

Abstract
We have recently shown that Toll-like receptor (TLR) signaling exacerbates pancreatic fibro-inflammation and promotes carcinogenesis in mice. Paradoxically, inhibition of the TLR-MYD88 signaling pathway is pro-tumorigenic owing to the dendritic cell-mediated TH2-polarization of CD4+ T cells. TLR signaling appears to be central in pancreatic cancer-associated inflammation.
Keywords: cancer-associated inflammation, dendritic cells, innate immunity, MYD88, pancreatic cancer, pattern-recognition receptors, Toll-like receptors, tumor microenvironment
Pancreatic ductal adenocarcinoma (PDAC) is accompanied by a rich stromal component including both immune and mesenchymal cells. An intricate relationship between the epithelial and the stromal compartments exists in pancreatic cancer, which features bidirectional communication via cytokines, chemokines, growth factors and receptor-ligand interactions, and ultimately results in the growth of both compartments as well as in the resistance to most treatment modalities.
Since chronic inflammation is one of the established risk factors for the development of PDAC, we investigated the role of pattern-recognition receptors (PRRs) in pancreatic carcinogenesis. In two recent publication, we focused on the most well-known PRR family—Toll-like receptors (TLRs)—which recognize conserved microbial byproducts (pathogen-associated molecular patterns, PAMPs) as well as byproducts of sterile inflammation (damage-associated molecular patterns, DAMPs) and signal either via the TRIF (TLR3 and TLR4) or the MYD88 pathway (all TLRs except TLR3) to promote inflammation.1 We found that TLR activation can accelerate pancreatic cancer development and TLR inhibition exerts protective effect. However, directly blocking MYD88 signaling results in paradoxically pro-tumorigenic outcomes.2,3
We showed that TLR4 and TLR7 are overexpressed by pancreatic cancer cells in both mice and humans.2,3 Continuous TLR activation using exogenous ligands augmented pancreatic inflammation in models of acute and chronic pancreatitis, and—as expected—accelerated pancreatic carcinogenesis in mice, while TLR blockade delayed tumor progression.2,3 However, PRR signaling features a high degree of complexity, and the overall outcome of its manipulation may not be predictable. In an attempt to examine the role of the two main pathways ignited by TLR4 in the context of pancreatic cancer, we separately blocked TRIF and MYD88, using synthetic inhibitors. Although blocking the TRIF pathway protected from pancreatic tumorigenesis, inhibition of the MYD88 pathway surprisingly exacerbated pancreatic firbro-infammation and accelerated carcinogenesis (Fig. 1).
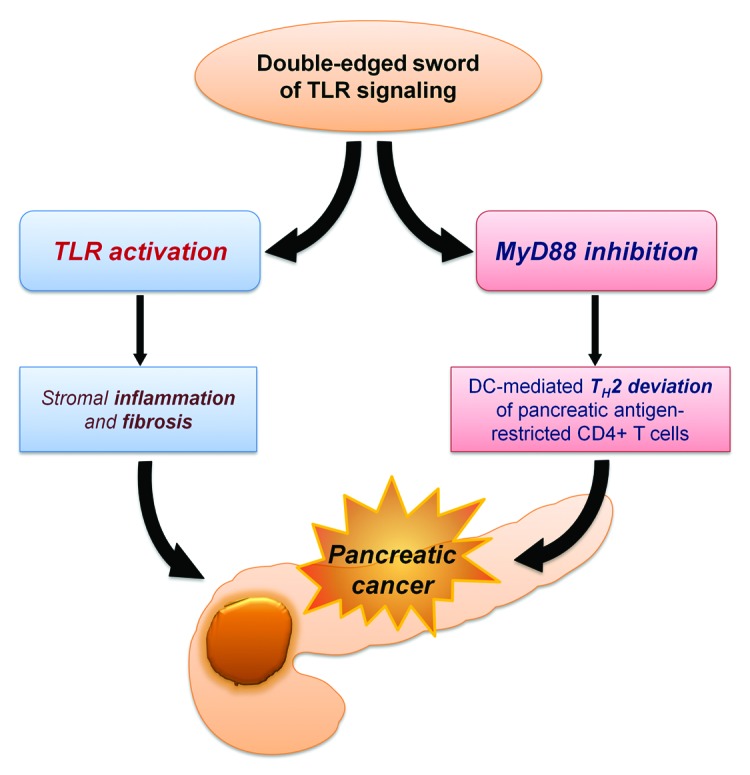
Figure 1. Model of the effects of Toll-like receptor (TLR) activation and MYD88 blockade on pancreatic carcinogenesis. DC, dendritic cell.
The complexity of PRR/MYD88 signaling may be considered at two different levels. First, at the subcellular level, PRRs can have differential inflammatory outcomes, depending on the adaptor molecules recruited and the downstream signaling cascades initiated. Furthermore, the simultaneous activation of more than one PRR, as well as other immune receptors that have convergent or antagonistic signaling pathways, can influence the biological outcome.4,5 Second, at the cellular level, PRR activation in different cell types can trigger different cellular behaviors, which can vary from cytotoxic and anti-carcinogenic to pro-inflammatory, tumor promoting effects. For example, TLR activation on dendritic cells (DCs) causes TH1 polarization of CD4+ T cells.6 For this reason, TLR agonists are being employed as adjuvants in an attempt to boost the antitumor response elicited by adoptive immunotherapy.7 However, TLR-induced DC activation also promotes the development of regulatory T cells (Tregs).6 On the contrary, blocking MYD88 in DCs leads to the TH2 polarization of CD4+ T cells.6 The latter phenomenon accounted for our intriguing findings in the setting of pancreatic cancer: MYD88 inhibition in DCs caused them to skew CD4+ T cells toward a TH2 profile, which in turn accelerated cancer progression by perpetuating inflammation.3 Depletion of CD4+ T cells rescued from the pro-carcinogenic effects of MYD88 blockade in the pancreas, although it did not protect from pancreatic cancer in the absence of MYD88 inhibition.3 Consistent with our findings, De Monte et al. have recently reported that DCs are found in high numbers in pancreatic cancer and in tumor draining lymph nodes, and that the amount of tumor-infiltrating TH2 cells inversely correlates with survival in human pancreatic cancer.8
In addition, TLRs and other immune receptors acting via the MYD88 pathway have the potential to influence signaling cascades within (pre)neoplastic cells that are important in tumorigenesis. In the case of pancreatic cancer, we showed that ligation of TLRs causes derangements in several tumor suppressor proteins (such as p16, p21, p27, p53 and pRb).2 At least in part, this seems to constitute an indirect effect mediated by stromal cells, as mice that were made chimeric with Tlr4−/− or Tlr7−/− bone marrows were partially protected from pancreatic carcinogenesis.2,3 Moreover, TLR agonists had no direct proliferative or mutagenic effects on pancreatic ductal epithelial cells.2,3
We found that TLR activation crosstalks with STAT3- and NOTCH-mediated signaling pathways, which have been recently demonstrated to transduce growth-promoting and apoptosis-resistance signals.2 Specifically, we showed that the activation of STAT3 and the upregulation of NOTCH receptors and ligands occurr in both the epithelial and the stromal component of pancreatic cancer, and—through NFκB and MAPK—contribute to the aggressive tumor phenotype induced by TLR7 activation. We speculate that all the aberrations induced by TLR ligation form a positive feedback loop between different components of the stroma as well as pancreatic epithelial cells, which includes both autocrine/paracrine communication (mediated by cytokines, chemokines and other soluble mediators) and contact-dependent signaling (e.g., NOTCH–NOTCH ligand interactions).
The importance of uncovering a pro-carcinogenic role of PRRs in pancreatic cancer is manifold. Since PRRs can promote pancreatic carcinogenesis, it is possible that their inhibition can serve as a therapeutic target, while our findings support that direct MYD88 inhibition would be catastrophic. Notably, we believe that TLR agonists should be used with great caution as vaccine adjuvants in patients with pancreatic cancer. Further, it will be very interesting to explore the natural ligands of PRRs in the context of pancreatic carcinogenesis. It is known that DAMPs released subsequently to cell damage and necrosis can activate several TLRs.9 We found that HMGB1 and S100A9—two DAMPs that bind TLR4—are present in the pancreas of PDAC patients.3 It is also possible that PAMPs originating from endogenous intestinal microbiota can reach the pancreas and contribute to pancreatic carcinogenesis in individuals at risk. Finally, polymorphisms that affect the functionality of PRRs or their downstream signal transducers may modulate the risk for pancreatic cancer development, and may also have prognostic value in PDAC patients. Further investigation is warranted to address these unanswered questions.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/22567
References
- 1.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 2.Ochi A, Graffeo CS, Zambirinis CP, Rehman A, Hackman M, Fallon N, et al. Toll-like receptor 7 regulates pancreatic carcinogenesis in mice and humans. J Clin Invest. 2012 doi: 10.1172/JCI63606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ochi A, Nguyen AH, Bedrosian AS, Mushlin HM, Zarbakhsh S, Barilla R, et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J Exp Med. 2012;209:1671–87. doi: 10.1084/jem.20111706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kondo T, Kawai T, Akira S. Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol. 2012;33:449–58. doi: 10.1016/j.it.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 5.O’Neill LA. When signaling pathways collide: positive and negative regulation of toll-like receptor signal transduction. Immunity. 2008;29:12–20. doi: 10.1016/j.immuni.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 6.Kapsenberg ML. Dendritic-cell control of pathogen-driven T-cell polarization. Nat Rev Immunol. 2003;3:984–93. doi: 10.1038/nri1246. [DOI] [PubMed] [Google Scholar]
- 7.Galluzzi L, Vacchelli E, Eggermont A, Fridman WH, Galon J, Sautès-Fridman C, et al. Trial Watch: Experimental Toll-like receptor agonists for cancer therapy. Oncoimmunology. 2012;1:699–716. doi: 10.4161/onci.20696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, et al. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med. 2011;208:469–78. doi: 10.1084/jem.20101876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–88. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]