GM130 levels, we performed immunofluorescence and flow cytometry assays. FRTL5 cells were grown in basal medium (−TSH) over 24–36 h, and then induced with TSH (for 24 h). TSH addition increases NIS (Kogai et al., 1997), as well as Rab1b and GM130 levels (Figure 7) as revealed by an increase of their mean fluorescence intensity assessed by flow cytometry (Figure 7, right panels), suggesting that TSH stimulates not only NIS synthesis but also Rab1b and GM130 Golgi proteins required for transport.
FIGURE 7:

NIS, GM130, and Rab1b increase after TSH stimulation in FRTL5 cells. Left panels, immunofluorescence analysis in basal (−TSH) or stimulated (+TSH) situations; images for each marker were acquired under the same setting conditions. Right panels, flow cytometry analysis to measure fluorescence intensity to detect NIS, GM130, and Rab1b using a secondary antibody labeled with Alexa Fluor 647. Histograms represent profiles in basal (−TSH, dark gray) or stimulated (+TSH, black) conditions for the proteins indicated on the left panel. A total of 5 × 105 cells were analyzed in each situation. Controls (light gray) were performed by incubating cells with only secondary antibody. The geometric mean fluorescence intensity value is indicated on top of each histogram.
To investigate whether a Rab1b increase was required for the TSH-mediated NIS stimulation, we tested whether Rab1b depletion caused any effect on NIS. RAB1B expression was knocked down using RNA interference (RNAi). Specific 21-mer-long RAB1B RNA duplexes were transfected to inhibit RAB1B expression in FRTL5, and an equivalent concentration of scrambled small interfering RNA (siRNA) was used as a negative control. Immunofluorescence analyses were used to test the Rab1b-depletion effect (Figure 8A). As it was previously shown in HeLa cells (Monetta et al., 2007), RAB1B RNAi transfection in FRTL5 cells induces a major decrease in Rab1b signal (Figure 8A, red) and Golgi disruption, as indicated by the appearance of a punctate GM130 pattern (Figure 8A, green). This punctate GM130 pattern was used to identify Rab1b-depleted cells after siRNA transfection. We then tested whether inhibition of Rab1b activity was able to block the increase of GM130 and NIS induced by TSH stimulation (Figure 8B). FRTL5 cells were first transfected with siRNA and then incubated in basal medium (−TSH) for 36 h. After a 24-h TSH stimulation, control cells (transfected with scrambled siRNA) showed an increase in the GM130 (Figure 8B, red) and NIS (Figure 8B, green) immunofluorescence signals. In contrast, in Rab1b-depleted cells (detected by their disrupted Golgi complex), showed no significant change in GM130 and NIS signals (confirmed by flow cytometry analysis; unpublished data), indicating that Rab1 inhibition blocked the TSH-stimulated GM130 and NIS increase. This inhibitory effect of Rab1b depletion was not due to Rab1b participation in transport because inhibition of transport induced by brefeldin A treatment did not block the TSH-stimulated NIS increase (Figure S2).
FIGURE 8:
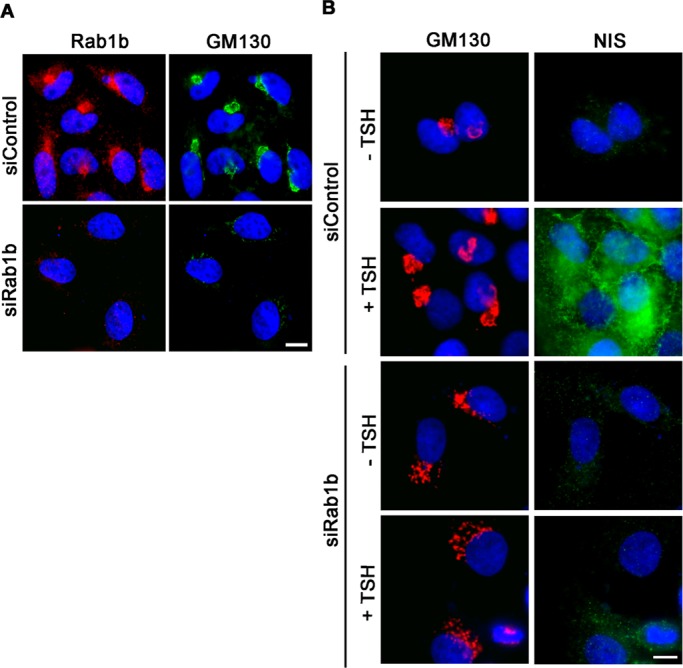
Rab1b depletion blocked GM130 and NIS increases induced by TSH stimulation. (A and B) Immunofluorescence analysis in Rab1b or control siRNA-transfected cells. (A) Rab1b and GM130 patterns after siRNA transfections in TSH-stimulated cells. (B) Effect of Rab1b siRNA transfection in basal condition (−TSH) or after TSH stimulation (+TSH). Images for each marker were acquired under the same setting conditions.
Considering that in FRTL5 cells TSH stimulus was able to increase Rab1b levels and that Rab1b inhibition abolished the increase in NIS levels induced by TSH, we postulated that Rab1b overexpression alone should elevate NIS levels in a TSH-independent manner. For testing this assumption, FRTL5 cells were transfected with GFP-Rab1bwt and, after 24 h of transfection, cells were divided into two plates and grown in complete medium (+TSH) for 16 h. Then one plate was incubated in basal medium (−TSH) for 48 h and subsequently stimulated with TSH (+TSH) for 24 h. The second plate was incubated in basal medium (–TSH) for 72 h. Immunofluorescence and cell cytometry assays were performed to test the effect of Rab1b overexpression on NIS expression in basal and TSH-stimulated conditions. GFP-Rab1bwt–transfected cells exhibited higher NIS signal than untransfected cells in both basal and TSH-stimulated conditions (Figure 9A). Quantification by cell cytometry indicated that NIS expression was 1.5 to ∼2 times higher in transfected cells than in untransfected cells in basal (−TSH) and stimulated (+TSH) conditions (Figure 9B). Controls performed with cells transfected with GFP empty vector indicated that NIS expression in transfected or untransfected cells was similar (unpublished data).
FIGURE 9:

NIS expression is higher in Rab1b-transfected cells than in untransfected cells. (A) Immunofluorescence analysis of GFP-Rab1b–transfected FRTL5 cells in basal (−TSH) or stimulated (+TSH) conditions. (B) Bar graphs showing mean fluorescence intensity (MFI) of NIS on untransfected (UNT) and GFF-Rab1b–transfected (T) cells in basal and stimulated conditions. The MFI value for each condition is indicated on top of each bar, and the ratio between samples for which the untransfected condition was taken as 1 is shown in parentheses. (C) Relative luciferase activity of the NIS promoter (pNUE) cotransfected with the indicated GFP constructs in FRTL5 cells, incubated in basal or stimulated conditions. The graphs show relative luciferase activities, numbers on the right of each bar indicate the average value of relative luciferase activity from two experiments, and the ratios between samples for the indicated conditions are shown in parentheses.
Taking into account that Rab1bwt modulates gene expression and GM130, KDELR, and JUN promoter activities (Figures 4–6), we tested whether high levels of Rab1b could modulate the promoter activity of NIS. Thus we assessed the effects of Rab1b overexpression on the activity of the rat NIS promoter region that includes the NIS upstream enhancer (NUE) located within −2495 and −2264 base pairs (Chun and Di Lauro, 2001). This NUE region is engaged in the most important aspect of NIS regulation and has a degenerate cAMP-response element (CRE-L; Chun and Di Lauro, 2001). A luciferase reporter vector, including the NUE region (pNUE; Nicola et al., 2010); GFP-Rab1bwt; and the control vector (Renilla reiniformis phRL-TK) were transiently cotransfected into FRTL-5 cells. After cotransfection, the pNUE luciferase activity was assayed in basal (−TSH) or stimulated (+TSH) conditions. Cotransfections with a GFP empty vector or a GFP-Rab1N121I construct instead of GFP-Rab1bwt were performed as controls. Cell sorting was performed to select cells transfected with the different GFP constructs and to exclusively determine pNUE luciferase activity in such cells. Relative luciferase activity of the pNUE promoter in basal condition (−TSH; Figure 9C) was similar in the context of GFP, GPF-Rab1b, or GFP-Rab1N121I overexpression. TSH induction (for 4 h) stimulated the pNUE luciferase activity in GFP-transfected cells compared with the basal condition (1.6-fold, from 15.71- to 22.29-fold). This stimulation is similar to that previously reported for nontransfected cells (Kogai et al., 1997). Interestingly, relative luciferase activity of the pNUE increased ∼2.3 fold (from 17.17 to 39.19) in GFP-Rab1b–expressing cells after TSH induction, whereas GFP-Rab1bN121I expression impeded TSH-induced pNUE activation. Rab1bwt effect on NIS promoter activity was evident at short times of TSH induction (4 h), while NIS promoter was equally high in both conditions after longer induction times (12–24 h; unpublished data), probably due to saturation of the promoter activity.
Taken together, these findings indicate that TSH-stimulated pathways increase the expression of Rab1b and GM130, both of which are Golgi proteins required for secretory transport. Furthermore, an increase in Rab1b positively modulates NIS expression and NIS promoter activity, suggesting a role of Rab1b in the NIS activation pathway.
Our data strongly suggest that activation of secretion (or membrane transport) by a secretory stimulus induces an increase in Rab1b levels that trigger signaling circuits essential to coordinating the cellular adaptations required for the response to the secretory stimulus.
DISCUSSION
In the present study, we aimed to characterize the role of the high Rab1GTPase levels detected in secretory tissues. We first analyzed the long-term effect of Rab1b transfection in HeLa cells and then the impact of altered Rab1b levels in a thyroid secretory cell line. The most important new findings related to Rab1b function are described in the following sections.
Rab1b and Golgi enlargement
Involvement of Rab1b in Golgi structure after inhibition of Rab1b activity has frequently been analyzed. Different methods of Rab1b inhibition, such as expression of the dominant-negative construct Rab1bN121I, siRNA transfection (Monetta et al., 2007), or overexpression of its GAP (Haas et al., 2007), caused fragmentation of the Golgi complex. Because Rab1b is required for the ER-to-Golgi transport it was always assumed that Golgi fragmentation after Rab1b inhibition was an indirect consequence of blocking forward membrane transport rather than a direct effect of Rab1b on Golgi structure. In this paper, we demonstrate that an increase in Rab1b expression induces an enlargement of the Golgi complex. Ultrastructural analysis performed in Rab1b-transfected cells (Figure 1) indicates that the Golgi ribbon goes through an elongation process, while the number of cisternae per stack and the cisternal surface area seem to remain unchanged. Golgi ribbon elongation occurs during cell growth and differentiation and takes place through growth of additional ministacks after an increased synthesis of Golgi complex proteins (Sengupta and Linstedt, 2011). In agreement, Rab1b overexpression increases GM130/GOLGA2 mRNA and protein levels (Table 2 and Figure 3, respectively). GM130 is a Rab1b effector (Moyer et al., 2001; Weide et al., 2001) that also interacts with the Golgi reassembly stacking protein, GRASP65 (Barr et al., 1997), a protein required for the formation of the Golgi ribbon. Moreover, GRASP55 (named “GORASP2” in Table 2), a GRASP65 homologue (Shorter et al., 1999) that also binds Rab1 but with less affinity than GRASP65, was detected by microarray analyses as one of the significantly up-regulated genes. Although GRASP proteins play multiple cellular roles (Vinke et al., 2011), the elongation of the Golgi ribbon observed in Rab1b-expressing cells strongly correlates with the higher levels of GM130, GRASP65, and GRASP55 and their role in linking, by lateral fusion, the Golgi cisternae during Golgi ribbon formation (Puthenveedu et al., 2006; Feinstein and Linstedt, 2008). Furthermore, cells transfected with Rab1b have increased mRNA expression of other Golgi proteins encoding genes, such as the Golgins 160 and 97 (named GOLGA3 and GOLGA1, respectively); the soluble N-ethylmaleimide–sensitive factor attachment protein receptors, SEC22L1 (Hay et al., 1998), BET1 (Zhang et al., 1997), and STXBP1 (Burgoyne and Morgan, 2007); and the Yip1-interacting factor homologue, YIF1 (Yoshida et al., 2008). Essentially, most of the 24 trafficking up-regulated genes (except for RAB26 and RAB11FIP2) encode proteins located at the ER–Golgi interface and have been shown to play a role in it. Only five trafficking genes were down-regulated. Some of them are members of the Ras superfamily of GTPases, such as Ras-related GTP-binding C (RRAGC), GTP-binding protein overexpressed in skeletal muscle (GEM), RAB32 (Maguire et al., 1994; Cohen-Solal et al., 2003; Bui et al., 2011); and others are functionally linked to the same family, such as syntaxin3A (STX3A; Martin-Martin et al., 1999) and cytohesin-1 (or pleckstrin homology, Sec7, and coiled-coil domains 1 [PSCD1; Geiger et al., 2000]).
Rab1b and gene expression modulation
The enlargement of the Golgi prompted us to think that a Rab1b increase stimulates a wide cellular response, including gene expression modulation. Microarray analyses were conducted to investigate global transcriptional changes, while eliminating any nonspecific response (such as a homeostatic effect) induced by the simple fact of overexpression. To do this, we performed microarrays on HeLa cells transfected with GFP, GFP-Rab1bwt, GFP-Rab1Q67L, and GFP-Rab7Q67L.
Functional classification of probe sets exclusively modified by Rab1b expression resulted in probe sets that were categorized in 21 functions (Tables 1 and S1 and Figure 2B), with 70% of the probe sets included in only 10 different functions. This is a small variety for a global approach. It is remarkable that we found a strong correlation between the Golgi enlargement phenotype and the function of the category of genes that were most significantly modified by Rab1b overexpression. In agreement with these observations, the dynamics of BFA-WO indicated that Rab1b-transfected cells were able to recover their Golgi pattern faster than untransfected cells, suggesting Golgi enlargement and the increase in levels of membrane transport protein positively regulate flux of membranes in secretory transport.
To further modulate Rab1b expression levels, we established HeLa cell lines that stably expressed Rab1b-myc constructs (Alvarez et al., 2003) in a tetracycline-inducible manner (T-Rex Rab1b cells; Figure 3, A and B). Tetracycline induced high levels of Rab1bwt and Rab1N121I (Figure 3B) and Rab1Q67L (unpublished data) expression after 48 h of treatment. Western blot analysis indicated that only cells expressing Rab1bwt exhibited changes in GM130, KDELR, and c-Jun expression (Figure 3B), indicating that mRNA changes detected in the microarray influenced protein expression levels in the same manner. The fact that overexpression of the dominant-negative (Rab1N121I) or the positive (Rab1Q67L) Rab1b constructs did not impact GM130, KDELR, and JUN expression levels also indicates that their modifications are a specific response to the Rab1bwt overexpression and not a general stress response induced by protein overexpression.
By cloning specific 5′-flanking sequences of the human GM130 and KDELR3 genes into the luciferase reporter pGL3 vector (Figure 4, A and B), we identified the promoter regions modulated by the Rab1b increase. Moreover, pGL-GM130 and pGL-KDELR luciferase activity increases in a time-dependent manner after tetracycline addition, indicating that such promoter activity was dependent on Rab1b levels (Figure 4, C and D). The fact that tetracycline addition in T-Rex Rab1bN121I cells (Figure 3B) induced expression of the Rab1b dominant-negative mutant without modifying GM130, KDELR or c-Jun expression strongly suggests that activation of their promoters requires normal Rab1b activity and is not a nonspecific response to protein overexpression or to tetracycline addition.
Although p38 MAPK, ERK 1/2, and JNK phosphorylation increase after Rab1b induction (Figure 5, C and D), the GM130, KDELR, and JUN promoter activity and protein expression modifications require p38 MAPK but not ERK activation. In addition, phosphorylation of JNK was required only for JUN expression (Figure 5, A and B). Time-course analysis of kinase activation (Figure 5E) indicates that p38 MAPK is activated first and its phosphorylation increases after 30 min of Rab1b induction. Our data suggest that changes in the levels of a Golgi-localized GTPase might elicit a MAPK signaling cascade that modulates the expression of a variety of genes. Association of Rabs in signaling to the nucleus was reported by the interaction of Rab5 with APPL1 and APPL2, proteins involved in modulating cell proliferation (Miaczynska et al., 2004). However, endosomes were the endomembrane platform for this role. Interestingly, it has been shown that p38 MAPK stimulates Rab5 activation and enhances endocytosis rate (Cavalli et al., 2001); however, our data indicate that an increase in Rab1b triggers p38 MAPK activation (Figure 4B).
To identify the 5′-flanking regions of the GM130 and KDELR3 genes involved in the Rab1b-induced stimulation, we studied several deletions of both promoters. The regions including CREB consensus binding site, were responsible for the Rab1b stimulatory effect. Consequently, we show that CREB phosphorylation increases after Rab1b induction with a similar kinetic to p38MAPK phosphorylation and that a p38MAPK inhibitor, but not the ERK or JNK inhibitors, was able to inhibit CREB phosphorylation (Figure 6, C and D). Moreover, expression of CREB in T-Rex Rab1b cells transfected with pGL3-GM130 or pGL3-KDELR induced analogous promoter regulation to the one observed by Rab1b induction. These results suggest that CREB is required for the Rab1b-dependent response. In agreement, the CREB consensus binding site appears to act effectively as a functional mediator of the Rab1b stimulation of GM130 and KDELR expression, as demonstrated here by site-directed mutagenesis (Figure 6A). Even though sequence analyses of a number of 5′-flanking regions of Rab1b-modulated genes indicate that the CREB consensus site was present in many of them, we cannot assume that the mechanism described above is controlling the expression of all the genes identified by our microarray analysis. Interestingly, the CREB-binding motif on the GM130 and KDELR promoters is similar to the CrebA binding site confirmed to be functional in a variety of secretory pathway genes in Drosophila (Fox et al., 2010). Because the CREB-like proteins Creb3L1 and Creb3L2 are the closest mammalian orthologues to Drosophila CrebA, we cannot rule out that other transcription factors, such as a CREB-like protein, could interact with the CREB consensus binding site on the GM130 or KDELR promoter regions.
How does the increase in Rab1b, a Golgi GTPase, elicit a MAPK signaling cascade that modulates the expression of a variety of genes? p38 MAPK is typically activated after cellular stress induced by a variety of extracellular stimuli and has an important role in a number of physiological processes as well. Furthermore, convincing evidence indicates that endomembranes, including the Golgi complex, can also be the platform to trigger a signaling cascade using classical signaling molecules (Mor and Philips, 2006; Farhan and Rabouille, 2011). It is clear that, through these signaling molecules, endomembranes can perform two functions: 1) elaborate and relay signaling initiated at the plasma membrane, and 2) initiate new signaling in response to stimuli originating from the endomembranes themselves. The increase of Rab1b is an endogenous event that could be considered “a cellular stress on the endomembrane system.” One of the central points of this study is to show that the increase in Rab1b levels induces the alteration of gene expression of a number of genes by regulating their promoter activity. We present here novel data showing that phosphorylation of the p38 MAPK and the CREB consensus binding site is required for this Rab1b activity. Although we believe that additional studies may be needed to further define the molecular mechanisms by which Rab1b activates p38 MAPK, we consider that it is important to first show that this novel Rab1b function is applicable to a physiological system in which Rab1b increase is inherent to the cell function.
Rab1b in thyroid cells
As has been previously reported, TSH addition induces NIS expression (Chun and Di Lauro, 2001). In this work, we found that TSH also induced Rab1b and GM130 expression (Figure 7). These findings indicate that the specific thyroid secretory stimulus can raise the expression of a cell type–specific protein (NIS) together with ubiquitous proteins required in the secretory pathway. Moreover, inhibition of Rab1b by siRNA transfection blocks the GM130 and NIS expression induced by TSH (Figure 8). This effect was not due to the classical Rab1b transport role, as blocking ER–Golgi transport with BFA, an inhibitor of the activity of the Rab1b-recruited GBF1 (GEF of ARF1) at the ER–Golgi interface, did not block NIS expression (Figure S2). These results suggest that, in thyroid cells, Rab1b expression and/or induction is required for the TSH-dependent induction of NIS. It is well understood that, in thyroid cells, binding of TSH to its receptor, TSHR, elicits the activation of the heterotrimeric G proteins, mainly Gαs and Gαq, which activate protein kinase A (PKA) and protein kinase C, respectively. Activated PKA phosphorylates a variety of proteins, changing their activities to promote growth and differentiation. The most classical target of PKA is the transcription factor CREB, which is required for the activation of the NUE region on the NIS gene promoter that includes, among other binding sites, a degenerate CRE-L (Chun et al., 2004). We cannot exclude the possibility that the CREB-binding site on the GM130 promoter is required by the TSHR/c-AMP pathway; however, the fact that Rab1b inhibition blocks the GM130 increase induced by TSH strongly suggests that Rab1b activity is required for the TSH-induced GM130 response. Interestingly, TSH also increases Rab5 and Rab7 but not Rab8 expression in a c-AMP–dependent manner (Croizet-Berger et al., 2002). Sequence analyses identified a potential CRE-L in the promoter sequences of RAB5A and RAB7 (Croizet-Berger et al., 2002), as well as in RAB1B. Further studies would be needed to further confirm the functional role of these sequences in thyroid cells.
We show here that FRTL5 cells transfected with GFP-Rab1bwt displayed higher NIS levels (Figure 9), while control cells transfected with GFP alone did not show the same effect (unpublished data). However, it is important to remark that TSH induced more NIS expression than Rab1b overexpression alone (Figure 9B, MIF: 387.51 and 104.94, respectively). This result indicates that Rab1b overexpression by itself does not bypass the need of TSH and is not capable of triggering an induction of NIS similar to TSH. Taken together, these results indicate that Rab1b is required but not sufficient to promote NIS expression. TSH addition increased NIS promoter activity 2.3-fold and 1.6-fold in GFP-Rab1bwt–overexpressing cells and in control cells expressing the GFP, respectively. These data indicate stimulation with TSH is enough to induce NIS promoter activity, and TSH induces higher NIS promoter activity in GFP-Rab1b–overexpressing cells than in control (GFP) cells. Therefore Rab1bwt overexpression can enhance the NIS promoter response to TSH. In FRTL5 cells, PKA is required to activate p38 that regulates the TSH-mediated NIS expression (Pomerance et al., 2000). PKA also phosphorylates CREB in a p38-independent manner. Speculating that Rab1b increase is cAMP/PKA-dependent, we propose that, in FRTL5, Rab1b is acting upstream of the p38 phosphorylation step and is required for activation of NIS expression. Our results show, for the first time, that changes in Rab1b levels modulate gene transcription and strongly suggest that a Rab1b increase is required to elicit a secretory response.
Interestingly, the intracellular signaling pathway activated by an increase in Rab1b can be compared with the unfolded protein response (UPR) pathway (activated by ER stress), as they both trigger an extensive transcriptional response. Activation of the UPR pathway selectively activates the transcription of genes encoding proteins (most of them chaperones) that cope with ER stress (Bernales et al., 2006). The Rab1b-induced response fundamentally impacts genes encoding proteins required for membrane transport or Golgi structure, and we postulate that this mechanism assists the cell to respond to a high demand for secretion. In addition, a recent report showed that the KDELR (Pulvirenti et al., 2008) acts as a signaling receptor that senses an increase in protein cargo in the ER and activates a phosphorylation cascade that promotes intra-Golgi trafficking.
This is the first report showing that variations in Rab1b levels participate in a signal transduction pathway modulating gene expression, and that p38 MAPK, as well as the CREB consensus DNA-binding site are required for this function. Taken together, our data strongly suggest that a secretory stimulus induces an increase in Rab1b levels that then triggers signaling circuits essential for the synthesis of molecules necessary to coordinate the flux of membrane transport along the secretory pathway. Future studies will focus on characterizing the transducers of the Rab1b-induced response to further elucidate the mechanisms involved in the regulation of transport and cellular adaptation after a secretory stimulus.
MATERIALS AND METHODS
DNA constructs and antibodies
Full-length Rab1b and mutated Rab1b sequences cloned into the pEF6/Myc-His B vector (Invitrogen, Carlsbad, CA) and GFP-Rab1 versions (cloned in pEGFP) have been previously described (Alvarez et al., 2003). For generation of stable cell lines, Rab1 constructs were subcloned from the pEF6/Myc-His B vector into pcDNA4/TO/myc-His B (Invitrogen). GFP-Rab7Q67L was provided by Marisa Colombo (Universidad de Cuyo, Argentina). For cloning KDELR3 or GM130 5’ promoter regions, the human GM130 (NM_004486) and KDELR3 (NM_006855) 5’-flanking regulatory regions corresponding to nucleotides −454 to −80 and −291 to +136 (relative to the transcription start site), respectively, were PCR-amplified from human whole-blood genomic DNA using the following specific primers: GM130: forward 5′-AGGAGTCAGGAAAGAAACTGTGGAG-3′, and reverse 5′- GTAACCAGGGCGATACTGGAAAGCTTGCG-3′; KDELR (isoform3): forward 5′- TACAGATGAGGAAACTGAGGCAGAG-3′, and reverse 5′- GTCCAGCCAGTCAGTCGTGAAGCTTCGC-3′. PCRs were performed in a Bio-Rad thermocycler (Hercules, CA) using Pfu polymerase (Invitrogen), with an initial denaturation cycle of 3 min at 95°C, followed by 30 cycles of 30 s at 95°C, 30 s at 59°C, and 45 s at 68°C, with a final extension cycle of 5 min at 68°C. The amplified products were cloned into the pCR 2.1 Topo vector (Invitrogen), digested with XhoI and HindIII, and then cloned into the pGL3-basic vector (Promega, Madison, WI). All the constructs were sequenced and verified. Fragments encompassing different lengths of the 5′-flanking region of the GM130 gene were obtained by PCR using as a template the pCR 2.1 Topo-GM130 to obtain the versions pGL3-GM130-1 and pGL3-GM130-2. The forward primer for pGL3-GM130-1 was 5′-CCTGGGGTCCGAGGCCC-3′ and for pGL3-GM130-2 was 5′-GCGACCTCTCAGGTGCACCG-3′. The reverse primer for both constructs was the same as the one described above for pGL3-GM130. PCR products were cloned into pCR 2.1 Topo and then subcloned into pGL3-Basic vector using XhoI and HindIII. The construct pGL3-GM130-3 was obtained from the GM130 cloned in pCR 2.1 Topo by performing enzyme restriction treatments to subclone it into the pGL3-Basic vector. Furthermore, fragments of different lengths encompassing the 5′-flanking region of the KDELR3 gene were obtained by PCR, using as a template the pCR 2.1 Topo-KDELR to obtain the versions pGL3-KDELR-1 and pGL3-KDELR-2, or by enzyme restriction treatment to obtain pGL3-KDELR-3. The forward primer for pGL3-KDELR-1 was 5′-GGCAGGCCTCCAGTTTCTGCGG-3′, and for pGL3-KDELR-2 was 5′-CGCCCAGTCCGGGAGCCG-3′. The reverse primer for both constructs was the same one as that described above for KDELR (isoform 3). PCR products for pGL3-KDELR-1 and pGL3- KDELR -2 were cloned into pCR 2.1 Topo and then subcloned into pGL3-Basic vector using XhoI and HindIII. The construct pGL3- KDELR-3 was obtained from the KDELR cloned in the Topo vector by performing the following steps: first, digestion with NotI, then ligation, and finally digestion with HindIII and XhoI was performed to subclone it into the pGL3-Basic vector. pGL3-GM130-1M and pGL3-KDELR-1M are mutant variants of their respective versions, in which the CREB-binding consensus site (TGACGT) was mutated for (AGATCT). Mutations were performed with the Quick Change Site-Directed Mutagenesis Kit according to manufacturer's protocol (Stratagene, La Jolla, CA), using pGL3-GM130-1 and pGL3-KDELR-1 as templates. The human c-JUN 5’-flanking regulatory region was previously reported (Wei et al., 1998).
The following antibodies were used: anti-KDELR from Calbiochem (San Diego, CA), anti-Rab1b and anti–c-JUN antibodies from Santa Cruz Biotechnologies (Santa Cruz, CA). Anti-Myc antibody from Invitrogen (San Diego, CA). Anti-GM130 antibody from BD Transduction Laboratories (San Jose, CA). Anti–phospho-p38 MAPK; anti–phospho-SAPK/JNK; anti–phospho-p44/42 MAPK (P-ERK1/2); anti–phospho-CREB; anti-p38 MAPK; anti-SAPK/JNK; anti-p44/42 MAPK (ERK1/2) and anti-CREB antibodies were from Cell Signaling (Danvers, MA). Anti-calreticulin antibody was from Affinity BioReagents (Golden, CO). Secondary antibodies conjugated to horseradish peroxidase, mouse, and rabbit were obtained from Zymed (San Diego, CA). Anti-rabbit Alexa Fluor 594 or 647 and anti-mouse Alexa Fluor 488 or 647 antibodies were from Invitrogen. Anti-human HLA (MHCI) and GalNAc-T2 were from Sigma-Aldrich (St. Louis, MO).
RNA isolation and microarray analysis
Cells expressing different GFP constructs were obtained using flow cytometry and cell sorting. The total RNA was extracted from 1 × 106 GFP-sorted cells, and the quality was evaluated using a previously established sample-processing method (Dumur et al., 2004). For preparation of RNA, the TRIZOL reagent was used (Invitrogen Life Technologies) following the manufacturer's protocol. RNA purity was judged by spectrophotometry at 260, 270, and 280 nm. RNA integrity, as well as cDNA and cRNA synthesis products, were assessed by running 1 μl of every sample in RNA 6000 Nano LabChips on a 2100 Bioanalyzer (Agilent Technologies, Foster City, CA).
The RNA samples were analyzed on HG-U133A 2.0 arrays following the standardized Affymetrix protocol, as described elsewhere (Dumur et al., 2004). Briefly, starting with 1 μg total RNA from every sample, we generated double-stranded cDNA using a 24-mer oligodeoxythymidylic acid primer with a T7 RNA polymerase promoter site added to the 3′ end (Superscript cDNA Synthesis System; Life Technologies, Rockville, MD). After second-strand synthesis, in vitro transcription was performed using the Enzo BioArray HighYield RNA Transcript labeling kit (Enzo Diagnostics, Farmingdale, NY) to produce biotin-labeled cRNA. cDNA and cRNA synthesis products were prepared and rigorously evaluated for quality to ensure the generation of good microarray data, using a sample processing method previously established in our laboratory. Fifteen micrograms of the cRNA product was fragmented, and 10 μg of this was hybridized for 18–20 h into HG-133A 2.0 microarrays containing 22,277 probe sets. Every chip was scanned at a high resolution, with pixelations ranging from 2.5 μm down to 0.51 μm, using the Affymetrix GeneChip Scanner 3000 according to the GeneChip Expression Analysis Technical Manual procedures (Affymetrix, Santa Clara, CA). After scanning, the raw intensities for each probe were stored in electronic files (in .DAT and .CEL formats) by the GeneChip Operating Software (GCOS version 1.4; Affymetrix). The overall quality of each array was assessed by monitoring the 3′/5′ ratios for two housekeeping genes (GAPDH and β-actin), and the percentage of “present” genes (%P) was calculated. Furthermore, box-and-whisker plots were used to assess and compare the intensity distribution across all the arrays included in this study, using the boxplot function from the Bioconductor affy package run on R 1.9.1.
Statistical analysis
Background correction, normalization, and estimation of probe set expression summaries was performed using the log-scale robust multiarray analysis method. Identification of altered gene expression among each GFP construct–transfected cell was assessed by using the significance score (S-score) method The S-score method uses an error-based model to determine the variances for probe pair signals and follows a normal standard distribution. The procedure produces scores centered around 0 (no change) with an SD of 1. Thus scores >2 or <−2 from a single comparison have, on average, a 95% chance of being significant hybridization changes, at a univariate level, corresponding to a p value of <0.05.
Interaction networks and functional analysis
Gene ontology and gene interaction analyses were performed using the Ingenuity Pathways Analysis tools 3.0 (www.ingenuity.com). The gene lists containing probe set IDs as gene identifiers, as well as fold-change values from corresponding supervised analyses, were mapped onto their corresponding gene object in the Ingenuity Pathways Knowledge Base (IPKB). These so-called focus genes were then used in the network-generation algorithm, based on the list of molecular interactions in IPKB. Significance for the enrichment of the genes in a network with particular biological functions was determined by the right-tailed Fisher's exact test, using a list of all the genes in the array as a reference set.
Biological networks were ranked by score, with the score corresponding to the likelihood of a set of genes being found in the networks due to random chance; that is, a score of 3 indicated that there was a 1/1000 chance that the focus genes were in a network due to random chance. Therefore we selected genes involved in networks with scores of 3 or higher to be at least 99.9% confident that they had not been generated by random chance alone.
RT-qPCR
A real-time quantitative reverse transcriptase PCR (RT-qPCR) was used to assess gene expression levels of selected genes using TaqMan chemistry. Probes and primer sets for detection of KDELR3, ARF4, RABAC1 (PRA1), ARL1, YIF1, GOLGA2 (GM130), GPR126, EGR1, IL-8, and c-JUN transcripts were obtained from inventoried assays (Applied Biosystems, Foster City, CA). Thus gene-specific probes labeled in the 5′ end with 6-carboxyfluorescein and in the 3′ end with a dark quencher were used for all the target genes of interest. For all samples, cyclophilin A from the Predeveloped TaqMan Assay Reagents (Applied Biosystems) was used as the endogenous control gene. The experiments were performed on the ABI Prism 7500 Sequence Detection System, using the TaqMan One-Step PCR Master Mix Reagents Kit (Applied Biosystems). All the samples were tested in triplicate. The cycling conditions were 48ºC for 30 min; 95ºC for 10 min; 40 cycles at 95ºC for 15 s; and 60ºC for 1 min.
Statistical analysis for RT-qPCR
The 2−ΔΔCt method was used to calculate the fold changes in the expression levels of the genes of interest. Pearson's r was used to examine the relationship between the microarray and RT-qPCR results, and correlations were considered to be statistically significant if the p value was <0.05.
Generation of T-Rex Rab1b cells
The above-mentioned, pcDNA4/TO/myc-His B plasmid containing Rab1b or Rab1N121I was transfected into T-Rex HeLa cells (T-Rex System, Invitrogen) for stable integration and subsequent inducible expression of Rab1b constructs. Cells were grown in Advanced DMEM (Life Technologies/Invitrogen) supplemented with 2% fetal bovine serum, and penicillin/streptomycin. Transfection was performed using Transit LTI (Mirus, Madison, WI) according to the manufacturer's instructions. After 48 h of transfection, cells were selected by using a culture medium containing 50 μg/ml Zeocin and 2.5 μg/ml blasticidin for 30 d. Zeocin-resistant clones were isolated, grown in 24-multiwell plates, and purified by plating at a low density in 10-cm plates in order to generate new clones (at the same antibiotic concentration). Clones were then expanded by continuous selection using 25 μg/ml Zeocin and 1.25 μg/ml blasticidin. For myc construct induction, tetracycline (10 μg/ml final concentration) was added to the medium.
For preparation of cell extracts, cells were collected with trypsin and rinsed with medium and then phosphate-buffered saline. Extracts were pelleted and resuspended in RIPA buffer containing protease inhibitors for 30 min on ice, before being pelleted again at 13,000 × g for 15 min. Supernatants were separated by SDS–PAGE and analyzed by Western blot. The blots were scanned, and the density of the bands was quantified using ImageJ software (www.rsb.info.nih.gov/ij).
BFA-WO
HeLa cells were transfected with GFP-Rab1bwt (or GFP alone or CFP-ManII as controls) and after 42 h of expression, were treated with BFA for 30 min. BFA-WO was carried out by removing, rinsing, and incubating cells with fresh medium for 30, 60, 90, and 120 min at 37ºC. Immunofluorescence assays were done at each time point by labeling cells with anti-GFP and anti–GalNAc-T2 antibodies. Results were analyzed in a fluorescence microscope, and patterns of GalNAc-T2 after BFA-WO were classified into four groups: ER (no WO effect), scattered punctated (SP, early WO effect), compact punctated (CP, late WO effect), and Golgi (complete WO effect; Figure S1A). The number of cells (transfected and nontransfected) showing each pattern was counted at each time point of BFA-WO. All patterns were observed at each time point, but the amount of cells exhibiting each pattern was different according the effect of BFA-WO. For comparing the consequence of Rab1b transfection on BFA-WO dynamics, 60 min of BFA-WO was chosen as the representative time point, because it is the one at which the higher diversity of GalNAc-T2 patterns was found.
Transient transfection of cells and reporter gene assays
Each reporter construct (firefly luciferase, 100 ng) in combination with a control vector (R. reiniformis phRL-TK, 10 ng) was transfected in the T-Rex Rab1bwt stable cells plated in a 96-well plate (18,000 cells/well), using 0.22 μl of Lipofectamine 2000 (Invitrogen). Twelve hours after transfection, Rab1bwt expression was induced by adding tetracycline (10 μg/ml) for 48 h. The luciferase activity was normalized to R. reiniformis and expressed as the fold increase with respect to the values obtained with the pGL3-basic empty vector, which was arbitrarily set to 1. In addition, the ratio (fold change) of relative luciferase activity between samples with or without tetracycline was calculated.
For analyzing the effect of the MAPK inhibitors, T-Rex cells were grown at 2% serum during 24 h. This serum was gradually reduced in concentration to reach 0% in ∼48–72 h. These cells were plated (without serum) and 24 h later, different reporter constructs were transfected. Ten hours later, cells were incubated with MAPK-specific inhibitors for 2 h before Rab1b expression was induced by adding tetracycline. The luciferase activity was measured after further 24 h. Inhibitors of p38 (SB 203580) and MEK1-2 (UO126) were used at 25 μM; JNK inhibitor (SP600125) was used at 50 μM. Inhibitors were resuspended in dimethyl sulfoxide (DMSO) and control samples (with or without tetracycline) contained a similar volume of DMSO.
FRTL-5 cell induction, transfection, and immunofluorescence assays
FRTL-5 cells (ATCC CRL 8305; Van Heuverswyn et al., 1984) were grown in DMEM/F12 medium (Life Technologies, Grand Island, NY) supplemented with 5% calf serum (Life Technologies), 1 mU/ml bovine TSH, 10 μg/ml bovine insulin, 5 mg/ml bovine transferrine (Sigma-Aldrich), and penicillin/streptomycin. For testing the effect of TSH stimulation, cells were grown at 70% of confluence and shifted to medium without TSH but containing 0.2% of calf serum (basal medium), and maintained for 36–48 h. Then cells were stimulated by the addition of TSH (0.5 mU/ml) and grown for 24–48 h. Control cells were kept in the presence of basal medium for 96 h.
When FRTL5 cells were transfected with the GFP-Rab1 constructs, this was performed 12 h before TSH deprivation (with basal medium). Cells were incubated in basal medium for 36 h, and then TSH (0.5 mU/ml) was added for 24 h. For immunofluorescence, cells were grown on glass coverslips, and fixation and staining of cells were performed as previously described (Alvarez et al., 2003). Immunofluorescence was analyzed using a Nikon Eclipse TE 2000-U (New York, NY) microscope.
Cell cytometry
NIS, Rab1b, and GM130 were measured by using the BD Cytofix/Cytoperm Plus Kit, following the manufacturer's protocol. Cells were incubated with the primary antibody (described above) and then with anti-mouse (for GM130) or anti-rabbit antibody (for NIS and Rab1b) labeled with Alexa Fluor 647. Cells were acquired in a FACSCanto II flow cytometer and analyzed using FlowJo (Tree Star, Ashland, OR) software. In all cases, control was performed by incubating cells with only the secondary antibody.
Supplementary Material
Acknowledgments
We thank J. L. Bocco and members of his lab and V. Andreoli and A. Saka (Universidad Nacional de Córdoba, Córdoba, Argentina) for c-JUN and CREB constructs and helpful discussions. We also thank R. Gehrau and D. D'Astolfo (Universidad Nacional de Córdoba, Córdoba, Argentina) for promoter activity assays assistance, A. Gamarnik (Instituto Leloir, Buenos Aires, Argentina) for letting us use equipment, and A. Massini and her lab (Universidad Nacional de Córdoba, Córdoba, Argentina) for providing us with FRTL5 cells and reagents and advice for their manipulation. Finally, we thank G. Panzetta-Dutari (Universidad Nacional de Córdoba, Córdoba, Argentina) and E. Sztul (University of Alabama at Birmingham, Birmingham, AL) for their important comments after reading the manuscript. This work was supported by FONCYT (Bid 1728, PICT-10867 and PICT 925), SeCYT (UNC), and CONICET (fellowships to P.M., N.R., and I.S.). M.A.D.M. acknowledges the support of Telethon and AIRC.
Abbreviations used:
- BFA-WO
brefeldin A wash-out
- CLEM
correlative light electron microscopy
- COP
coatomer protein
- CREB
cAMP-responsive element binding protein
- CRE-L
cAMP-response element
- DMSO
dimethyl sulfoxide
- EGR1
early growth response 1
- ER
endoplasmic reticulum
- ERK
extracellular signal–regulated protein kinase
- GFP
green fluorescent protein
- GM130
Golgi matrix protein of 130 kDa
- GPR126
G protein–coupled receptor 126
- IL8
interleukin 8
- IPKB
Ingenuity Pathways Knowledge Base
- JNK
c-Jun NH2-terminal kinase
- KDELR
KDEL-receptor
- MAPK
mitogen-activated protein kinase
- NIS
sodium iodide symporter
- NUE
NIS upstream enhancer
- PKA
protein kinase A
- RNAi
RNA interference
- RT-qPCR
quantitative reverse-transcriptase PCR
- siRNA
small interfering RNA
- S-score
significance score
- TG
thyroglobulin
- TSH
thyroid-stimulating hormone
- TSHR
TSH to its receptor
- UPR
unfolded protein response
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-07-0530) on January 16, 2013.
*These authors contributed equally to this work.
REFERENCES
- Allan BB, Moyer BD, Balch WE. Rab1 recruitment of p115 into a cis-SNARE complex: programming budding COPII vesicles for fusion. Science. 2000;289:444–448. doi: 10.1126/science.289.5478.444. [DOI] [PubMed] [Google Scholar]
- Alvarez C, Garcia-Mata R, Brandon E, Sztul E. COPI recruitment is modulated by a Rab1b-dependent mechanism. Mol Biol Cell. 2003;14:2116–2127. doi: 10.1091/mbc.E02-09-0625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr FA, Puype M, Vandekerckhove J, Warren G. GRASP65, a protein involved in the stacking of Golgi cisternae. Cell. 1997;91:253–262. doi: 10.1016/s0092-8674(00)80407-9. [DOI] [PubMed] [Google Scholar]
- Bernales S, Papa FR, Walter P. Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol. 2006;22:487–508. doi: 10.1146/annurev.cellbio.21.122303.120200. [DOI] [PubMed] [Google Scholar]
- Bucci C, Chiariello M. Signal transduction gRABs attention. Cell Signal. 2006;18:1–8. doi: 10.1016/j.cellsig.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Bui M, Gilady SY, Fitzsimmons RE, Benson MD, Lynes EM, Gesson K, Alto NM, Strack S, Scott JD, Simmen T. Rab32 modulates apoptosis onset and mitochondria-associated membrane (MAM) properties. J Biol Chem. 2011;285:31590–31602. doi: 10.1074/jbc.M110.101584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne RD, Morgan A. Membrane trafficking: three steps to fusion. Curr Biol. 2007;17:R255–R258. doi: 10.1016/j.cub.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Cavalli V, Vilbois F, Corti M, Marcote MJ, Tamura K, Karin M, Arkinstall S, Gruenberg J. The stress-induced MAP kinase p38 regulates endocytic trafficking via the GDI:Rab5 complex. Mol Cell. 2001;7:421–432. doi: 10.1016/s1097-2765(01)00189-7. [DOI] [PubMed] [Google Scholar]
- Cavenagh MM, Whitney JA, Carroll K, Zhang C, Boman AL, Rosenwald AG, Mellman I, Kahn RA. Intracellular distribution of Arf proteins in mammalian cells. Arf6 is uniquely localized to the plasma membrane. J Biol Chem. 1996;271:21767–21774. doi: 10.1074/jbc.271.36.21767. [DOI] [PubMed] [Google Scholar]
- Cheng KW, et al. The RAB25 small GTPase determines aggressiveness of ovarian and breast cancers. Nat Med. 2004;10:1251–1256. doi: 10.1038/nm1125. [DOI] [PubMed] [Google Scholar]
- Chun JT, Di Dato V, D'Andrea B, Zannini M, Di Lauro R. The CRE-like element inside the 5′-upstream region of the rat sodium/iodide symporter gene interacts with diverse classes of b-Zip molecules that regulate transcriptional activities through strong synergy with Pax-8. Mol Endocrinol. 2004;18:2817–2829. doi: 10.1210/me.2004-0020. [DOI] [PubMed] [Google Scholar]
- Chun JT, Di Lauro R. Characterization of the upstream enhancer of the rat sodium/iodide symporter gene. Exp Clin Endocrinol Diabetes. 2001;109:23–26. doi: 10.1055/s-2001-11021. [DOI] [PubMed] [Google Scholar]
- Cohen-Solal KA, Sood R, Marin Y, Crespo-Carbone SM, Sinsimer D, Martino JJ, Robbins C, Makalowska I, Trent J, Chen S. Identification and characterization of mouse Rab32 by mRNA and protein expression analysis. Biochim Biophys Acta. 2003;1651:68–75. doi: 10.1016/s1570-9639(03)00236-x. [DOI] [PubMed] [Google Scholar]
- Croizet-Berger K, Daumerie C, Couvreur M, Courtoy PJ, van den Hove MF. The endocytic catalysts, Rab5a and Rab7, are tandem regulators of thyroid hormone production. Proc Natl Acad Sci USA. 2002;99:8277–8282. doi: 10.1073/pnas.122187699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumur CI, Nasim S, Best AM, Archer KJ, Ladd AC, Mas VR, Wilkinson DS, Garrett CT, Ferreira-Gonzalez A. Evaluation of quality-control criteria for microarray gene expression analysis. Clin Chem. 2004;50:1994–2002. doi: 10.1373/clinchem.2004.033225. [DOI] [PubMed] [Google Scholar]
- Farhan H, Rabouille C. Signalling to and from the secretory pathway. J Cell Sci. 2011;124:171–180. doi: 10.1242/jcs.076455. [DOI] [PubMed] [Google Scholar]
- Feinstein TN, Linstedt AD. GRASP55 regulates Golgi ribbon formation. Mol Biol Cell. 2008;19:2696–2707. doi: 10.1091/mbc.E07-11-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox RM, Hanlon CD, Andrew DJ. The CrebA/Creb3-like transcription factors are major and direct regulators of secretory capacity. J Cell Biol. 2010;191:479–492. doi: 10.1083/jcb.201004062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger C, et al. Cytohesin-1 regulates beta-2 integrin-mediated adhesion through both ARF-GEF function and interaction with LFA-1. EMBO J. 2000;19:2525–2536. doi: 10.1093/emboj/19.11.2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurkan C, Lapp H, Alory C, Su AI, Hogenesch JB, Balch WE. Large-scale profiling of Rab GTPase trafficking networks: the membrome. Mol Biol Cell. 2005;16:3847–3864. doi: 10.1091/mbc.E05-01-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas AK, Yoshimura S, Stephens DJ, Preisinger C, Fuchs E, Barr FA. Analysis of GTPase-activating proteins: Rab1 and Rab43 are key Rabs required to maintain a functional Golgi complex in human cells. J Cell Sci. 2007;120:2997–3010. doi: 10.1242/jcs.014225. [DOI] [PubMed] [Google Scholar]
- Hay JC, Chao DS, Kuo CS, Scheller RH. Protein interactions regulating vesicle transport between the endoplasmic reticulum and Golgi apparatus in mammalian cells. Cell. 1997;89:149–158. doi: 10.1016/s0092-8674(00)80191-9. [DOI] [PubMed] [Google Scholar]
- Hay JC, Klumperman J, Oorschot V, Steegmaier M, Kuo CS, Scheller RH. Localization, dynamics, protein interactions reveal distinct roles for ER and Golgi SNAREs. J Cell Biol. 1998;141:1489–1502. doi: 10.1083/jcb.141.7.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogai T, Endo T, Saito T, Miyazaki A, Kawaguchi A, Onaya T. Regulation by thyroid-stimulating hormone of sodium/iodide symporter gene expression and protein levels in FRTL-5 cells. Endocrinology. 1997;138:2227–2232. doi: 10.1210/endo.138.6.5189. [DOI] [PubMed] [Google Scholar]
- Liu YJ, Wang Q, Li W, Huang XH, Zhen MC, Huang SH, Chen LZ, Xue L, Zhang HW. Rab23 is a potential biological target for treating hepatocellular carcinoma. World J Gastroenterol. 2007;13:1010–1017. doi: 10.3748/wjg.v13.i7.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bergami P, et al. Rewired ERK-JNK signaling pathways in melanoma. Cancer Cell. 2007;11:447–460. doi: 10.1016/j.ccr.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire J, Santoro T, Jensen P, Siebenlist U, Yewdell J, Kelly K. Gem: an induced, immediate early protein belonging to the Ras family. Science. 1994;265:241–244. doi: 10.1126/science.7912851. [DOI] [PubMed] [Google Scholar]
- Martin-Martin B, Nabokina SM, Lazo PA, Mollinedo F. Co-expression of several human syntaxin genes in neutrophils and differentiating HL-60 cells: variant isoforms and detection of syntaxin 1. J Leukoc Biol. 1999;65:397–406. doi: 10.1002/jlb.65.3.397. [DOI] [PubMed] [Google Scholar]
- Martincic I, Peralta ME, Ngsee JK. Isolation and characterization of a dual prenylated Rab and VAMP2 receptor. J Biol Chem. 1997;272:26991–26998. doi: 10.1074/jbc.272.43.26991. [DOI] [PubMed] [Google Scholar]
- Matern H, Yang X, Andrulis E, Sternglanz R, Trepte HH, Gallwitz D. A novel Golgi membrane protein is part of a GTPase-binding protein complex involved in vesicle targeting. EMBO J. 2000;19:4485–4492. doi: 10.1093/emboj/19.17.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miaczynska M, Christoforidis S, Giner A, Shevchenko A, Uttenweiler-Joseph S, Habermann B, Wilm M, Parton RG, Zerial M. APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment. Cell. 2004;116:445–456. doi: 10.1016/s0092-8674(04)00117-5. [DOI] [PubMed] [Google Scholar]
- Monetta P, Slavin I, Romero N, Alvarez C. Rab1b interacts with GBF1 and modulates both ARF1 dynamics and COPI association. Mol Biol Cell. 2007;18:2400–2410. doi: 10.1091/mbc.E06-11-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mor A, Philips MR. Compartmentalized Ras/MAPK signaling. Annu Rev Immunol. 2006;24:771–800. doi: 10.1146/annurev.immunol.24.021605.090723. [DOI] [PubMed] [Google Scholar]
- Mori Y, et al. Identification of genes uniquely involved in frequent microsatellite instability colon carcinogenesis by expression profiling combined with epigenetic scanning. Cancer Res. 2004;64:2434–2438. doi: 10.1158/0008-5472.can-03-3508. [DOI] [PubMed] [Google Scholar]
- Moyer BD, Allan BB, Balch WE. Rab1 interaction with a GM130 effector complex regulates COPII vesicle cis-Golgi tethering. Traffic. 2001;2:268–276. doi: 10.1034/j.1600-0854.2001.1o007.x. [DOI] [PubMed] [Google Scholar]
- Munro S. The Arf-like GTPase Arl1 and its role in membrane traffic. Biochem Soc Trans. 2005;33:601–605. doi: 10.1042/BST0330601. [DOI] [PubMed] [Google Scholar]
- Nicola JP, Nazar M, Mascanfroni ID, Pellizas CG, Masini-Repiso AM. NF-κB p65 subunit mediates lipopolysaccharide-induced Na+/I− symporter gene expression by involving functional interaction with the paired domain transcription factor Pax8. Mol Endocrinol. 2010;24:1846–1862. doi: 10.1210/me.2010-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelham HR. Recycling of proteins between the endoplasmic reticulum and Golgi complex. Curr Opin Cell Biol. 1991;3:585–591. doi: 10.1016/0955-0674(91)90027-v. [DOI] [PubMed] [Google Scholar]
- Pomerance M, Abdullah H, Kamerji S, Corrèze C, Blondeau J. Thyroid-stimulating hormone and cyclic AMP activate p38 mitogen-activated protein kinase cascade. J Biol Chem. 2000;275:40539–40546. doi: 10.1074/jbc.M002097200. [DOI] [PubMed] [Google Scholar]
- Pulvirenti T, et al. A traffic-activated Golgi-based signalling circuit coordinates the secretory pathway. Nat Cell Biol. 2008;10:912–922. doi: 10.1038/ncb1751. [DOI] [PubMed] [Google Scholar]
- Puthenveedu MA, Bachert C, Puri S, Lanni F, Linstedt AD. GM130 and GRASP65-dependent lateral cisternal fusion allows uniform Golgi-enzyme distribution. Nat Cell Biol. 2006;8:238–248. doi: 10.1038/ncb1366. [DOI] [PubMed] [Google Scholar]
- Segev N. GTPases in intracellular trafficking: an overview. Semin Cell Dev Biol. 2010;22:1–2. doi: 10.1016/j.semcdb.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta D, Linstedt AD. Control of organelle size: the Golgi complex. Annu Rev Cell Dev Biol. 2011;27:57–77. doi: 10.1146/annurev-cellbio-100109-104003. [DOI] [PubMed] [Google Scholar]
- Shibata D, et al. RAB32 hypermethylation and microsatellite instability in gastric and endometrial adenocarcinomas. Int J Cancer. 2006;119:801–806. doi: 10.1002/ijc.21912. [DOI] [PubMed] [Google Scholar]
- Shimada K, Uzawa K, Kato M, Endo Y, Shiiba M, Bukawa H, Yokoe H, Seki N, Tanzawa H. Aberrant expression of RAB1A in human tongue cancer. Br J Cancer. 2005;92:1915–1921. doi: 10.1038/sj.bjc.6602594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J, Watson R, Giannakou ME, Clarke M, Warren G, Barr FA. GRASP55, a second mammalian GRASP protein involved in the stacking of Golgi cisternae in a cell-free system. EMBO J. 1999;18:4949–4960. doi: 10.1093/emboj/18.18.4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- Van Heuverswyn B, Streydio C, Brocas H, Refetoff S, Dumont J, Vassart G. Thyrotropin controls transcription of the thyroglobulin gene. Proc Natl Acad Sci USA. 1984;81:5941–5945. doi: 10.1073/pnas.81.19.5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinke FP, Grieve AG, Rabouille C. The multiple facets of the Golgi reassembly stacking proteins. Biochem J. 2011;433:423–433. doi: 10.1042/BJ20101540. [DOI] [PubMed] [Google Scholar]
- Wagner AC, Strowski MZ, Goke B, Williams JA. Molecular cloning of a new member of the Rab protein family, Rab 26, from rat pancreas. Biochem Biophys Res Commun. 1995;207:950–956. doi: 10.1006/bbrc.1995.1277. [DOI] [PubMed] [Google Scholar]
- Wei P, Inamdar N, Vedeckis WV. Transrepression of c-jun gene expression by the glucocorticoid receptor requires both AP-1 sites in the c-jun promoter. Mol Endocrinol. 1998;12:1322–1333. doi: 10.1210/mend.12.9.0158. [DOI] [PubMed] [Google Scholar]
- Weide T, Bayer M, Koster M, Siebrasse JP, Peters R, Barnekow A. The Golgi matrix protein GM130: a specific interacting partner of the small GTPase rab1b. EMBO Rep. 2001;2:336–341. doi: 10.1093/embo-reports/kve065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, et al. YIPF5 and YIF1A recycle between the ER and the Golgi apparatus and are involved in the maintenance of the Golgi structure. Exp Cell Res. 2008;314:3427–3443. doi: 10.1016/j.yexcr.2008.07.023. [DOI] [PubMed] [Google Scholar]
- Zhang T, Wong SH, Tang BL, Xu Y, Peter F, Subramaniam VN, Hong W. The mammalian protein (rbet1) homologous to yeast Bet1p is primarily associated with the pre-Golgi intermediate compartment and is involved in vesicular transport from the endoplasmic reticulum to the Golgi apparatus. J Cell Biol. 1997;139:1157–1168. doi: 10.1083/jcb.139.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.