

Abstract
To optimally integrate targeted kinase inhibitors and immunotherapies in the treatment of melanoma, it will be critical to understand how BRAFV600E mutational status and BRAFV600E inhibition influence the expression of genes that govern antitumor immune responses. Because major histocompatibility complex (MHC) molecules are critical for interactions between tumor cells and lymphocytes, we investigated the impact of BRAFV600E-selective inhibitors on the expression of MHC molecules. We found that the treatment of A375 melanoma cells with vemurafenib enhances the induction of MHC Class I and Class II molecules by interferon γ and IFNα2b. Consistent with these findings, we observed that the forced overexpression of BRAFV600E has the opposite effect and can repress the baseline expression of MHC Class I molecules in A375 cells. Further studies utilizing eight other melanoma cell lines revealed that the vemurafenib-mediated enhancement of MHC induction by IFNγ only occurs in the context of homozygous, but not heterozygous, BRAFV600E mutation. These findings suggest that BRAFV600Eactivity directly influences the expression of MHC molecules and the response to Type I and Type II IFNs. Furthermore, our data suggest that the effect of vemurafenib on the expression of immune system-relevant genes may depend on the zygosity of the BRAFV600E mutation, which is not routinely assessed in melanoma patients.
Keywords: BRAF, melanoma, MHC, vemurafenib
Introduction
The treatment of metastatic melanoma is a significant challenge and there has been little change in the survival of melanoma patients over the past 20 years.1 Fortunately, recent insights into melanoma-relevant oncogenic signaling events have begun to provide novel therapeutic approaches to disrupt pathways that drive the growth and survival of these tumors.2,3 Most noticeably, the discoveries of compounds that inhibit hyperactivated mutants of BRAF, particularly BRAFV600E, have supported the notion that this oncogenic event not only is important for the pathogenesis of melanoma but also constitutes a viable therapeutic target.4 Unfortunately, while many patients whose tumors harbor the BRAFV600E mutation initially respond to kinase inhibitors such as vemurafenib and dabrafenib, the development of resistance is common and long-term complete responses (CRs) only occur in a small percentage of individuals.5 As such, additional approaches to treat advanced melanoma patients are still needed.
Another strategy to treat melanoma that has received significant attention relies on immunotherapy.6 Most recently, the blockade if immune checkpoints with the monoclonal antibody ipilimumab has been approved by the Food and Drug Administration (FDA) for the treatment of metastatic melanoma patients.7 Other immunotherapeutic approaches being used or evaluated to treat melanoma include the use of cytokines including Type I interferons and interleukin (IL)-2, vaccines and adoptive T-cell transfer.6,8 Various combinations of these strategies have also been evaluated and have shown encouraging results.9 The majority of the aforementioned immunotherapeutic approaches against melanoma focus on enhancing the development of tumor-specific CD8+ cytotoxic T lymphocytes (CTLs) and CD4+ T lymphocytes to generate a therapeutic cell-mediated antitumor immune response. Such a cell-mediated response requires that melanoma cells process antigenic peptides and present them in the context of MHC molecules, to allow for their recognition by CTLs and/or CD4+ T cells. Hence, approaches that enhance the expression of MHC molecules by tumor cells are being sought as a means to promote tumor cell immune recognition.10 Despite the challenges associated with immunotherapies, the potential of this approach has been demonstrated in multiple settings, including patients with advanced disease and large tumor burdens that underwent a significant rate of long-term CRs.11
While these recent advances have expanded the therapeutic options for melanoma patients, the overall prognosis for most individuals bearing metastatic melanoma remains poor, being measured in months.12 Thus, combinatorial regimens including kinase inhibitors and immunotherapeutic approaches constitute the logical next step for the treatment of melanoma, and clinical trials to test such combinations are underway (http://clinicaltrials.gov).13 However, to optimally combine kinase inhibitors with immunotherapy, it will be critical to understand how kinase inhibitors influence the expression of genes coding for immune regulators, especially those that govern the interaction between T lymphocytes and tumor cells. Since interferons (IFNs) are potent inducers of MHC expression, are present in the tumor microenvironment, and can be used therapeutically, we explored how BRAFV600E inhibitors influence the induction of MHC molecules by IFNs.14-16 We found that vemurafenib can enhance the induction of MHC Class I and Class II molecules by IFNγ and IFNα2b in A375 melanoma cells. Additional studies revealed that vemurafenib could enhance MHC induction by IFNγ in melanoma cells harboring a homozygous BRAFV600E mutation but neither in cell lines that are heterozygous for BRAFV600E nor those bearing wild-type BRAF codon 600. These data suggest that the inhibition of BRAFV600E can enhance MHC induction by IFNs in some cellular contexts and support the notion that the impact of vemurafenib on the expression of immune regulators can be influenced by the zygosity of the BRAFV600E mutation.
Results
PLX4720 enhances the induction of MHC Class I molecules by IFNγ in A375 melanoma cells
Increasing evidence suggests that the inhibition of BRAFV600E or mitogen-activate protein kinase (MAPK) signaling in melanoma increases the expression of melanocyte differentiation antigens (MDAs) as well as of genes involved in antigen presentation.17-19 However, it has been repeatedly reported that, when used alone, BRAFV600E inhibitors do not increase MHC Class I expression.17,18,20 This indicates that the inhibition of BRAFV600E activity does not functionally impact the baseline expression of MHC Class I molecules in melanoma cells. However, the expression of MHC Class I molecules is dynamic and can greatly vary in response to cytokines such as IFNγ. In addition, IFNγ has been shown to rescue defects of MHC expression in melanoma cells.21 Therefore, we sought to determine whether inhibitors of BRAFV600E could potentiate the effects of IFNγ on MHC expression. We selected A375 cells as a model cell line since they are known to respond to IFNγ and harbor the BRAFV600E mutation. To confirm that PLX4720 inhibits BRAFV600E signaling, we treated A375 cells with either vehicle (DMSO) or 10 μM PLX4720 and evaluated the levels of ERK phosphorylation (at T202 and Y204) as a read-out for activated MAPK signaling. As shown in Figure 1A, PLX4720 significantly reduced the levels of ERK phosphorylation. We next examined whether PLX4720 could influence the induction of MHC Class I molecules by IFNγ in A375 cells. As expected, the treatment of A375 cells with IFNγ lead to an upregulation of MHC Class I expression on the cell surface, as measured by flow cytometry (Figs. 1B and C). The pre-treatment of A375 cells with PLX4720 enhanced this induction, suggesting that BRAFV600E inhibition can influence the upregulation of MHC Class I molecules by IFNγ, at least in some melanoma models (Figs. 1B and C).
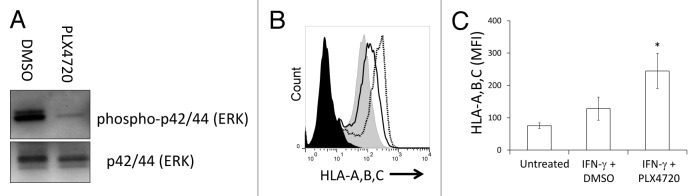
Figure 1. The BRAFV600E selective inhibitor PLX4720 enhances the induction of MHC Class I molecules in A375 melanoma cells. (A) A375 cells were pre-treated with vehicle (DMSO) or 10 μM PLX4720 60 min prior to the addition of 100 U/mL IFNγ. Whole cell lysates were prepared 24 h later and the levels of phospho-ERK (T202/Y204) and total ERK were analyzed by immunoblotting. A representative result is shown. (B) A375 cells were pre-treated with vehicle (DMSO, black line, unfilled) or 10 μM PLX4720 (black dotted line) for 60 min prior to the addition of 100 U/mL IFNγ. Control cells were left untreated (gray filled). Cell surface MHC Class I expression was analyzed by flow cytometry 72 h later, using an antibody that recognizes a shared epitope on HLA-A, B and C molecules. Cells stained with an isotype control antibody are shown (black filled). A representative flow cytometry histogram is shown. (C) Averaged mean fluorescence intensity (MFI) from three independent flow cytometry experiments is shown (y-axis) and treatments are indicated along the x-axis. Error bars represent the SEM (*p < 0.05; two-tailed paired Student’s t-test, as compared with cells treated with IFNγ and pre-treated with DMSO).
Vemurafenib enhances the induction of MHC Class I, β2 microblobulin and MHC Class II molecules by IFNγ in A375 melanoma cells
As the cellular response to IFNγ is dose-dependent,22 we next determined whether the effect of vemurafenib was influenced by the concentration of IFNγ. In addition, although PLX4720 is structurally related to vemurafenib, it is not used in the clinic. Therefore, we repeated the experiments described above using vemurafenib (which has been approved by the FDA for use in patients bearing BRAFV600E-positive melanoma) and included SKMEL-2 cells as a control, as these cells bear a variant of BRAF that is wild-type at codon 600 and hence should be insensitive to vemurafenib. Similar to PLX4720, vemurafenib decreased ERK phosphorylation levels in A375 cells, both alone and when combined with IFNγ (Fig. 2A). Consistent with what we observed with PLX4720, vemurafenib enhanced the induction of MHC Class I molecules and β2-microglobulin (B2M) by IFNγ in A375 cells (Fig. 2B). The IFNγ-mediated induction of MHC Class II molecules was also enhanced by vemurafenib. The effect of vemurafenib was greatest at the highest concentrations of IFNγ used in this assay (200 U/mL and 2000 U/mL). In contrast to A375 cells, vemurafenib had no effect on MHC Class I and II induction in SKMEL-2 cells, in spite of the fact that these cells responded to IFNγ with increases in the cell surface expression of MHC Class I, MHC Class II and B2M molecules (Fig. 2B). Because the induction of both MHC Class I and Class II molecules was enhanced by vemurafenib, we reasoned that this agent might increase the activity of IFNγ-responsive proteins that regulate both MHC Class I and Class II molecules, such as the MHC Class II transactivator CIITA.23,24 Moreover, we have previously found that the enhancement in MHC induction by epidermal growth factor receptor inhibitors (EGFRIs) is associated with an increased expression of CIITA.25 Consistent with previous observations, vemurafenib exacerbated the augmented in CIITA protein levels triggered by IFNγ (Fig. 2C). In addition, in the presence of IFNγ, vemurafenib increased the levels of the CIITA mRNA as well as those of the mRNA coding for the related transcriptional co-activator NLRC5 (Fig. 2D).26 The levels of MHC Class I (HLA-A) and Class II (HLA-DR)-coding mRNAs were also increased by vemurafenib (Fig. 2D), as were those of mRNA coding for gamma-interferon-inducible lysosomal thiol reductase (GILT), an enzyme involved in the processing of some MDAs including tyrosinase-related protein 1 (Fig. S1).27

Figure 2. The enhancement of MHC induction by vemurafenib increases with escalating concentrations of IFNγ and is associated with increased CIITA and NLRC5 expression. (A) A375 cells were treated with vehicle (DMSO) or 10 μM vemurafenib (VEM), alone or in combination with 2000 U/mL IFNγ. Whole cell lysates were prepared 24 h later and the levels of phospho-ERK (T202/Y204) and total ERK were analyzed by immunoblotting. A representative result is shown. (B) A375 (BRAFV600E homozygous) or SKMEL-2 (bearing wild-type BRAF codon 600) cells were pretreated with vehicle (DMSO, gray diamonds) or 10 μM (vemurafenib) VEM 60 min prior to the addition of IFNγ at concentrations indicated along the x-axis. Cell surface MHC Class I (HLA-A, B, C), MHC Class II (HLA-DR) and β2-microglobulin (B2M) levels were analyzed 72 h later by flow cytometry. Values represent the average mean fluorescence intensity (MFI) from three independent experiments, and error bars represent SEM (*p < 0.05, ***p < 0.001, repeated measures ANOVA, as compared with DMSO-pre-treated cells exposed to the same concentration of IFNγ) (C) Protein lysates were isolated from A375 cells 72 h after treatment with vehicle (DMSO) or VEM, given as the only treatment or 60 min prior to the addition of 2000 U/mL IFNγ. GAPDH levels were monitored as a loading control. A representative immunoblot is shown. (D) Induction of HLA-A-, HLA-DR-, CIITA- and NLRC5-coding mRNA in A375 cells. A375 cells were pre-treated with vehicle (DMSO) or 10 μM VEM 60 min prior to the addition of 2000 U/mL IFNγ. Control cells were treated with vehicle (DMSO). mRNA levels were measured using quantitative real-time RT-PCR 72 hours later and are expressed as fold over vehicle-treated cells. Error bars represent SEM from at least 3 independent experiments. (*p < 0.05, **p < 0.01, two-tailed paired Student’s t-test, as compared with cells treated with IFNγ and pre-treated with DMSO)
Since the anti-proliferative effects of vemurafenib on A375 cells is optimal at nanomolar concentrations, we repeated these experiments using serial dilutions of vemurafenib ranging from from 10 μM to 100 nM.28 Similar to what we observed with the 10 μM dose, lower concentrations of vemurafenib also enhanced the induction of MHC Class I, MHC Class II and B2M molecules in response to IFNγ (Fig. 3A and B). In this model system, the peak effect on MHC induction was observed using vemurafenib concentrations of 312 nM and 625 nM, though 100 nM was still active in this regard. These concentrations are presumably relevant in clinical settings, as patients normally receive 960 mg vemurafenib twice daily and its steady-state plasma concentration has been reported to be 86 μM ± 32μM.29 Since vemurafenib is bound to proteins for > 99%, free concentrations in patients are expected to be within the concentration ranges used in our in vitro experiments, involving 10% fetal bovine serum.30 We also tested the effects of the kinase inhibitor sorafenib, dacarbazine and temozolomide, all of which have been investigated for their antineoplastic properties against metastatic melanoma. Sorafenib appeared to enhance the induction of MHC Class I molecules by IFNγ while limiting the induction of the MHC Class II molecule HLA-DR, yet none of these differences were statistically significant upon repeated measures ANOVA testing. No effect on MHC expression was seen with dacarbazine or temozolomide (Fig. 3B). Thus, in our model system, nanomolar concentrations of vemurafenib enhanced the IFNγ−mediated induction of MHC Class I, MHC Class II and B2M molecules.

Figure 3. Nanomolar concentrations of vemurafenib enhance the induction of MHC Class I, β2-microglobulin and MHC Class II molecules on A375 cells. (A) Representative flow cytometry histograms are shown for cell surface expression of MHC Class I (HLA-A,B,C; left panel), β2-microglobulin (B2M, middle panel) or MHC Class II (HLA-DR, right panel) molecules on A375 cells treated with vehicle alone (DMSO, gray filled), with vehicle 60 min prior to the addition of 2000 U/mL IFNγ (black line), or 625 nM vemurafenib 60 min prior to the addition of 2000 U/mL IFNγ (black dotted line). Cells stained with an isotype control antibody are shown (black filled). (B) A375 cells were treated with vehicle (DMSO) alone (1st bar) or 60 min prior to the addition of 2000 U/mL IFNγ (2nd bar). The 3rd through the 9th bars represent cells pre-treated with vemurafenib at the concentrations indicated along the x-axis 60 min prior to IFNγ. Cells pre-treated with 20 μM dacarbazine (DTIC), 10 μM sorafenib (SOR), and 10 μM temozolomide (TEMO) 60 min prior to IFNγ are shown in the 10th-12th bars, as indicated along the x-axis. The average MFI from 3 independent flow cytometry experiments is shown along the y-axis. Error bars represent the SEM (*p < 0.05, **p < 0.01, ***p < 0.001, repeated measures ANOVA, as compared with DMSO-pre-treated cells exposed to the same concentration of IFNγ) (C) A375 cells were transiently transfected with a plasmid encoding BRAFV600E and green fluorescent protein (GFP) or the empty vector encoding GFP alone. Flow cytometry was used to select transfected (GFP+) and non-transfected (GFP-) cells and MHC Class I levels were measured on these two cell populations. Average values from three independent experiments are shown. MHC Class I levels are expressed as the % of MHC Class I on control cells (cells non-transfected using the empty vector plasmid). (***p < 0.001, repeated measures ANOVA, as compared with non-transfected cells) (D) Representative flow cytometry histograms are shown for untreated A375 cells (gray filled), or A375 cells pre-treated with vehicle (DMSO, solid black line) or 500 nM vemurafenib (dotted black line) 60 min prior to the administration of 909 U/mL IFNα2b. Cells were stained for MHC Class I (left panel, HLA-A,B,C) or MHC Class II (right panel, HLA-DR) expression 72 h following the addition of IFNα2b. Cells stained with an isotype control antibody are shown (black filled line). (E) The average MFI from five independent experiments is shown for A375 cells pre-treated with vehicle (gray diamonds) or 500 nM vemurafenib (black squares) for 60 min prior to the addition of IFNα2b at the doses shown along the x-axis. Error bars represent the SD (***p < 0.001, repeated measures ANOVA, as compared with cells treated with the same concentration of IFNα2b and vehicle).
Forced overexpression of BRAFV600E reduces the levels of MHC Class I molecules
We next wanted to determine if the overexpression of BRAFV600E would have the opposite effect of vemurafenib on the expression of MHC Class I molecules. To this aim, A375 cells were transfected with a plasmid encoding BRAFV600E and green fluorescent protein (GFP) on the same transcript, or the parental plasmid encoding GFP alone (empty vector). As shown in Figure 3C, in cells that were successfully transfected with the BRAFV600E-coding construct, there was a significant decrease in cell surface MHC Class I expression. Thus, the forced overexpression of BRAFV600E can repress baseline expression levels of MHC Class I molecules even in cells that already harbor the BRAFV600E mutation.
Vemurafenib enhances the induction of MHC molecules by IFNα2b
We next sought to determine if vemurafenib can influence MHC induction in response to Type I interferons, since—similar to IFNγ (a Type II interferon)—these cytokines are potent inducers of MHC Class I expression. In addition, from a therapeutic standpoint, Type I IFNs are highly relevant to melanoma, as IFNα2b is the only FDA-approved adjuvant therapy for this disease.16 Therefore, to determine if vemurafenib can enhance MHC Class I induction by IFNα2b, we pre-treated A375 cells with either vehicle (DMSO) or vemurafenib, and then exposed them to increasing concentrations of IFNα2b. We found that vemurafenib enhances the induction of MHC Class I molecules by IFNα2b at all the doses employed, which ranged from 0.09 to 909 U/mL (Figs. 3D and E). Of note, IFNα2b had no impact on the expression of MHC Class II molecules when used alone, yet it increased MHC Class II expression in A375 cells exposed to vemurafenib (Fig. 3D and Fig. S2). Thus, in some cellular models of melanoma, vemurafenib can enhance the IFNα2b-induction of MHC molecules.
MHC induction by IFNγ is enhanced by vemurafenib in cell lines that are homozygous, but not in those that heterozygous, for the BRAFV600E mutation
To determine if the results obtained with A375 cells could be reproduced in other cellular contexts, we repeated the experiments described above with additional melanoma cell lines. A list of the cell lines used in this study is reported in Table S1. These models included another cell line bearing wild-type BRAF (MeWo), as well as cell lines that are known to harbor the BRAFV600E mutation including MALME-3M, SKMEL-3, SKMEL-5, SKMEL-28, HT-144 and UACC-62 cells. We selected these cellular models since they were all commercially available and because information regarding their mutational status was publicly available at the Wellcome Trust Sanger Institute database (http://www.sanger.ac.uk). We confirmed the BRAF mutational status of these cell lines by pyrosequencing BRAF codon 600 and included an epidermoid carcinoma cell line (A431) as a control. Please note that, in this context, the terms wild-type, heterozygous and homozygous only refer to the stutus of BRAF codon 600. Specifically, the term wild-type refers to cells in which all copies of codon 600 are wild-type (i.e., encode V), the term heterozygous refers to cells in which both wild-type and mutant (i.e., encoding E) sequences are detected, and the term homozygous referes to cells in which all copies of codon 600 are mutated. Consistent with the information contained in the Wellcome Trust Sanger Institute database, we found that MALME-3M, SKMEL-3, and SKMEL-5 cells all are heterozygous for the BRAFV600E mutation (Fig. 4). In contrast, in A375, SKMEL-28 and HT-144 cells, only the mutant sequence at codon 600 could be detected, i.e., they are all homozygous for the BRAFV600E mutation (Fig. 4). Thus, we investigated how vemurafenib influences MHC induction by IFNγ in these cells. In line with previous observations, vemurafenib had no impact on the induction of MHC Class I and Class II molecules in MeWo cells, which are wild-type at BRAF codon 600 (Fig. 5A). Surprisingly, vemurafenib also had no effects on MHC induction in MALME-3M, SKMEL3, or SKMEL5 cells, all of which are heterozygous for BRAFV600E (Fig. 5A). In contrast, similar to what we observed with A375 cells, the induction of MHC Class I molecules in BRAFV600E homozygous SKMEL-28, HT-144 and UACC-62 cells was enhanced by vemurafenib (Fig. 5A and Fig. S3). This effect was not due to underlying differences in how heterozygous and homozygous cell lines respond to IFNγ. Indeed, the fold increase of MHC Class I levels by IFNγ was the same (around 4-fold) in both BRAFV600E homozygous and heterozygous cell lines (Fig. 5B). However, vemurafenib enhanced the response to IFNγ only in cells homozygous for BRAFV600E. Since we found that vemurafenib enhances the IFNγ-mediated induction of the CIITA mRNA in A375 cells (Fig. 2D), which are homozygous for BRAFV600E, we assessed whether this held true in another BRAFV600E homozygous cell line. Indeed, vemurafenib aggravated the IFNγ-mediated upregulation of CIITA mRNA levels also in SKMEL-28 cells (Fig. 5C). The effect of vemurafenib was most pronounced 48 hours after the addition of IFNγ, a finding that is consistent with what we observed in keratinocytes responding to EGFRIs.25
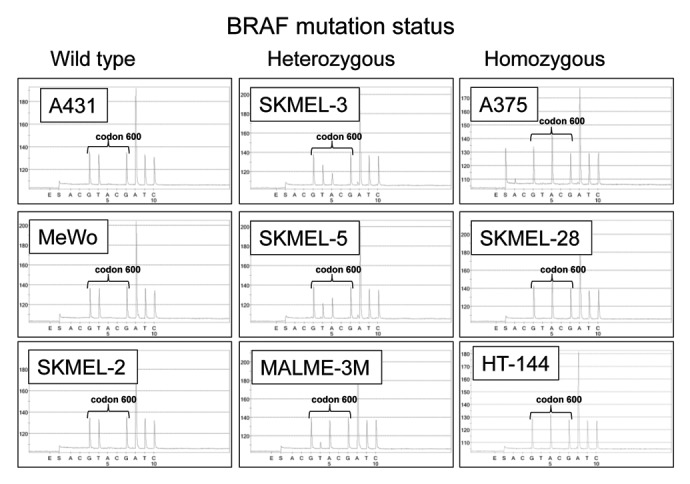
Figure 4.BRAF codon 600 sequence analysis of melanoma cell lines. Genomic DNA was isolated from the indicated cell lines indicated and BRAF codon 600 was amplified using PCR and sequenced. In each panel, the sequence of codon 600 is indicated with brackets. The wild-type BRAF codon 600 sequence is GTG whereas the BRAFV600E codon sequence is GAG. A431 cells are wild-type for BRAFV600E as are MeWo and SKMEL-2 cells. Cell lines heterozygous for BRAFV600E (SKMEL-3, SKMEL-5, and MALME-3M) contain a mixture of GTG- and GAG-containing sequences, whereas cell lines harboring only BRAFV600E (A375, SKMEL-28, and HT-144) display only the GAG sequence at codon 600.

Figure 5. Vemurafenib enhancement of MHC induction occurs in cell lines harboring homozygous but not heterozygous BRAFV600E mutations. (A) The indicated cell lines were pre-treated with either vehicle (DMSO, gray diamonds) or 500 nM vemurafenib (black squares) and then treated with IFNγ at the concentrations indicated along the x-axis. Cell surface MHC Class I (HLA-A, B and C, top panels) and MHC Class II (HLA-DR, lower panels) levels were measured 72 h later by flow cytometry. The y-axis represents average MFI for two or three independent experiments. Error bars represent SEM (*p < 0.05, **p < 0.01, repeated measures ANOVA, as compared with DMSO-pre-treated cells exposed to the same concentration of IFNγ). (B) Fold induction of cell surface MHC Class I molecules was calculated by dividing averaged MFI values of cells treated as indicated along the x-axis by averaged MFI values of cells treated with vehicle (DMSO) alone. Fold inductions for heterozygous cell lines (MALME-3M, SKMEL-3, and SKMEL-5) were averaged together (gray bars) as were the fold inductions for three homozygous (white bars) cell lines (A375, HT-144, and SKMEL-28). (**p < 0.01, ***p < 0.001, repeated measures ANOVA, as compared with identically treated BRAFV600E heterozygous cells; ††p < 0.01, †††p < 0.001, repeated measures ANOVA, as compared with BRAFV600E homozygous cells pre-treated with DMSO and exposed to the same concentration of IFNγ) (C) SKMEL-28 cells were pre-treated with vehicle (DMSO) or 500 nM vemurafenib for 60 min prior to the addition of 20 U/mL IFNγ. CIITA mRNA levels were assessed at the time points indicated along the x-axis and are expressed as fold induction over cells treated only with vehicle (DMSO). (***p < 0.001, repeated measures ANOVA, as compared with DMSO-pre-treated cells exposed to the same concentration of IFNγ) (D) DMSO-treated cells (gray diamonds) from panel (A) are compared with cells treated with 500 nM BEZ235 (black triangles or squares) for MHC Class I and Class II expression, assessed as detailed for (A).
To determine whether this effect was unique to inhibitors targeting the MAPK pathway, we examined how the induction of MHC Class I and Class II molecules by IFNγ is altered in the presence of an inhibitor of the phosphoinositide-3-kinase (PI3K) pathway, which is known to be important in the pathogenesis of melanoma.31 We used BEZ235 a dual PI3K/mammalian target of rapamycin (mTOR) inhibitor, as it is currently being tested in clinical trials (http://clinicaltrials.gov). While BEZ235 reduced the phosphorylation of AKT (on S473) in our model system, consistent with its ability to inhibit mTOR and PI3K signaling (Fig. S4), it failed to enhance the induction of MHC molecules by IFNγ. Rather, BEZ235 attenuated the IFNγ-mediated MHC induction in some of the cell lines examined (Fig. 5D and Fig. S3).
Discussion
The data presented herein demonstrate that BRAFV600E has a repressive effect on MHC expression and that, in some cellular contexts, the inhibition of BRAFV600E activity can augment the induction of MHC Class I and Class II molecules by IFNγ, a cytokine that presumably is present in the tumor microenvironment, and IFNα2b, an approved immunotherapeutic for melanoma.14,16,32 Our findings are relevant for several reasons. Most notably, the expression of MHC Class I molecules has been shown to predict the clinical response of melanoma patients to immunotherapy.33,34 Thus, our results suggest that, in some settings, BRAFV600E inhibition alone or combined with IFNα2b may be a valid pharmacologic approach to enhance the expression of MHC Class I molecules on melanoma cells. Potentially, this may enhance the response to immunotherapeutic approaches against melanoma. In addition, our findings support the notion that combining IFNα2b and BRAFV600E inhibition may offer a novel approach to promote the recognition of tumor cells by CTLs in the adjuvant setting. In addition to their canonical role in antigen presentation and their ability to influence tumor cell recognition by the immune system, MHC Class I molecules have recently been suggested to exert direct tumor suppressor properties.35 This provides further rationale to develop approaches that enhance the expression of MHC Class I molecules on melanoma cells.10,36
Our findings gather to those of several other studies in support of the notion that inhibitors of BRAFV600E have immunological effects that are relevant for antitumor immune responses (Fig. 6). For example, it has recently been demonstrated that BRAFV600E inhibitors can increase the expression of MDAs, block the production of immunosuppressive cytokines such as IL-1α/β, and increase the recruitment of CD8+ T cells into tumors.17,37,38 Indeed, it is becoming apparent that—in spite of the fact that the driving force behind the development of these therapeutic approaches had little to do with tumor immunology—the impact of targeted therapies on the expression of immune regulators may directly influence antitumor immune responses.39 Our data also suggest that—in addition to providing a growth advantage to tumor cells—the genetic amplification of BRAFV600E may also promote tumor cell immune escape by attenuating the baseline expression levels of MHC Class I molecules.
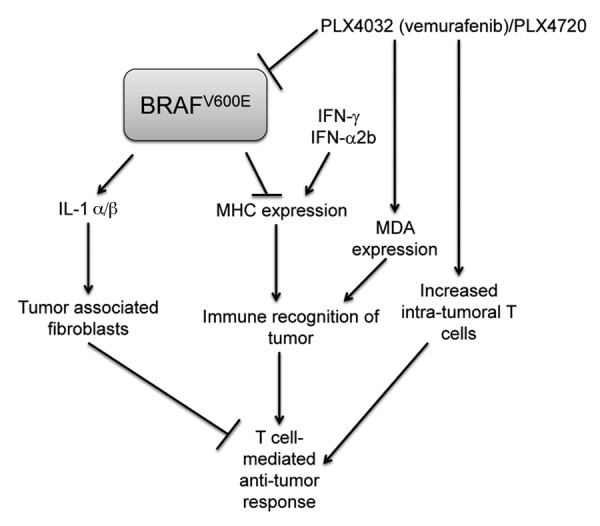
Figure 6. Model of the interactions between BRAFV600E, immune gene expression and antitumor immune responses. BRAFV600E has a repressive effect on MHC expression so that the induction of MHC molecules by IFNγ or IFNα2b can be enhanced in the presence of BRAFV600E inhibitors. Increases in MHC expression are likely complemented by increases in melanocyte differentiation antigens (MDAs), which are also induced by inhibitors of BRAFV600E. Enhanced MHC expression can increase the recognition of neoplastic cells by intratumoral T cells, which are augmented in the setting of vemurafenib therapy. BRAFV600E also promotes the expression of cytokines such as interleukin (IL)-1 α/β, that can activate the immunosuppressive effects of tumor-associated fibroblasts. These effects are also sensitive to BRAFV600E inhibitors.
Another important aspect of our study relates to the impact of BRAFV600E zygosity on the the ability of vemurafenib to influence the expression of immune regulators that are likely to be relevant for antitumor immunity. Because of its pivotal role in regulating cell proliferation, the prevailing therapeutic paradigm regarding the BRAFV600E mutation in melanoma has centered on its presence, irrespective of zygosity. If the BRAFV600E mutation is present, as determined by sequencing or mutation-specific PCR-based assays, patients are eligible to receive a BRAFV600E-selective inhibitor such as vemurafenib or dabrafenib. The scarce attention given so far to the zygosity of the BRAFV600E mutation presumably reflects the absence of a clinical need to differentiate BRAFV600E heterozygous and homozygous tumors or to understand how the zygosity of the mutation influences tumor biology. While the percentage of individuals harboring tumors with homozygous BRAFV600E mutations is unclear, accumulating evidence suggests that it is not a rare event among melanoma patients. In particular, one study reported that roughly half of patients bearing a BRAFV600E-positive melanoma, as determined by Sanger dideoxysequencing, were homozygous.40 Another study suggested that while the BRAFV600E is a ‘stable’ mutation, as assessed using metachronous melanoma metastases from different body sites, its zygosity can change over time from heterozygous to homozygous.41
At present, molecular tests for the use of inhibitors that are selective for the BRAFV600E mutation center on the detection of BRAFV600E in DNA isolated from patient material. Typically, this is biopsy material that has been fixed in formalin and embedded in paraffin. A specific test for this setting has been approved by the FDA. However, in these assays, the zygosity of the BRAFV600E mutation is not routinely assessed. Our data raise the possibility that this parameter may influence how melanoma cells respond to vemurafenib (or other targeted inhibitors of BRAFV600E) with regards to the expression of MHC molecules.34,42 This is particularly important as combination therapies utilizing both targeted kinase inhibitors and immunotherapeutic approaches are being evaluated in patients with advanced melanoma. Further, it suggests that BRAFV600E zygosity may be a relevant biomarker for therapies using BRAFV600E-specific kinase inhibitors alone or in combination with immunotherapeutic regimens (such as IL-2 or ipilimumab). However, it is important to note that it remains unclear how BRAFV600E inhibition influences the expression of IFNγ-responsive genes coding for proteins with immunosuppressive functions such as indoleamine 2,3-dioxygenase.15 In addition, the enhancement of MHC Class II expression on tumor cells by vemurafenib may not necessarily be beneficial, since clinical data on the prognostic value of this parameter among melanoma patients are contradictory.43,44
In summary, we have demonstrated that BRAFV600E can influence basal MHC Class I expression and that inhibitors of BRAFV600E can potentiate the induction of MHC molecules by IFNγ and IFNα2b. This effect is mediated by a mechanism that is influenced by the zygosity of the BRAFV600E mutation. We recognize that there may be exceptions to our model, implying that the treatment of BRAFV600E homozygous cells with a BRAFV600E-selective inhibitor may not always enhance the induction of MHC molecules by IFNγ and IFNα2b and that some BRAFV600E heterozygous cells will respond to vemurafenib with increase in an MHC induction by IFNs. Nevertheless, our data support other results from studies suggesting that BRAFV600E inhibition should be viewed in a broader context, including effects on immune gene expression and antitumor immune responses. Moreover, our findings suggest that the assessment of BRAFV600E zygosity may warrant further examination as a biomarker for patients bearing BRAFV600E-positive melanoma. In addition, these studies support the notion that vemurafenib alone or in combination with IFNα2b may represent a novel approach to enhance MHC Class I expression on melanoma cells and—in so doing so—potentiate antitumor immune responses.
Materials and Methods
Cell lines
All cell lines except UACC-62 cells were purchased directly from the American Type Culture Collection within the past 12 mo. UACC-62 cells were obtained from the National Cancer Institute within the same time period. Cells were grown in Dulbecco’s minimal essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (HyClone, Inc.), penicillin (50 U/mL), streptomycin (50 μg/mL), and L-glutamine (1 mM) (Life Technologies) and maintained at 37°C and 5% CO2 atmosphere.
Reagents and treatments
Human IFNγ (Peprotech) was re-suspended in DMEM (200 μg/mL) and stored at -80°C. Each unit of IFNγ as indicated in the text corresponds to 50 pg/mL. IFNα2b was purchased from Sigma-Aldrich re-suspended in DMEM (20 μg/mL) and stored at -80°C. Each unit of IFNα2b represents 110 pg/mL. PLX4720 was purchased from B-Bridge International and vemurafenib was purchased from LC Laboratories. Sorafenib was purchased from LC laboratories, dacarbazine was purchased from ThermoFisher Scientific and temozolomide was purchased from LKT Laboratories. All the aforementioned chemicals were dissolved in DMSO and stored at -80°C in aliquots until use.
RNA isolation and real-time PCR
RNA isolation and reverse transcription were performed as previously described.45 Quantitative real-time PCR was performed using a CFX96 thermal cycler (Bio-Rad) and measuring SYBR green incorporation into double stranded amplicons. Reactions were performed in a volume of 25 μl containing forward and reverse primers at a final concentration of 100 nM. Primer sequences were as follows: CIITA fwd 5′- CTGAAGGATGTGGAAGACCTGGGAAAGC-3′, CIITA rev 5′- GTCCCCGATCTTGTTCTCACTC-3′; HLA-A fwd 5′-CCGTGGATAGAGCAGGAG-3′, HLA-A Reverse 5′-CGTCGCAGCCATACATTATC-3′; HLA-DRα fwd 5′-GAGTTTGATGCTCCAAGCCCTCT-3′, HLA-DRα rev 5′-CAGAGGCCCCCTGCGTTCTGCTGCATT-3′; NLRC5 fwd 5′-ACCTTGGACCCTGAACAGAGAG-3′ and NLRC5 rev 5′-CTGGTGAACCCATCATCATAGCC-3′; and GAPDH fwd 5′−GAAGGTGAAGGTCGGAGTCA-3′, GAPDH rev 5′GAAGATGGTAGATGGGATTTCC-3′.
Immunoblotting
Cell lysates were prepared by washing adherent cells with cold (4°C) phosphate buffered saline (PBS) and resuspending cell pellets in urea lysis buffer (8 M urea, 50 mM Tris pH 8.0, 0.15 M β-mercaptoethanol) or utilizing a commercial extraction kit from Thermo Fisher Scientific. Primary antibodies recognizing ERK (p44/p42), phospho-ERK (p44/p42, Thr202/Tyr204) and CIITA were purchased from Cell Signaling, while those recognizing GAPDH were purchased from Abcam.
Plasmids and transient transfection of A375 cells
The PCR using a high fidelity polymerase from Thermo Fisher Scientific was used to amplify the BRAFV600E-coding region (from Addgene plasmid 17544).46 Primers used were 5′- GACCCCGGGATAAGATGGCGGCGCTGA-3′ and 5′- CCTTGCGGCCGCCTCAGTGGACAGGAAACGCA-3′ and the PCR product was cloned as a XmaI/EagI fragment into the plasmid pIRES-hrGPF II (Agilent Technologies) cut with XmaI/NotI. The plasmid was confirmed by restriction digestion and sequencing. A375 cells were transfected using Lipofectamine 2000 (Life Technologies/Invitrogen) per manufacturer’s protocol.
Flow cytometry
Cells were trypsinized, washed in FACS buffer (2 mM EDTA, 1% bovine serum albumin in phosphate buffered saline), and pelleted by centrifugation. Cell pellets were then resuspended in anti-HLA-ABC (clone G46–2.6, BD Biosciences) conjugated to fluorescein isothiocyanate (FITC), anti-HLA-DR (clone L203, R and D Systems) conjugated to peridinin-chlorophyl-protein-complex (PerCP), anti-Β2Μ (clone 2M2, Biolegend) conjugated to FITC, or isotype control antibodies. Cells were incubated on ice for 30 min washed in FACS buffer and resuspended in 0.5 mL of FACS buffer containing 0.5% paraformaldehyde. Surface protein expression of HLA-DR, HLA-ABC or B2M was measured using a FACScalibur (BD Biosciences) flow cytometer and MHC expression analyzed on ungated cells using the Flowjo software (Tree Star). For the analysis of cells transiently transfected with a BRAFV600E-coding plasmid, 48–72 h after transfection cells were treated as above except that cells were stained using an anti-HLA-A,B,C antibody conjugated to PerCP/Cy5.5 (clone W6/32, Biolegend Inc.). MHC Class I analysis of transfected and non-transfected cells was performed by gating on GFP-positive and GFP-negative cells, respectively.
Assessment of BRAFV600E mutational status
Genomic DNA was isolated from all cell lines using a genomic DNA isolation kit per the manufacturer’s protocol (Promega). Pyrosequencing to detect the BRAFV600E mutation was performed as follows. Briefly, PCR was performed using the following conditions: 200 nM fwd primer 5′-TGAAGACCTCACAGTAAA AATAGG-3′, 200 nM rev primer Biotin-TCCAGACAACTGTTCAAACTG-3′, 12 μL HotStar Mastermix (Qiagen), and 25 ng DNA in a final volume of 25 μL. Thermal cycling was performed as indicated: 95°C for 15 min, 42 cycles of 95°C for 10 sec, and 55°C for 20 sec, 72°C for 20 sec, followed by a hold at 72°C for 5 min. PCR products were immediately subjected to pyrosequencing on a PyroMark Q96 ID instrument (Qiagen) according to manufacturer’s protocols. The sequencing primer used was 5′-TGATTTTGGTCTAGCTACA-3′. Pyrosequencing was performed with the following dispensation order ACGTACGATC. Sequence analysis was performed using the PyroMark ID software set for single nucleotide polymorphism allelic quantification.
Statistical analyses
All statistics were performed using InStat (GraphPad Software Inc.). Either a paired Student’s t-test or a repeated measures ANOVA were performed as indicated in the text. Tests for Gaussian (normal) distribution were performed using the Kolmogorov-Smirnov test.
Supplementary Material
Acknowledgments
We are grateful to Jack L. Arbiser, Keith A. Delman, Jacques Galipeau, Susanna F. Greer, David H. Lawson, Jodi Osborn, Robert A. Swerlick, Kellie J. White and Michael R. Rossi for their valuable comments on this manuscript. We appreciate the technical assistance of Cora Foulks and Vincent Maffei. This work was supported by a research grant from the Melanoma and Skin Cancer Fund and a Developmental Research Award from the SPORE in Head and Neck Cancer both from the Winship Cancer Institute.
Conflicts Of Interest
Charles E. Hill serves on the scientific advisory board for Roche Diagnostics.
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/22890
References
- 1.Pollack LA, Li J, Berkowitz Z, Weir HK, Wu XC, Ajani UA, et al. Melanoma survival in the United States, 1992 to 2005. J Am Acad Dermatol. 2011;65(Suppl 1):S78.e1–S78.e10. doi: 10.1016/j.jaad.2011.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–47. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 3.Bastian BC, LeBoit PE, Hamm H, Bröcker EB, Pinkel D. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Res. 1998;58:2170–5. [PubMed] [Google Scholar]
- 4.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. BRIM-3 Study Group Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–90. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zito CR, Kluger HM. Immunotherapy for metastatic melanoma. J Cell Biochem. 2012;113:725–34. doi: 10.1002/jcb.23402. [DOI] [PubMed] [Google Scholar]
- 7.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364:2119–27. doi: 10.1056/NEJMoa1012863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lampen MH, van Hall T. Strategies to counteract MHC-I defects in tumors. Curr Opin Immunol. 2011;23:293–8. doi: 10.1016/j.coi.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 11.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199–206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blank CU, Hooijkaas AI, Haanen JB, Schumacher TN. Combination of targeted therapy and immunotherapy in melanoma. Cancer Immunol Immunother. 2011;60:1359–71. doi: 10.1007/s00262-011-1079-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kline J, Zhang L, Battaglia L, Cohen KS, Gajewski TF. Cellular and molecular requirements for rejection of B16 melanoma in the setting of regulatory T cell depletion and homeostatic proliferation. J Immunol. 2012;188:2630–42. doi: 10.4049/jimmunol.1100845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zaidi MR, Merlino G. The two faces of interferon-γ in cancer. Clin Cancer Res. 2011;17:6118–24. doi: 10.1158/1078-0432.CCR-11-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lawson DH. Choices in adjuvant therapy of melanoma. Cancer Control. 2005;12:236–41. doi: 10.1177/107327480501200405. [DOI] [PubMed] [Google Scholar]
- 17.Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010;70:5213–9. doi: 10.1158/0008-5472.CAN-10-0118. [DOI] [PubMed] [Google Scholar]
- 18.Donia M, Fagone P, Nicoletti F, Andersen RS, Høgdall E, Thor Straten P, et al. BRAF inhibition improves tumor recognition by the immune system. OncoImmunology. 2012;1:0–7. doi: 10.4161/onci.21940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kono M, Dunn IS, Durda PJ, Butera D, Rose LB, Haggerty TJ, et al. Role of the mitogen-activated protein kinase signaling pathway in the regulation of human melanocytic antigen expression. Mol Cancer Res. 2006;4:779–92. doi: 10.1158/1541-7786.MCR-06-0077. [DOI] [PubMed] [Google Scholar]
- 20.Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res. 2012;72:3928–37. doi: 10.1158/0008-5472.CAN-11-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White CA, Thomson SA, Cooper L, van Endert PM, Tampe R, Coupar B, et al. Constitutive transduction of peptide transporter and HLA genes restores antigen processing function and cytotoxic T cell-mediated immune recognition of human melanoma cells. Int J Cancer. 1998;75:590–5. doi: 10.1002/(SICI)1097-0215(19980209)75:4<590::AID-IJC16>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 22.Kortylewski M, Komyod W, Kauffmann ME, Bosserhoff A, Heinrich PC, Behrmann I. Interferon-gamma-mediated growth regulation of melanoma cells: involvement of STAT1-dependent and STAT1-independent signals. J Invest Dermatol. 2004;122:414–22. doi: 10.1046/j.0022-202X.2004.22237.x. [DOI] [PubMed] [Google Scholar]
- 23.Martin BK, Chin KC, Olsen JC, Skinner CA, Dey A, Ozato K, et al. Induction of MHC class I expression by the MHC class II transactivator CIITA. Immunity. 1997;6:591–600. doi: 10.1016/S1074-7613(00)80347-7. [DOI] [PubMed] [Google Scholar]
- 24.Beresford GW, Boss JM. CIITA coordinates multiple histone acetylation modifications at the HLA-DRA promoter. Nat Immunol. 2001;2:652–7. doi: 10.1038/89810. [DOI] [PubMed] [Google Scholar]
- 25.Pollack BP, Sapkota B, Cartee TV. Epidermal growth factor receptor inhibition augments the expression of MHC class I and II genes. Clin Cancer Res. 2011;17:4400–13. doi: 10.1158/1078-0432.CCR-10-3283. [DOI] [PubMed] [Google Scholar]
- 26.Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, et al. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci U S A. 2010;107:13794–9. doi: 10.1073/pnas.1008684107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rausch MP, Hastings KT. GILT modulates CD4+ T-cell tolerance to the melanocyte differentiation antigen tyrosinase-related protein 1. J Invest Dermatol. 2012;132:154–62. doi: 10.1038/jid.2011.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sala E, Mologni L, Truffa S, Gaetano C, Bollag GE, Gambacorti-Passerini C. BRAF silencing by short hairpin RNA or chemical blockade by PLX4032 leads to different responses in melanoma and thyroid carcinoma cells. Mol Cancer Res. 2008;6:751–9. doi: 10.1158/1541-7786.MCR-07-2001. [DOI] [PubMed] [Google Scholar]
- 29.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Comin-Anduix B, Chodon T, Sazegar H, Matsunaga D, Mock S, Jalil J, et al. The oncogenic BRAF kinase inhibitor PLX4032/RG7204 does not affect the viability or function of human lymphocytes across a wide range of concentrations. Clin Cancer Res. 2010;16:6040–8. doi: 10.1158/1078-0432.CCR-10-1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madhunapantula SV, Robertson GP. The PTEN-AKT3 signaling cascade as a therapeutic target in melanoma. Pigment Cell & Melanoma Research. 2009;22:400–19. doi: 10.1111/j.1755-148X.2009.00585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dighe AS, Richards E, Old LJ, Schreiber RD. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity. 1994;1:447–56. doi: 10.1016/1074-7613(94)90087-6. [DOI] [PubMed] [Google Scholar]
- 33.Carretero R, Romero JM, Ruiz-Cabello F, Maleno I, Rodriguez F, Camacho FM, et al. Analysis of HLA class I expression in progressing and regressing metastatic melanoma lesions after immunotherapy. Immunogenetics. 2008;60:439–47. doi: 10.1007/s00251-008-0303-5. [DOI] [PubMed] [Google Scholar]
- 34.Carretero R, Wang E, Rodriguez AI, Reinboth J, Ascierto ML, Engle AM, et al. Regression of melanoma metastases after immunotherapy is associated with activation of antigen presentation and interferon-mediated rejection genes. Int J Cancer. 2012;131:387–95. doi: 10.1002/ijc.26471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garrido C, Paco L, Romero I, Berruguilla E, Stefansky J, Collado A, et al. MHC class I molecules act as tumor suppressor genes regulating the cell cycle gene expression, invasion and intrinsic tumorigenicity of melanoma cells. Carcinogenesis. 2012;33:687–93. doi: 10.1093/carcin/bgr318. [DOI] [PubMed] [Google Scholar]
- 36.Garrido F, Cabrera T, Aptsiauri N. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: implications for immunotherapy. Int J Cancer. 2010;127:249–56. doi: 10.1002/ijc.25270. [DOI] [PubMed] [Google Scholar]
- 37.Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012;18:1386–94. doi: 10.1158/1078-0432.CCR-11-2479. [DOI] [PubMed] [Google Scholar]
- 38.Khalili JS, Liu S, Rodríguez-Cruz TG, Whittington M, Wardell S, Liu C, et al. Oncogenic BRAF(V600E) Promotes Stromal Cell-Mediated Immunosuppression Via Induction of Interleukin-1 in Melanoma. Clin Cancer Res. 2012;18:5329–40. doi: 10.1158/1078-0432.CCR-12-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pollack BP. EGFR inhibitors, MHC expression and immune responses : Can EGFR inhibitors be used as immune response modifiers? Oncoimmunology. 2012;1:71–4. doi: 10.4161/onci.1.1.18073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rubinstein JC, Sznol M, Pavlick AC, Ariyan S, Cheng E, Bacchiocchi A, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med. 2010;8:67. doi: 10.1186/1479-5876-8-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sigalotti L, Fratta E, Parisi G, Coral S, Maio M. Stability of BRAF V600E mutation in metastatic melanoma: new insights for therapeutic success? Br J Cancer. 2011;105:327–8. doi: 10.1038/bjc.2011.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cohen EP, Kim TS. Neoplastic cells that express low levels of MHC class I determinants escape host immunity. Semin Cancer Biol. 1994;5:419–28. [PubMed] [Google Scholar]
- 43.van Duinen SG, Ruiter DJ, Broecker EB, van der Velde EA, Sorg C, Welvaart K, et al. Level of HLA antigens in locoregional metastases and clinical course of the disease in patients with melanoma. Cancer Res. 1988;48:1019–25. [PubMed] [Google Scholar]
- 44.Anichini A, Mortarini R, Nonaka D, Molla A, Vegetti C, Montaldi E, et al. Association of antigen-processing machinery and HLA antigen phenotype of melanoma cells with survival in American Joint Committee on Cancer stage III and IV melanoma patients. Cancer Res. 2006;66:6405–11. doi: 10.1158/0008-5472.CAN-06-0854. [DOI] [PubMed] [Google Scholar]
- 45.Pollack BP, Sapkota B, Boss JM. Ultraviolet radiation-induced transcription is associated with gene-specific histone acetylation. Photochem Photobiol. 2009;85:652–62. doi: 10.1111/j.1751-1097.2008.00485.x. [DOI] [PubMed] [Google Scholar]
- 46.Hao H, Muniz-Medina VM, Mehta H, Thomas NE, Khazak V, Der CJ, et al. Context-dependent roles of mutant B-Raf signaling in melanoma and colorectal carcinoma cell growth. Mol Cancer Ther. 2007;6:2220–9. doi: 10.1158/1535-7163.MCT-06-0728. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.