

Abstract
In the last years post-transcriptional regulation (PTR) of gene expression has been increasingly recognized to be a powerful and general determinant of the quantitative changes in proteomes, and therefore a driving force for cell phenotypes. By means of networks of trans-factors on one hand, and cis-elements found primarily in untranslated regions (UTRs) of mRNA on the other hand, mRNA availability to translation and translation rates are tightly controlled and can be rapidly tuned according to the changing state of the cell. A number of dedicated resources and tools, including databases and predictive algorithms, have been proposed as bioinformatics aids for the study of this fundamental layer of gene expression regulation. Their use, however, is rendered difficult by heterogeneity and fragmentation.
This review aims to locate these resources in their proper space, classifying them according to their goals, limitations and integration capabilities and, in the end, to provide the user with an initial toolbox for the bioinformatic analysis of post-transcriptional regulation of gene expression. The accompanying website, available at www.ptrguide.org, lists all resources, provides summary and features for each one and will be regularly updated in the future.
Keywords: post-transcriptional regulation, translation, UTR, database, tool, RBP, ncRNA, miRNA, cis-element, trans-factor
Introduction
Post-transcriptional regulation (PTR) of gene expression is the process responsible for modulating mRNA levels and the related amount of protein. Initially thought to have a limited impact on cell phenotype, it has become increasingly recognized as a powerful and general determinant of the quantitative changes in proteomes.1 Untranslated regions of mRNAs (UTRs) are the fundamental mediators of this process, because they bear sequence and structure patterns preferentially bound by regulators which influence nuclear export, localization, stability of mRNAs and ultimately their translation rates,2 as well as capping, alternative splicing and polyadenylation of the transcribed pre-mRNA. One of the most important classes of post-transcriptional regulatory factors are the RNA-binding proteins (RBPs), whose human genome complement is at least 800 genes3, 4, 5 and which are characterized by the presence of different functional domains6 among which the most represented are, according to the latest release of Ensembl (Ensembl 68), the zinc-finger C2H2 domain (787 genes), the RNA-recognition motif (RRM, 233 genes), the sterile α motif (SAM, 93 genes) and the K-homology domain (KH, 38 genes). RBPs bind to the 5′UTR of a transcript often to modulate translation initiation, and to its 3′UTR usually to influence its stability or translatability;3 but they have also being well characterized for modulating splicing of the pre-mRNA, mRNA export and mRNA localization in the cytoplasm.7 Another major group of actors in PTR are non-coding RNAs (ncRNAs). Among them are various classes of long ncRNAs (lncRNAs), the intensively studied micro-RNAs (miRNAs), and then siRNAs (small-interfering RNAs), piRNAs (piwi-interacting RNAs), snoRNAs (small nucleolar RNAs), snRNAs (small nuclear RNAs), and several other types.8 miRNAs (around 1,500 are currently annotated in the human genome) bind to the 3′UTR of a transcript by means of short regions of perfect sequence complementation (which leads to increased transcript degradation) or with some mismatches (which promotes translational repression and increased degradation).9 Both RBPs and ncRNAs bind mRNAs in the so-called cis-elements, found primarily in 5′ and 3′ UTRs. These elements can be represented as recurring RNA sequences or secondary structures shared by a number of transcripts and defined by a pattern, to which the trans factors bind to exert a control over the mRNA. A well-known example of cis-regulatory elements are the AU-Rich Elements (ARE),10 motifs rich in Us with some interspersed As or Gs shared by several thousand 3′UTRs and bound by a large number of RBPs of which at least 23 are known.10 Another well characterized class of UTR cis-elements are the Iron Response Elements (IREs), which help in coordinating cellular iron homeostasis at the translational level.11
The last years have seen a rapid increase in resources dedicated to the analysis of these factors and elements to unravel associated mechanisms of gene expression regulation. Available databases are focused mainly on UTRs annotation,12,13 RBP-target interactions,14,15 ncRNAs,16,17,18,19,20,21,22,23,24 of which miRNA-target interactions are the greater part,16,17,18,19,20 with a limited number of resources focusing on lncRNAs,22,23 and cis-elements.25,26,27,28,29,30 Furthermore, a small number of resources integrating different data types is available.12,13,31 Predictive tools also exist, in particular for cis-elements pattern-based search32,33 and ncRNAs.34,35,36,37,38,39 This review will first introduce the foremost available resources, excluding those related to splicing and the no longer updated ones, and will catalog them in three categories: RBP-oriented, ncRNA-oriented and cis-element-oriented, with a number of resources falling in more than one category (Fig. 1). We will highlight also further features of these resources, as integrating different data types or being predictive. We will then proceed to illustrate a tentative pipeline combining several of these tools to enable the discovery of regulatory mechanisms. Eventually, we will present a biological use-case in which these resources are employed to identify potential regulatory circuits. We conclude with a short discussion on the future directions to be pursued in order to enhance the usefulness and completeness of this toolbox for the analysis of circuits of post-transcriptional regulation of gene expression. We provide an accompanying website to this review, available at www.ptrguide.org. The website lists all the cited resources, providing a summary and details on features and availability of the resources; it will be regularly updated with new resources and updates of existing ones, with the aim of providing a one-stop catalog for available PTR mining tools (Table 1).

Figure 1. Venn diagram showing the classification of the analyzed resources according to their biological focus. Symbols next to the resource name correspond to each set (triangles for RBPs, squares for ncRNAs and circles for cis-elements) and further highlight the presence of a limited number of integrative tools, with most of the resources being confined to only one kind of regulatory element.
Table 1. PTR resources presented in the review.
| Name | Ref | Last update | Batch mode | Data download | Organisms | Link |
|---|---|---|---|---|---|---|
| ARED |
25 |
Mar 2011 |
v |
x |
HSA, MMU |
http://brp.kfshrc.edu.sa/ARED/ |
| AREsite |
26 |
Nov 2010 |
x |
v |
HSA, MMU |
http://rna.tbi.univie.ac.at/AREsite |
| AURA |
13 |
Nov 2011 |
x |
v |
HSA |
http://aura.science.unitn.it/ |
| CLIPZ |
15 |
Jan 2011 |
v |
v |
HSA, MMU, CEL |
http://www.clipz.unibas.ch/ |
| DIANA-miRPath |
38 |
Mar 2012 |
v |
x |
HSA, MMU |
http://www.microrna.gr/miRPathv2 |
| doRiNa |
31 |
May 2012 |
v |
v |
HSA, MMU, DME, CEL |
http://dorina.mdc-berlin.de |
| IRESite |
27 |
Apr 2011 |
x |
x |
HSA, MMU, RNO, DME, SCE and 4 more |
http://iresite.org/ |
| lncRNAdb |
22 |
Jul 2011 |
x |
v |
HSA, MMU, DME, CEL, ATH, XLA, SCE and 53 more |
http://lncrnadb.com/ |
| miRanda |
36 |
Nov 2010 |
v |
V |
HSA, MMU, RNO, DME, CEL |
http://www.microrna.org/microrna/home.do |
| MAGIA2 |
24 |
Apr 2012 |
v |
X |
HSA, MMU, RNO, DME |
http://gencomp.bio.unipd.it/magia2 |
| miRConnX |
39 |
Jul 2011 |
v |
v |
HSA, MMU |
http://mirconnx.csb.pitt.edu/ |
| miRecords |
18 |
Nov 2010 |
x |
v |
HSA, MMU, RNO, DME, CEL, GGA, DRE, OAR, CFA |
http://mirecords.biolead.org/ |
| miRGator |
23 |
Jan 2011 |
v |
x |
HSA |
http://mirgator.kobic.re.kr |
| miRNAMap |
19 |
Jul 2007 |
x |
v |
HSA, MMU, RNO, DME, CEL, XTR and 4 more |
http://mirnamap.mbc.nctu.edu.tw/ |
| miRTarBase |
17 |
Oct 2011 |
x |
v |
HSA, MMU, RNO, DME, CEL, ATH, XLA and 7 more |
http://mirtarbase.mbc.nctu.edu.tw/ |
| NONCODE |
21 |
Jan 2012 |
x |
v |
HSA, MMU, DME, CEL, ATH, XLA and 1233 more |
http://www.noncode.org |
| NRED |
24 |
Sep 2008 |
x |
v |
HSA, MMU |
http://jsm-research.imb.uq.edu.au/nred |
| PicTar |
35 |
Mar 2007 |
x |
v |
HSA, MMU, DME, CEL |
http://pictar.mdc-berlin.de/ |
| PITA |
37 |
Aug 2008 |
v |
v |
HSA, MMU, DME, CEL |
http://genie.weizmann.ac.il/pubs/mir07/mir07_data.html |
| RBPDB |
14 |
Jan 2011 |
x |
v |
HSA, MMU, DME, CEL |
http://rbpdb.ccbr.utoronto.ca/ |
| Rfam |
30 |
Jun 2011 |
x |
v |
HSA, MMU, DME, CEL, ATH, SCE and 3104 more |
http://rfam.sanger.ac.uk/ |
| SelenoDB |
28 |
Sep 2007 |
x |
v |
HSA, MMU, DME, CEL, SCE and 3 more |
http://www.selenodb.org/ |
| SIREs |
33 |
Jan 2010 |
v |
x |
any |
http://ccbg.imppc.org/sires/ |
| starBase |
16 |
Sep 2011 |
x |
v |
HSA, MMU, CEL, ATH, OSA, VME |
http://starbase.sysu.edu.cn/ |
| TargetScan |
34 |
Mar 2012 |
x |
v |
HSA, MMU, CEL, DRE |
http://www.targetscan.org/ |
| Transterm |
32 |
Oct 2011 |
v |
x |
any |
http://mrna.otago.ac.nz/ |
| UTRdb/UTRsite | 12 | Oct 2009 | v | v | HSA, MMU, DME, CEL, ATH, XLA and 73 more | http://utrdb.ba.itb.cnr.it/ |
The table shows the list of databases and tools presented in the review: for each of them we report the last update (or publication date when the former is not available) along with the reference number in the manuscript, the resource website address, the organisms for which the resource provides data (listed by their three-letters code), the possibility to do a batch analysis (searching for more than one gene/element at a time) and to download the whole database.
Resources
Databases and tools can be classified according to their main focus and purpose. They can be RBP-oriented when they deal with RBPs and the effect these exert on mRNAs, ncRNA-oriented when they analyze regulation by the various families of these RNAs (as miRNAs and lncRNAs); and cis-oriented whenever a cis element is annotated and characterized in its occurrences throughout expressed exons.
RBP-Oriented
Despite the increasingly recognized importance of these factors in PTR of gene expression, only five resources are available which focus on RBPs, completely or even only partially. RBPDB12 and CLIPZ15 are built exclusively around RBPs: RBPDB offers a literature-curated collection of RBP binding sites and motifs, searchable by species or by protein domain and including logos or position-weight matrices where available. It allows the user to input sequences that can be searched for the presence of binding sites of the included RBPs. It also has predictive capabilities, albeit limited: indeed, it allows the user to match an input sequence vs. position weighted matrices (PWMs) contained in the database to identify possible RBP binding sites. CLIPZ is instead an analysis environment of RNA binding sequences by RBPs derived from the high-throughput techniques for cross-linking based mRNA footprinting, including CLIP,40 PAR-CLIP41 and iCLIP42 followed by RNA-seq. It contains analytical tools to let the user load and analyze the own CLIP-seq data, identify binding sites and annotate them on the reference genome. UTRdb/UTRsite,12 AURA13 and doRiNA31 hold RBP-related data as the two resources described above, but they differ in still keeping a broader perspective on post-transcriptional regulation. UTRdb/UTRsite contains data about UTRs in a number of species, annotating them with a specific subset of RBP binding sites, cis-regulatory sequence patterns, miRNAs and SNP data. It provides UTR sequence data along with conserved elements, visually arranged in a linear fashion. AURA annotates human UTRs with RBPs, ncRNAs, cis-elements, phylogenetic conservation and sequence variation obtained from 10 different databases, and includes literature curation. This database has its strength in committing to experimentally inferred interactions; it allows displaying UTRs in a genome browser like view, with calculated UTR secondary structures, and experimental mRNA and protein levels; visualization of joint gene expression data of targets and associated regulators can also aid inference of regulatory events. doRiNA31 integrates RBP and miRNA binding sites, by including only high-throughput assays-derived data sets for RBPs and a set of predictions for miRNAs. It exploits the UCSC database genome viewer annotated with binding sites, offering various query possibilities: by specifying a specific list of RBPs and miRNA one can obtain subsets of UTRs regulated by common groups of RBPs and miRNAs, thus guiding the discovery of novel PTR networks. By including high-throughput techniques-derived data, AURA and doRiNA provide a great wealth of information on RBP binding sites: the user need however to be aware that, as these data are available for only a limited number of RBPs, the resulting network will be biased toward these factors, providing a potentially incomplete or misleading picture of the PTR phenomena at study.
ncRNA-Oriented
A wealth of resources focused on noncoding RNAs are available: these can be differentiated by the data they hold, either experimentally validated or predicted. miRecords,17 mirTarBase18 and miRNAMap19 aim to collect miRNAs annotations and miRNA-target interactions. miRecords and miRNAMap contain both experimentally validated and predicted data (which are obtained by merging the output of 11 prediction algorithms for miRecords and 3 for miRNAmap), while miRTarBase includes only experimentally validated data. All three databases link out to various miRNA reference annotation sources such as miRBase, with mirTarBase and miRNAMap also displaying pre-miR secondary structure and miRNA expression levels in various normal and diseased tissues. miRGator20 also focuses on this class of ncRNAs, trying to give a broader overlook on the miRNA functional role by means of several auxiliary annotations: it integrates predicted miRNA-mRNA interactions, paired miRNA-mRNA expression profiles and miRNA disease signatures. Through their association analysis feature, exploiting the various expression profiles contained in the database, a miRNA can be associated to a particular tissue, a disease state or to anti-coexpressed genes. User expression profiles cannot be uploaded, although miRNA sets can be tested for enrichments through the miR set analysis tool. starBase16 is quite unique in its kind as it is dedicated to the annotation of experimentally validated Argonaute binding sites, derived from CLIP-seq and Degradome-seq43 assays: these sites, hallmark of miRNA-mediated regulation, are then merged with the output of various miRNA-target prediction tools in order to infer several thousands of miRNA-mRNA relationships. The experimental data-based tool is MAGIA2,24 an analysis platform allowing to upload your own miRNA and mRNA expression data sets, combine them with transcription factor binding sites and miRNA target predictions, and eventually infer regulatory networks from the integrated data. A wealth of tools is instead available to computationally predict miRNA targets: among these we consider TargetScan,34 PicTar,35 miRanda,36 PITA,37 DIANA-mirPath38 and miRConnX.39 TargetScan predicts interaction by requiring seed match conservation in five species and by filtering false positives through comparable abundance hexamers control; along the same line, based on sequence information, DIANA-mirPath combines predictions with experimentally verified targets, employing artificial neural networks or sequence-based 38-bases sliding windows to identify true positive miRNA binding sites in human and model organisms 3′UTRs. Users can also exploit data on SNPs in miRNA binding sites and related pathways information. PicTar instead identifies seed matches by keeping into account free energy of the miRNA-mRNA hybrid and by using a combination of scores to evaluate match goodness; miRanda also employs hybrid free energy but also phylogenetic conservation and seed matching, complemented with non-uniform distribution of target sites and 5′−3′ asymmetry constraints. PITA is the last of the tools keeping secondary structure free energy into account: it scores the sequence seed matches according to the gain in free energy obtained when the miRNA binds to the target, compared with the energy needed to open the structure of the target in that portion and thus promoting binding. miRConnX takes advantage of a pre-computed network of predicted mRNA-miRNA, transcription factor-gene and transcription factor-miRNA relationships, supplemented with literature data, combined with a dynamic networks based on user-provided gene expression data (both mRNA and miRNA). The user data network is built by various correlation measures (following the guilt-by-association principle) and integrated with the pre-computed one through a weighted sum integration function. The resulting integrated network can then be browsed, exported or analyzed in several ways, such as searching for network motifs. Users need to keep in mind that the data set size required for such an approach to produce meaningful results is quite high (in the order of tens, if not hundreds, of samples). NONCODE,21 lncRNAdb22 and NRED23 are reference databases for ncRNAs and related expression information. Long-noncoding RNAs have been mostly regarded as chromatin-associated, and thus transcription-related, factors. However, some evidence of their involvement in PTR of gene expression is emerging (for examples, see refs. 46 and 47), and we therefore include them in our review. NONCODE offers a wealth of expression and functional data concerning all kinds of ncRNAs: data are predominantly experimental, and the database includes a novel classification system based on cellular function. lncRNAdb and NRED are connected and aim, on one side to comprehensively list experimentally inferred lncRNAs described to have biological function in eukaryotes, and on the other side to provide gene expression information for thousands of these lncRNAs in human and mouse. lncRNAdb includes sequence and structure information along with links to the UCSC genome browser,44 literature sources and data from the NRED database: these are obtained primarily by microarray or in situ hybridization analysis and are complemented by auxiliary annotations, such as phylogenetic conservation and secondary structure evidence.
Cis-Oriented
Most of the resources in this category are focused on one specific type of cis-elements; still, among them four databases are more general and aim at considering or predicting a great deal of these: Transterm32 containing various patterns of cis-regulatory elements in mRNA UTRs: input sequences can be selected among the sets provided on the website or provided by the user: all instances matching the patterns or just the ones of the user-selected pattern will be reported. The UTRscan feature of UTRdb/UTRsite12 works in the same way, predicting instances of cis-elements. AURA13 contains instead annotated instances of elements like AREs (predicted) and mRNA-editing data (experimentally validated). The last general resource, Rfam,30 annotates and lists, organizing them in clans and families, currently known cis-elements found in 5′UTRs and 3′UTRs. On a wider perspective, this database also aims at cataloging all ncRNAs by means of sequence alignment and statistical profile models. ARED24 and AREsite25 are two databases devoted to AU-rich elements (AREs), a widely studied cis-element type found in 3′ UTRs. ARED is built by searching in GenBank mRNA and EST records for a single 13-base pairs pattern, and the results are then classified according to ARE classes.10 Every ARE-containing mRNA is then linked to the related UniGene and Gene Ontology annotation. AREsite works along the same line, but allows the user to screen UTRs for eight different ARE patterns, corresponding to types extracted from the literature. Along with ARE localization on the UTR, it displays information about the structural context of the motif and its level of phylogenetic conservation. IRESite27 contains experimentally validated Internal Ribosome Entry Sites (IRESs) found in 5′UTRs. These are listed with related gene and mRNA details; furthermore, the user can input its own sequence or secondary structure to search for matches with all IRESs contained in the database. SIREs33 is instead a web server for the prediction of IREs11: it takes into account both sequence and secondary structure constraints known to characterize this kind of elements. Structure analysis, folding data and quality indications are provided for each prediction output. SelenoDB28 aims at annotating all selenoproteins and SECIS (SEleno Cysteine Insertion Sequence) elements45 found in the 3′UTRs of the mRNAs coding for these proteins. These cis-elements are predicted in selenoprotein 3′UTRs by means of a computational tool, and annotated with sequence, position and related gene data. Finally, 3′-UTR SIRF,29 lists all computationally predicted short-interspersed repeats in 3′UTRs. Motifs can be searched alone or in combination to identify genes whose 3′UTRs bear these putatively co-occurring repeats.
Designing a Discovery Pipeline
Choosing which resources to use among the ones presented here may be far from trivial, especially for non-computational biologists. We thus propose a pipeline to empower the discovery of potential post-transcriptional regulatory mechanisms by exploiting some of the available tools. This is, of course, just one of the many possible combinations of instruments that can be used to reach this goal, and is offered as an example to illustrate the concepts behind an effective discovery workflow. Figure 2 reports the steps composing our pipeline. It starts with the identification of a set of interesting genes or mRNAs (1) and related UTRs: in a common setting these may represent differentially expressed genes obtained through a case-control microarray or RNA-seq experiment, although the UTR list can come in whatever other way. In the next step the workflow splits in two parallel branches: on one side, UTRs are searched for known binding sites of trans-factors (2). These are both experimentally validated (A) for RBPs and miRNAs coming from AURA13 and miRecords18 respectively, and computationally predicted (B) by applying RBPDB12 and TargetScan.34 In the other branch (3) we scan our UTRs in order to identify cis-elements that may be contained therein. Again, we employ both experimentally validated data (A), coming from IRESite27 and possibly other sources, and computationally predicted annotations, obtained through UTRsite,12 AREsite,26 SIREs33 and others. Once data collection is completed, we can move to the next step (4): building a network including our initial genes and all the factors identified until this point as regulators. Such construction can be done by means of software like Cytoscape48 and can be automated through a scripting language such as Python. Visual inspection of the resulting network will highlight hub nodes, that is, highly controlled mRNAs or widespread regulators of the mRNAs of interest. More rigorous statistical analysis can be performed on the network nodes. As regulatory factors like RBPs may post-transcriptionally control hundreds of different mRNAs, it is worth looking for enrichment of a potential regulator in the set of mRNAs under analysis: this may be done by applying a Fisher test, as it is commonly done for the over-representation of ontology terms in gene lists.49 This test will be associated to a p-value testifying for the hypothesized enrichment. In order to discriminate between general factors and potential aspecific interactions, it can be useful to also generate a control network to compare with the one under study: to do so, one can select a comparable number of UTRs at random (from the data produced by the same experiment) and reapply the pipeline to this new data set. The two resulting networks can then be compared, and factors present or enriched in both of these be excluded from further analysis: these may indeed represent widespread regulatory mechanisms, most probably not responsible for the differential expression of this group of genes and difficult to target. The last step of our pipeline leaves the in silico world and goes back to the bench: in order to understand and validate the regulatory mechanism we have hypothesized and prioritized, a classical array of methods are available. In case of cell studies, gene silencing through RNA interference, gene overexpression through transfection or viral infection, and target gene expression probing through real-time PCR or high throughput mRNA quantification methods are the most common choices. This will eventually provide data concerning the effect of the depletion or enrichment of our potential regulator(s) over target genes, and, on a wider perspective, over the network we are characterizing.
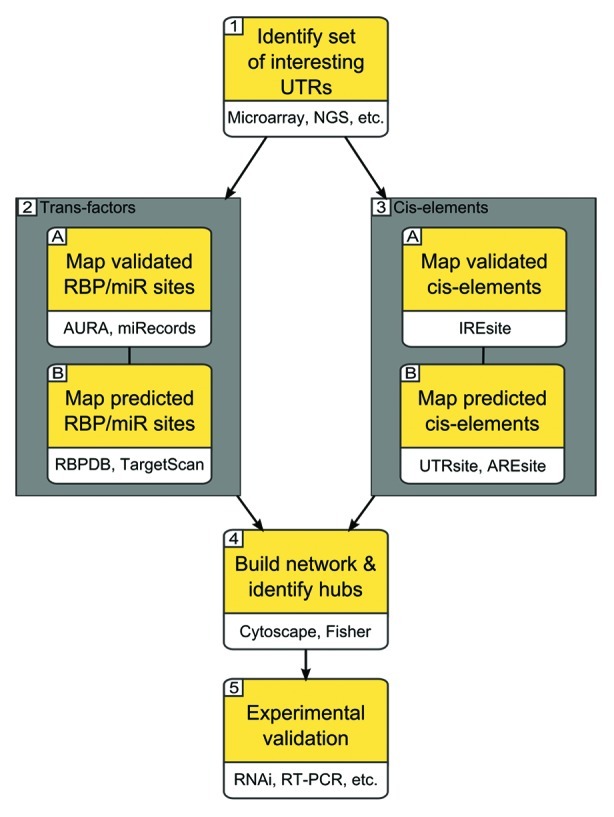
Figure 2. A possible discovery pipeline for post-transcriptional regulatory mechanisms. The workflow starts by the selection of interesting UTRs: these may, for instance, come from high-throughput experiments done by the microarrays or next generation sequencing technologies. The pipeline then proceeds by searching for both experimentally validated and computationally predicted trans-factors binding sites and cis-elements over these UTRs. The resulting interactions are then collected into a network: important nodes are identified by enrichment tests such as the Fisher test. Interesting leads are eventually subjected to experimental validation by various methods of targeted gene expression perturbation, as RNA interference.
A Case
We now proceed to apply the proposed pipeline to a set of differentially expressed genes, in order to provide a practical example of how it could work. We downloaded the GSE11324 data set51 from GEO52: in this data set, the transcriptome of MCF7 cells is profiled under estrogen stimulation at several time points (0 to 12hrs). By means of GEO2R,52 we computed differentially expressed genes between 0hr and 12hr of estrogen stimulation. We then selected the 50 mRNAs with the highest absolute fold change, corresponding to 43 genes (obviously this is an arbitrary choice, we presume that the highest fold changes indicate the most relevant biological changes, even this cannot be necessarily the case, and there are other ways of prioritizing the genes). The gene list (a) and a summary of the most prominent findings (b-c) are shown in Figure 3. From (b), reporting only trans-factors and cis-elements shared by at least 10% of the mRNAs (in which the size of the circle is proportional to the percentage of controlled mRNAs), it is evident that the relevant genes share AU-rich elements and AGO binding sites: in particular, AREs are predicted to be present in the 3′UTR of 86% of the mRNAs (enrichment p-value = 1.3E-10), while AGO binding sites in 83% of these(enrichment p-value = 4.9E-10). Other potentially involved factors are the IGF-binding proteins IGFBP1/2/3, having experimentally determined binding sites in 80% of the mRNAs (enrichment p-value = 0.48); PUM2, whose binding elements are found for 44% of the mRNAs (enrichment p-value = 1.37E-08); along with predicted IRES (Internal Ribosome Entry Site, 88% of the mRNAs), MBEs (Musashi binding elements, 67% of the mRNAs) and K-Boxes, GY-Box and PAS (Poly-adenylation signal) at a lower frequency. Two families of miRNAs (mir-15 and mir-16) are predicted by Targetscan34 to control at least ten genes of our set. In order to understand if these factors are specific to our DEGs network, we randomly selected another 50 mRNAs from the data set and reapplied the pipeline to these (network not shown): while AGO and IGFBP1/2/3 sites are again found in many UTRs (90% and 75% respectively), leading us to consider them non-relevant findings, ARE and PUM2 sites are found in lower proportions (54% and 28% of the UTRs); predicted IRES involve only 26% of the randomly selected mRNA, while MBEs are found in the same proportion as in the top DEGs (67%). Among microRNAs, mir-15 and mir-16 are not predicted to control many of our mRNAs: miR-590 and miR-30 seem to control instead 15 or more genes of our random set, with miR-23 predicted for 13 genes. Other elements are found with low frequencies (less than 10% of the mRNAs) and are thus not considered as relevant. We can thus confirm some of the involved factors as specific for our DEGs network, avoiding to focus on possibly general regulatory mechanisms. These findings are obviously biased by the still low number of available transcriptome-wide CLIP experiments, which provide much more data than literature annotations, and therefore emerge in the results. Enrichment p-values are computed for experimentally validated data by means of a Fisher test, as previously stated. The resulting post-transcriptional network of RBP, miRNAs and cis-elements, shown in (c) and built via a Python script into the Cytoscape48 platform, offers a complex landscape for further validation.

Figure 3. Selected genes and results obtained by the application of the proposed pipeline. (A) Is the list of genes selected for the case example. (B) Shows the post-transcriptional interactions prioritized through the pipeline: orange circles represent experimentally validated interactions while cyan circles represent predicted interactions. Size of the circles is proportional to the fraction of genes controlled by the element which name labels the circle (RBP, ncRNA or cis-element). Percentage of controlled genes is shown under the factor name. (C) Displays the post-transcriptional regulatory network composed of RBPs, miRNAs and cis-elements obtained by the application of the pipeline. Yellow squares represent our genes of interest, while light blue circles are the different factors controlling these genes. Oriented arrows pointing toward a gene represent an observed regulatory event (binding site or cis-element).
Future Directions
This review has highlighted the main tools of the steadily increasing number of resources available on networks of regulation at the post-transcriptional level, as one of the indicators of the growing interest in the topic. In particular, a wealth of databases and algorithms is offered focusing on miRNA-mRNA interactions, both for experimentally validated data and computational prediction, mirroring the exceptional interest raised by these controllers of gene expression in the research community. A more limited variety of resources dedicated to RBPs, cis-elements and others ncRNAs is also available. Only three tools, among the ones we analyzed, attempt to integrate different component of these networks: RBPs and miRNA binding sites only,31 or including also predicted RNA secondary structures and cis-elements.12,13 While these resources considerably ease the task of hypothesizing the existence of new networks, they still contain just a fraction of the data really available in the literature, and obviously are affected by the small number of trans-factor experimentally tested in a high throughput way with respect to the annotated ones. Moreover, the majority of the tools still does not allow online batch or programmatic analysis, forcing the user willing to work on a medium-to-big sized data set to download and replicate the database locally, and write ad hoc scripts. Integrating these tools into an automatic or semi-automatic pipeline is thus time consuming, if not impossible. Future developments should go toward this direction, providing a one-stop, truly integrated, comprehensive and multi-faceted PTR analysis toolset. Availability of such a tool will consistently empower the mapping of post-transcriptional and specifically translational networks, reaching the level of service already offered by resources focusing on the analysis of transcriptional regulation. Nevertheless, this will require a substantial effort of implementation and update, which could be eased by coordination between the available resources and integration with major genome databases such as the UCSC Genome Browser44 and Ensembl.50 Furthermore, we think that at least two additional features are currently missing but definitely needed. First, a systematic literature-derived annotation of the molecular downstream and phenotypic effects of a given interaction would provide more grounded clues, orienting the experimental validation. Second, more tailored statistical methods for enrichment of cis-elements or trans-factor, as those for ontology terms enrichment,49 would be beneficial to avoid generation of a large number of false positives as an effect of the high multiplicity of action of several studied trans-factors.
Glossary
Abbreviations:
- PTR
post-transcriptional regulation
- UTR
untranslated region of mRNA
- RBP
RNA-binding protein
- RRM
RNA-recognition motif
- KH
K-homology domain
- dsRBD
double strand RNA binding domain
- ncRNA
non-coding RNA
- lncRNA
long non-coding RNA
- miRNA
micro-RNA
- siRNA
small interfering RNA
- piRNA
piwi-interacting RNA
- snoRNA
small nucleolar RNA
- snRNA
small nuclear RNA
- ARE
AU-rich element
- IRE
iron response element
- IRES
internal ribosome entry site
- SECIS
seleno-cysteine insertion sequence
- SIRF
short interspersed repeats fragment
- CLIP
cross-linking immunoPrecipitation
- PAR-CLIP
photoactivatable-ribonucleoside-enhanced crosslinking and immunoprecipitation
- iCLIP
individual-nucleotide resolution UV cross-linking and immunoprecipitation
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/22035
References
- 1.Moore MJ. From birth to death: the complex lives of eukaryotic mRNAs. Science. 2005;309:1514–8. doi: 10.1126/science.1111443. [DOI] [PubMed] [Google Scholar]
- 2.Mignone F, Gissi C, Liuni S, Pesole G. Untranslated regions of mRNAs. Genome Biol. 2002;3:reviews0004.1. doi: 10.1186/gb-2002-3-3-reviews0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glisovic T, Bachorik JL, Yong J, Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008;582:1977–86. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 5.Baltz AG, Munschauer M, Schwanhäusser B, Vasile A, Murakawa Y, Schueler M, et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell. 2012;46:674–90. doi: 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 6.Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol. 2007;8:479–90. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andreassi C, Riccio A. To localize or not to localize: mRNA fate is in 3’UTR ends. Trends Cell Biol. 2009;19:465–74. doi: 10.1016/j.tcb.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 8.Costa FF. Non-coding RNAs: Meet thy masters. Bioessays. 2010;32:599–608. doi: 10.1002/bies.200900112. [DOI] [PubMed] [Google Scholar]
- 9.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102–14. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 10.Barreau C, Paillard L, Osborne HB. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 2005;33:7138–50. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Pantopoulos K. Regulation of cellular iron metabolism. Biochem J. 2011;434:365–81. doi: 10.1042/BJ20101825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grillo G, Turi A, Licciulli F, Mignone F, Liuni S, Banfi S, et al. UTRdb and UTRsite (RELEASE 2010): a collection of sequences and regulatory motifs of the untranslated regions of eukaryotic mRNAs. Nucleic Acids Res. 2010;38(Database issue):D75–80. doi: 10.1093/nar/gkp902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dassi E, Malossini A, Re A, Mazza T, Tebaldi T, Caputi L, et al. AURA: Atlas of UTR Regulatory Activity. Bioinformatics 2011; doi: 10.1093/bioinformatics/btr608 [DOI] [PubMed] [Google Scholar]
- 14.Cook KB, Kazan H, Zuberi K, Morris Q, Hughes TR. RBPDB: a database of RNA-binding specificities. Nucleic Acids Res. 2011;39(suppl 1):D301–8. doi: 10.1093/nar/gkq1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khorshid M, Rodak C, Zavolan M. CLIPZ: a database and analysis environment for experimentally determined binding sites of RNA-binding proteins. Nucleic Acids Res. 2011;39(suppl 1):D245–52. doi: 10.1093/nar/gkq940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang JH, Li JH, Shao P, Zhou H, Chen YQ, Qu LH. starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 2011;39(Database issue):D202–9. doi: 10.1093/nar/gkq1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu SD, Lin FM, Wu WY, Liang C, Huang WC, Chan WL, et al. miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011;39(Database issue):D163–9. doi: 10.1093/nar/gkq1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiao F, Zuo Z, Cai G, Kang S, Gao X, Li T. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009;37(Database issue):D105–10. doi: 10.1093/nar/gkn851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu SD, Chu CH, Tsou AP, Chen SJ, Chen HC, Hsu PW, et al. miRNAMap 2.0: genomic maps of microRNAs in metazoan genomes. Nucleic Acids Res. 2008;36(Database issue):D165–9. doi: 10.1093/nar/gkm1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cho S, Jun Y, Lee S, Choi HS, Jung S, Jang Y, et al. miRGator v2.0: an integrated system for functional investigation of microRNAs. Nucleic Acids Res. 2011;39(Database issue):D158–62. doi: 10.1093/nar/gkq1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bu D, Yu K, Sun S, Xie C, Skogerbø G, Miao R, et al. NONCODE v3.0: integrative annotation of long noncoding RNAs. Nucleic Acids Res. 2012;40(Database issue):D210–5. doi: 10.1093/nar/gkr1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amaral PP, Clark MB, Gascoigne DK, Dinger ME, Mattick JS. lncRNAdb: a reference database for long noncoding RNAs. Nucleic Acids Res. 2011;39(Database issue):D146–51. doi: 10.1093/nar/gkq1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dinger ME, Pang KC, Mercer TR, Crowe ML, Grimmond SM, Mattick JS. NRED: a database of long noncoding RNA expression. Nucleic Acids Res. 2009;37(Database issue):D122–6. doi: 10.1093/nar/gkn617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bisognin A, Sales G, Coppe A, Bortoluzzi S, Romualdi C. MAGIA²: from miRNA and genes expression data integrative analysis to microRNA-transcription factor mixed regulatory circuits (2012 update) Nucleic Acids Res. 2012;40(Web Server issue):W13-21. doi: 10.1093/nar/gks460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bakheet T, Williams BR, Khabar KS. ARED 3.0: the large and diverse AU-rich transcriptome. Nucleic Acids Res. 2006;34(Database issue):D111–4. doi: 10.1093/nar/gkj052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gruber AR, Fallmann J, Kratochvill F, Kovarik P, Hofacker IL. AREsite: a database for the comprehensive investigation of AU-rich elements. Nucleic Acids Res. 2011;39(Database issue):D66–9. doi: 10.1093/nar/gkq990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mokrejs M, Masek T, Vopálensky V, Hlubucek P, Delbos P, Pospísek M. IRESite--a tool for the examination of viral and cellular internal ribosome entry sites. Nucleic Acids Res. 2010;38(Database issue):D131–6. doi: 10.1093/nar/gkp981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Castellano S, Gladyshev VN, Guigó R, Berry MJ. SelenoDB 1.0 : a database of selenoprotein genes, proteins and SECIS elements. Nucleic Acids Res. 2008;36(Database issue):D332–8. doi: 10.1093/nar/gkm731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andken BB, Lim I, Benson G, Vincent JJ, Ferenc MT, Heinrich B, et al. 3′-UTR SIRF: a database for identifying clusters of whort interspersed repeats in 3′ untranslated regions. BMC Bioinformatics. 2007;8:274. doi: 10.1186/1471-2105-8-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gardner PP, Daub J, Tate J, Moore BL, Osuch IH, Griffiths-Jones S, et al. Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Res. 2011;39(Database issue):D141–5. doi: 10.1093/nar/gkq1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anders G, Mackowiak SD, Jens M, Maaskola J, Kuntzagk A, Rajewsky N, et al. doRiNA: a database of RNA interactions in post-transcriptional regulation. Nucleic Acids Res. 2012;40(Database issue):D180–6. doi: 10.1093/nar/gkr1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jacobs GH, Chen A, Stevens SG, Stockwell PA, Black MA, Tate WP, et al. Transterm: a database to aid the analysis of regulatory sequences in mRNAs. Nucleic Acids Res. 2009;37(Database issue):D72–6. doi: 10.1093/nar/gkn763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Campillos M, Cases I, Hentze MW, Sanchez M. SIREs: searching for iron-responsive elements. Nucleic Acids Res. 2010;38(Webserver issue):W360-7. doi: 10.1093/nar/gkq371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 35.Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 36.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39:1278–84. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 38.Vlachos IS, Kostoulas N, Vergoulis T, Georgakilas G, Reczko M, Maragkakis M, et al. DIANA miRPath v.2.0: investigating the combinatorial effect of microRNAs in pathways. Nucleic Acids Res. 2012;40(Webserver issue):W498-504. doi: 10.1093/nar/gks494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang GT, Athanassiou C, Benos PV. mirConnX: condition-specific mRNA-microRNA network integrator. Nucleic Acids Res. 2011;39(Webserver issue):W416-23. doi: 10.1093/nar/gkr276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–5. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- 41.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–41. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.König J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol. 2010;17:909–15. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.German MA, Pillay M, Jeong DH, Hetawal A, Luo S, Janardhanan P, et al. Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat Biotechnol. 2008;26:941–6. doi: 10.1038/nbt1417. [DOI] [PubMed] [Google Scholar]
- 44.Dreszer TR, Karolchik D, Zweig AS, Hinrichs AS, Raney BJ, Kuhn RM, et al. The UCSC Genome Browser database: extensions and updates 2011. Nucleic Acids Res. 2012;40(D1):D918–23. doi: 10.1093/nar/gkr1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoffmann PR, Berry MJ. Selenoprotein synthesis: a unique translational mechanism used by a diverse family of proteins. Thyroid. 2005;15:769–75. doi: 10.1089/thy.2005.15.769. [DOI] [PubMed] [Google Scholar]
- 46.Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature. 2011;470:284–8. doi: 10.1038/nature09701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoon JH, Abdelmohsen K, Srikantan S, Yang X, Martindale JL, De S, et al. LincRNA-p21 Suppresses Target mRNA Translation. Mol Cell. 2012;47:648–55. doi: 10.1016/j.molcel.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–2. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 50.Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, et al. Ensembl 2012. Nucleic Acids Res. 2012;40(D1):D84–90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–97. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 52.Barrett T, Troup DB, Wilhite SE, Ledoux P, Evangelista C, Kim IF, et al. NCBI GEO: archive for functional genomics data sets--10 years on. Nucleic Acids Res. 2011;39(Database issue):D1005–10. doi: 10.1093/nar/gkq1184. [DOI] [PMC free article] [PubMed] [Google Scholar]