

Abstract
High expression of autotaxin in cancers is often associated with increased tumor progression, angiogenesis and metastasis. This is explained mainly since autotaxin produces the lipid growth factor, lysophosphatidate (LPA), which stimulates cell division, survival and migration. It has recently become evident that these signaling effects of LPA also produce resistance to chemotherapy and radiation-induced cell death. This results especially from the stimulation of LPA2 receptors, which depletes the cell of Siva-1, a pro-apoptotic signaling protein and stimulates prosurvival kinase pathways through a mechanism mediated via TRIP-6. LPA signaling also increases the formation of sphingosine 1-phosphate, a pro-survival lipid. At the same time, LPA decreases the accumulation of ceramides, which are used in radiation therapy and by many chemotherapeutic agents to stimulate apoptosis. The signaling actions of extracellular LPA are terminated by its dephosphorylation by a family of lipid phosphate phosphatases (LPP) that act as ecto-enzymes. In addition, lipid phosphate phoshatase-1 attenuates signaling downstream of the activation of both LPA receptors and receptor tyrosine kinases. This makes many cancer cells hypersensitive to the action of various growth factors since they often express low LPP1/3 activity. Increasing our understanding of the complicated signaling pathways that are used by LPA to stimulate cell survival should identify new therapeutic targets that can be exploited to increase the efficacy of chemo- and radio-therapy. This article is part of a Special Issue entitled Advances in Lysophospholipid Research.
Keywords: Ceramide, Lipid phosphate phosphatase, Metastasis, Phospholipase D, Sphingosine kinase, LPA2
1. Introduction and overview
The involvement of the lipid mediator lysophosphatidate (LPA) in regulating tumor progression, angiogenesis and metastasis has been increasingly recognized over the last decade. Much of the impetus for this started with the identification of autotaxin (ATX) as an extracellular lysophospholipase D, whose major signaling effect is to generate extracellular LPA [1–3]. ATX was originally identified in 1992 in the media derived from A2058 human melanoma cells. Stracke et al. showed that these melanoma cells secreted an autocrine motility factor, which they identified and named autotaxin [4]. ATX is a member of a pyrophosphatase/phosphodiesterase family and it is also known as ecto-nucleotide pyrophosphatase/phosphodiesterase family member 2 (ENPP2). This family of enzymes consists of cell-surface or secreted proteins, which were initially described as degrading nucleotide phosphates. More recent work shows that they have more diverse functions.
Increased autotaxin expression in tumors is associated with enhanced tumor progression, aggressiveness, increased angiogenesis, metastasis and chemo-resistance [5–9]. Tumor progression is a multistep process that arises from accumulating mutations leading to oncogene activation and tumor suppressor inactivation ultimately resulting in neoplastic transformation of cells. This is followed by a transition into an invasive (metastatic) tumor with an ability to migrate and penetrate through basement membranes and invade surrounding tissue. Neoplastic transformation, in addition to oncogene activation, often requires other interactive pathways such as the ATX–LPA signaling axis to promote proliferation and support metastasis [10].
This action of LPA is readily understood based on the knowledge that was gained over the last two decades concerning the signaling effects of LPA, which are mediated through at least nine G-protein coupled receptors (GPCR) [11]. Most of these signaling events lead to the promotion of cell division, survival and migration. Stimulation of these processes is necessary for the physiological regulation of tissue remodeling, vasculogenesis and wound healing. The importance of autotaxin in embryogenesis is demonstrated by the observation that autotaxin knockout mice die in utero at day 9.5 with defective blood vessel formation and neural tube defects [3]. These functions of autotaxin and LPA signaling become dysfunctional in cancers. However, there is at present no cancer therapy that targets autotaxin activity or LPA signaling. We will discuss the background evidence that increased LPA signaling promotes tumor progression, metastasis and particularly the resistance of cancers to treatment with chemotherapeutic agents or radiotherapy.
2. Role of autotaxin and lysophosphatidate in tumor biology
Lysophosphatidylcholine (LPC) is the most abundant phospholipid in blood plasma where it reaches concentrations of about 200 μM in human beings [12]. The liver secretes unsaturated LPC [13] and presumably, this depends on the activity of a phospholipase A1. It is probable that other organs also have this ability to secrete unsaturated LPC. Saturated LPC in the circulation is produced mainly by lecithin: cholesterol acyltransferase acting on the phosphatidylcholine in high density lipoproteins. This is achieved by transferring the unsaturated fatty acid (mainly linoleate) to cholesterol [14]. These reactions provide a continuous supply of LPC that is readily accessible to most tissues. It was thought several years ago that LPC was a bioactive signaling molecule. However, more recent work shows that inhibiting ATX activity abrogates most of the effects of LPC on cell survival and migration [15–17]. Therefore, many biological effects of LPC can now be attributed to LPA, especially in the context of cell migration and protection from apoptosis. This means that autotaxin acts as a “gatekeeper” to control many of the signaling effects that are generated from LPC.
The action of ATX produces most of the LPA that is present in the circulation (Fig. 1). This is illustrated by experiments where ATX activity is inhibited and this leads to a rapid and dramatic fall in circulating LPA [17–19]. ATX activity is probably partially regulated by feedback inhibition from LPA, or by sphingosine 1-phosphate (S1P), which is the sphingolipid analogue of LPA [20].
Fig. 1.
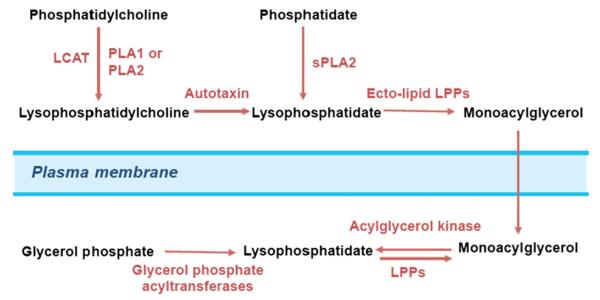
Regulation of extracellular lysophosphatidate turnover and cell signaling. Most of the extracellular LPA is produced by autotaxin from the abundant LPC that is present in extracellular fluids. Additional LPA is produced from phosphatidylcholine or phosphatidate by various phospholipases. Extracellular LPA is degraded by the ecto-activities of the lipid phosphate phosphatases, which are expressed on the plasma membrane of cells. Extracellular LPA can activate signal transduction events through up to nine specific plasma membrane GPRCR. Most of these receptors provide signals for cell growth, migration and survival.
ATX contains somatomedin B–like (SMB) domains as indicated from its crystal structure [21]. The SMB2 domain of ATX binds to β1- and β3-integrins, which increases the concentration of ATX on the surface of platelets [22]. Fulkerson et al. [22] proposed that this interaction localizes ATX on the surface of cells, thus delivering LPA in the vicinity of its receptors. Such an interaction could localize ATX to aggressive tumors and produce a microenvironment for increased LPA production. It is also suggested that this interaction will restructure the SMB domains, which regulate the LPA exit site and expedite product release [23]. Hence, the ATX–integrin β3 interaction could be especially important considering that many aggressive tumors such as melanoma and carcinomas of the prostate, breast, cervix and pancreas express the integrin αvβ3 subtype, which promotes proliferation, survival, migration, invasion and chemo-resistance [24–26]. In summary, the major role of autotaxin in cancer progression is explained by LPA production and also by its interaction with target cells, which facilitates delivery of LPA locally to its receptors. In addition, other functions of ATX have been proposed which are not directly related to LPA production and are mediated by ATX effects on cell adhesion [27].
The catalytic and nuclease-like domains of ATX were recently shown to be covalently linked by an essential disulfide bridge between Cys(413) and Cys(805) [28]. Residues 829–850 of ATX are within the nuclease-like domain and they are involved in the secretion of ATX, whereas Lys(852) is required for the expression of catalytic activity. Jansen et al. [28] proposed that the nuclease-like domain of ATX is crucial for catalysis and it is a possible target to generate ATX inhibitors.
3. Other routes of LPA formation
Apart from the actions of autotaxin, other pathways can produce extracellular LPA (Fig. 1). One such pathway involves secretory phospholipase A2 (sPLA2), which produces LPA from phosphatidate (PA). This latter lipid is present in micro-vesicles released during inflammation reactions [29]. Also, group VIA phospholipase A2 (Ca2+ independent phospholipase A2β, iPLA2β) produces extracellular LPA in human epithelial ovarian cancer cells [30,31]. The importance of this reaction in ovarian cancer biology is illustrated by the observation that tumorigenesis and ascites formation are decreased in iPLA2β(−/−) null mice compared to wild-type mice [31]. LPA and LPC concentrations are decreased in the tumor microenvironment of iPLA2β(−/−) mice to about 80% of that in wild-type mice. The authors showed that LPA, but not LPC, stimulated cell migration and invasion when iPLA2β expression was knocked down in vitro. LPA, but not LPC, also enhances ascites formation by about 5-fold and tumorigenesis in iPLA2β(−/−) mice [31].
LPA can also be produced inside cells from monoacylglycerol through a kinase reaction. Alternatively, it can be made by the first reaction of the Kennedy pathway in which sn-glycerol 3-phosphate is converted to LPA by the glycerophosphate acyltransferases. Intracellular LPA can bind to nuclear LPA1 receptors to modulate pro-inflammatory signaling [32,33]. LPA can also induce PPARγ signaling [34,35]. Drugs that activate PPARγ have been used in clinical trials of several different cancers [36–38]. It is unclear how LPA, which is a polar lipid, moves out of the cell.
4. Turnover of extracellular LPA through the ecto-activities of lipid phosphate phosphatases
Plasma LPA concentrations are normally <1 μM, but they have been reported to reach >10 μM in ovarian cancer and this depends mainly on LPA production by ATX [8]. LPA is also degraded rapidly as expected for an important bioactive compound (Fig. 1). This is achieved through the actions of a family of three enzymes that are called lipid phosphate phosphatases (LPPs) [39]. These enzymes dephosphorylate a large variety of bioactive lipid phosphates and pyrophosphates, including LPA [40]. The location of the active sites of the LPPs is on the outer leaflet of the plasma membrane of cells and this provides the necessary orientation for the degradation of extracellular lipid phosphates [41]. This was first shown in experiments with fibroblasts where increasing the expression of LPP1 enhanced their ability to degrade LPA, phosphatidate (PA) and ceramide 1-phosphate [42]. The effect of the ecto-LPP activity on LPA is to convert it to monoacylglycerols, which terminates signaling since monoacylglycerols are normally not signaling molecules. The exception to this is 2-monoarachidonoylglycerol, which is an agonist for the cannabinoid receptors (CB1 and CB2). This route of 2-mono-arachidonoylglycerol generation has not been investigated systematically and we do not know how important the LPPs are in promoting cannabinoid signaling.
The dephosphorylation of LPA in the circulation is relatively rapid and LPA has a half-life of about 3 min in the blood of mice [17,43]. The LPP isoform, LPP1, is an important contributor to the degradation of circulating LPA since the half-life for LPA was increased to about 12 min in LPP1 hypomorph mice (Ppap2atr/tr). These transgenic mice show decreases of 35–95% in LPP activity in most tissues. The exception to this is the brain where LPP expression was not significantly altered [43]. Plasma LPA concentrations in the LPP1 hypomorph mice are significantly increased compared to control mice. However, to obtain these results the authors needed to control very carefully for the age and gender of the mice. Otherwise, the variations in LPA concentrations among mice tended to obscure the significance of the differences. Previous work with mice that over-expressed LPP1 did not show the expected decrease in circulating LPA [44]. It is concluded from this combined work that LPP1 has a profound effect in balancing the kinetics of LPA formation by autotaxin. This balance of LPA turnover is responsive to the gender, age and physiological status of the animals.
The ecto-activity of LPP1 and LPP3 can probably also contribute to the degradation of extracellular S1P [45], which is a sphingolipid analogue of LPA that activates five GPRCs [46]. The degradation of extracellular S1P by the LPPs leads to increased sphingosine uptake by cells followed by the generation of intracellular S1P [47]. Increased S1P production and secretion is associated with increased chemo-resistance and a stimulation of angiogenesis for the growing tumor [48]. Therefore, LPP1 and LPP3 could also function to regulate the effects of S1P on these processes.
Significantly, the expression of LPP1 and LPP3 is very low in several cancer cells [49–52] and this is a contributing factor to the increased plasma LPA concentrations that are associated with ovarian cancers [46,51]. Total LPP activity was about 4-times lower in MCF-7, MDA-MB-231 and MDA-MB-435 cells compared to non-transformed breast epithelial cells (MCF10A). These decreases were accounted for by lower expressions of LPP1/3 relative to LPP2 (G. Venkatraman and DN Brindley, unpublished work). In addition, gonadotropin-releasing hormone increases ecto-LPP expression in ovarian cancer cells and this has an anti-proliferative effect [50]. LPP3 over-expression decreases growth and colony-formation by ovarian cancer cells by degrading extra-cellular LPA. It was concluded that increasing LPP activity and decreasing LPA signaling in cancer cells could be used as a target for cancer therapy [51,53].
Consequently, the combination of high ATX and low LPP exposes cancer cells to a microenvironment with elevated extracellular LPA concentrations [9]. The low expression of LPP1 and LPP3 in some cancer cells also makes them hypersensitive to the effects of growth factors that promote tumor progression, angiogenesis, metastasis and chemo-resistance [8].
5. Role of lipid phosphate phosphatases in regulating cell signaling downstream of the activation of growth factor receptors
The LPPs are expressed on internal membranes including the endoplasmic reticulum [42,54] and the Golgi network [55]. It is assumed that the catalytic sites face the lumenal side of these membranes. This internal LPP activity appears to regulate the turnover of lipid phosphates that are formed downstream of the activation of GPCR, including the LPA receptors, as well as receptor tyrosine kinases. This action controls the concentrations of the lipid phosphates relative to their dephosphorylated products, which are often also bioactive [56]. For example, the LPPs were assumed to metabolize the PA to diacylglycerol (DG) following the activation of phospholipase D (PLD) (Fig. 2).
Fig. 2.

Lipid phosphate phosphatases attenuate signaling downstream of GPCRs and receptor tyrosine kinases. This effect depends on the phosphatase activity demonstrating that the blocking of signaling depends on the metabolism of a lipid phosphate formed in response to activation of the receptors. The concept is illustrated by the effect of the LPPs in decreasing the accumulation of phosphatidate following the activation of PLD. PA activates sphingosine kinase-1 (SK-1), Sos, Raf and ERK, mTOR, protein PKC-ζ and it inhibits protein phosphatase-1 (PP-1). These actions stimulate cell survival and migration. Many cancer cells show very low expression of LPP1 and LPP3. We propose that this makes them hypersensitive to growth factors such as LPA, S1P, EGF and PDGF, which contribute to chemoresistance.
Evidence that the LPPs control intracellular in addition to extracellular signaling came from studies which showed that they regulate ERK activation as a result of thrombin signaling [57]. Thrombin is not a substrate of LPPs and the ecto-LPP activity cannot be responsible for controlling extracellular signaling by degrading this protein. Overexpression of LPP1 and LPP2 decreases the accumulation of PA relative to DG and this is compatible with the abilities of the LPPs to convert PA to DG and to control ERK activity [58]. Subsequent work demonstrated that mouse embryonic fibroblasts that overexpressed LPP1 showed decreased stimulation of ERK-dependent cell migration by PDGF [59]. This result was attributed to increases in intracellular DG concentrations in the LPP1 overexpressing cells. Sustained increases in the concentration of intracellular DG were postulated to decrease the expression of classical protein kinase C isoforms, which are required for PDGF-induced migration [60]. Long et al. also showed that LPP2 and LPP3 regulate cell survival by controlling intracellular PA accumulation [61].
Increased expression of LPP1 also decreases cell migration that is stimulated by LPA and a phosphonate LPA analogue, which activates LPA1/3 receptors [62]. This LPA analogue cannot be hydrolyzed by LPP1 and therefore the attenuation of PLD and ERK activation by LPP1 cannot be caused by its ecto-phosphatase activity [62]. The authors demonstrated that the catalytic action of LPP1 decreased the effects of LPA in stimulating PLD activation and thus intracellular accumulation of PA (Fig. 2). LPA-induced migration of the fibroblasts was consequently decreased since this migration depended on PLD2 activity. In addition, LPP1 over-expression also decreases the activation of ERK and Rho, which are two other important proteins involved in LPA-induced fibroblast migration. By contrast, the effect of PDGF in stimulating the migration of fibroblasts was not affected by over-expression of LPP1 and there was no significant effect on PDGF-stimulated ERK activation. Although the activation of PLD by PDGF was decreased by LPP1 over-expression, PDGF-induced migration was not affected since this process does not require PLD activity. This combined work has implications for cancer biology because cell migration is one of the components of metastasis.
Several studies showed that the level of LPP expression regulates intracellular PA concentrations [44,58,61,63,64] and this review will specifically emphasize cell signaling by PA. Over-expression of LPP3 in Swiss 3T3 or HEK 293 cells increased the conversion of PA to DG during PLD activation [65]. LPP2 was proposed to be responsible for the dephosphorylation of PA formed from PLD1 stimulation. This effect on PA formation decreases the binding of sphingosine kinase-1 to perinuclear membranes [66]. Ras-transformed fibroblasts have lower LPP activities than the control fibroblasts and they show an increased ratio of PA:DG after stimulation of PLD activity [49]. Basal levels of PA concentrations also increased with time in culture in the Ras-transformed fibroblasts [67]. It was, therefore, concluded from this combined information that the decrease in PA concentrations was caused by the action of the LPPs in converting PA to DG leave in Fig. 2. However, there is another explanation; namely that the increased LPP expression attenuates PLD activation in response to the stimulation of GPRCs or receptor tyrosine kinases and that this is responsible for the observed decrease in PA accumulation [62]. It is now though likely that the lipins, which are specific phosphatidate phosphatases found in the cytosol of cells, are the second enzymes in the PLD pathway rather than the LPPs leave in Fig. 2 [36,37,42].
Modifying LPP expression could, therefore, have a profound effect on cell signaling through their effects in controlling the activity of PLD and the accumulation of PA. This lipid controls the actions of several signaling targets such as protein kinase C-ζ, mTORC1, Sos, Raf, ERK, phospholipase C-γ and sphingosine kinase-1 (Fig. 2) [8,45,47,68–71]. Significantly, PLD1 is a critical downstream mediator of H-Ras-induced tumor formation [72]. The effects of the LPPs on DG accumulation could also control the activation of various protein kinase Cs. Several of these signaling pathways are involved in promoting cell survival by growth factors. Therefore the low LPP activity in many tumor cells could enhance LPA-induced signaling through PLD and favor LPA-induced chemo-resistance [8,9].
Increased PA production is a component in increased cancer cell survival and increased PA concentrations are observed in Ras-transformed fibroblasts [49]. PLD expression is often elevated in human breast tumors [73,74]. Increased PLD activity in MCF-7 cells causes rapamycin resistance, whereas decreasing the high PLD activity in MDA-MB-231 cells increases rapamycin sensitivity [75]. PLD activation in MDA-MB-231 cells stabilizes mutant p53 expression, which suppresses apoptosis [76], whereas decreasing PLD expression inhibits the proliferation of MDA-MB-231 cells [77]. Increased PLD activity confers estrogen-independence for the survival of breast cancer cells [78]. Other work demonstrates that the PLD(2) gene is a susceptibility locus for colorectal cancer [79]. These combined observations support the hypothesis that PLD activation is an important component in cell survival and migration in breast and other cancers as illustrated in Fig. 2.
As such, this review will concentrate on the PLD pathway because of the evidence for its regulation by the LPPs. This emphasis does not lessen the importance of other survival pathways such as the PI3K and ERK pathways that are stimulated when cells are activated with LPA. Neither do we wish to exclude the importance of the family of DG kinases, which generate PA from DAG [80]. This PA could participate in the signaling pathways that are illustrated in Fig. 2 and contribute to the effects on carcinogenesis.
There are several potential intracellular substrates for LPP action that include LPA, ceramide 1-phosphate and S1P. In the case of S1P, there are also two specific S1P phosphatases [81] that could dephosphorylate this substrate in addition to the actions of the LPPs. The LPPs could determine the balance of intracellular signals that dictate various cellular processes such as inflammation by modulating the levels of ceramide 1-phosphate and S1P compared to their products, ceramide and sphingosine. Ceramide 1-phosphate and S1P are both involved in inflammatory signaling. Ceramide 1-phosphate promotes the activation of PLA2 to produce arachidonate, whereas S1P induces cyclooxygenase-2, which converts arachidonate to prostaglandin E2 [82].
Another possible function of the LPPs is to regulate the accumulation of intracellular LPA following the action of phospholipase A1 or A2 on PA. As mentioned above, LPA can bind to nuclear LPA1 receptors to modulate pro-inflammatory signaling [32,33] and it can also induce PPARγ signaling [34,35].
Further evidence for the role of LPP1 in controlling LPA signaling comes from experiments with human bronchial epithelial cells. Overexpression of LPP-1 partially attenuated LPA-induced increases in the intracellular Ca2+ concentrations, phosphorylation of inhibitory κB and translocation of nuclear factor-κB to the nucleus, and it almost completely prevented IL-8 secretion [83]. The importance of the catalytic activity of LPP1 was demonstrated since over-expression of the inactive LPP-1 (R217K) mutant partially attenuated LPA-induced IL-8 secretion without altering LPA-induced changes in intracellular Ca2+ concentrations, phosphorylation of inhibitory κB and translocation of nuclear factor-κB or IL-8 gene expression. The authors of this work concluded that the effects of LPP1 were downstream of LPA receptor activation since the effects could not be explained by the action of LPP1 on the extracellular degradation of LPA.
At present, little is known about the relative roles of the different LPPs in dephosphorylating various lipid phosphates in intact cells as opposed to the broad substrate specificity that is observed in cell free systems. The substrate preference in whole cells should depend on the accessibility of different lipid phosphates to the active sites of the LPPs, which could explain the different signaling effects that are regulated by the LPPs.
For example, LPP2, in contrast to LPP1 and LPP3, is important in regulating the cell cycle [84]. This is shown in experiments where knocking down LPP2 in fibroblasts delayed cyclin A accumulation and entry of the fibroblasts into S-phase. Conversely, over-expressing LPP2 caused a premature entry into S-phase due to premature cyclin A accumulation. This effect depended on the catalytic activity of LPP2, which indicates that these results depend on the degradation of an unknown lipid phosphate that contributes to the control of cell cycle progression.
LPP2 over-expression in the fibroblasts caused a subsequent arrest in G2/M of the cell cycle after 15 to 35 passages when the cells exhibited a senescent phenotype [84]. This phenotype is reminiscent of oncogenes like Ras and BRAF, which stimulate cell proliferation followed by premature senescence. This effect probably prevents the development of malignancy [85,86]. Interestingly, LPP2 knockout mice are fertile and unexpectedly exhibit no obvious phenotype [87]. However, LPP2 regulates the timing of entry into S-phase and it is not essential for cell cycle progression. Knockout of cyclins D1, D2, E1, and E2 in addition to cyclin-dependent kinases-2, -4 and -6, which are well-known to be involved in regulating entry into S-phase or late G1 phase, also causes no obvious change in phenotype [84].
Later work using gene microarrays identified LPP2 (PPAP2C) as one of the three potential novel targets, which are increased in expression in transformed compared to non-transformed human adult mesenchymal stem cells [88]. LPP2 expression is also increased in transformed fibroblasts, and the cancer cell lines MCF7, SK-LMS1, MG63, and U2OS. Knockdown of LPP2 impaired anchorage-dependent growth of the cancer cell lines and also decreased the growth of primary mesenchymal stem cells, but not of differentiated human fibroblasts. These authors [67] also confirmed previous work showing that knockdown of LPP2 delayed entry into the S-phase of the cell cycle and they showed that the transcription of PPAP2C is partly controlled by p53 [88]. This work established that decreasing LPP2 expression could be a therapeutic target for treating cancer.
By contrast, LPP1 and LPP3 activities are decreased in several cancer cell lines. This could make these cancer cells hypersensitive to survival and migratory signals [8]. First, the ability of cancer cells to degrade extracellular LPA and S1P would be attenuated. This should increase the exposure of the tumor to these extracellular messengers, which would result in greater stimulation of cell division, survival, migration and angiogenesis. Second, low expression of LPP activity would increase the responses of the cancer cells to agonists that activate GPRCs (e.g., those for LPA and S1P) and receptor tyrosine kinases (e.g., EGF and PDGF). This would favor intracellular signaling by lipid phosphates formed downstream of the activation of these receptors that could contribute to the development of chemo-resistance (Fig. 2).
6. Effects of LPA signaling in controlling the proapoptotic effects of ceramides versus the survival signals from sphingosine 1-phosphate
Many chemotherapeutic agents (including Taxol, doxorubicin and vincristine) and radiation therapy increase ceramide formation as part of their therapeutic actions in stimulating cell death (Fig. 3) [89–95]. Ceramides are sphingolipids that release cytochrome C from mitochondria and activate caspases to initiate apoptosis [93,96]. The increased balance of ceramide versus S1P signaling is considered to be an important “rheostat” that favors the death versus survival of cancer cells (Fig. 3) [97,98]. Consistent with this hypothesis, defects in ceramide generation have been proposed to be associated with chemo-resistance [99].
Fig. 3.

Effects of LPA signaling in decreasing the proapoptotic effects of ceramides and increasing the survival signals from sphingosine 1-phosphate. Most chemotherapeutic agents and radiation therapy increase ceramide formation to produce an apoptotic signal. Ceramides block phospholipase D (PLD) activation and thereby the activation of survival and migratory signals that are initiated by phosphatidate (PA). These effects are counteracted by survival signals from LPA, which activates PLD and phosphatidylinositol 3-kinase (PI3K). These actions decrease ceramide production and increase the formation of S1P. These effects decrease the ratio of ceramide to S1P and promote cell survival and chemo-resistance relative to cell death.
Growth factors, including LPA, counteract these apoptotic effects of ceramides by controlling the sphingolipid “rheostat”. For example, LPA increases ERK and PI3K activities, which provide survival signals. Another important aspect of LPA signaling is that it can decrease ceramide formation in breast cancer cells response to Taxol treatment by its stimulation of PI3K (Fig. 3) [16]. This lessens the proapoptotic effects of ceramides and also decreases the potential for ceramides to block PLD activation [100] and the pro-survival effects of LPA and S1P [100,101]. LPA also increases the synthesis and secretion of S1P from cancer cells [102]. This LPA-induced decrease in ceramide:S1P signaling provides an additional mechanism for producing resistance to the actions of chemotherapeutic agents [16].
7. Overexpression of LPA receptor subtypes in different types of cancers
LPA elicits many of its cellular actions via the stimulation of specific GPCRs. LPA activates two clusters of GPCR. To date, six GPCR have been validated as specific targets of LPA. These include three GPCR encoded by the Endothelial Differentiation Gene family and designated LPA1, LPA2, and LPA3. Three other GPCR that include LPA4(GPR23/P2Y9), LPA5(GPR92), and LPA6(P2Y5) are related to the purinergic family of GPCR. There are three other GPCR including GPR87 [103,104], P2Y10 [105], and GPR35 [106,107] that remain putative LPA receptors and will have to be further evaluated.
Several reports describe overexpression of one or more of these receptors in certain types of cancers. Most cancers overexpress multiple subtypes of LPA receptors [108]. Specifically, LPA1 has been shown to be a regulator of cancer cell motility and metastasis [53,102,109,110] and MMP expression [111]. The role of LPA1 in tumor cells is not entirely clear because there is evidence that overexpression of this receptor subtype can lead to increased apoptosis due to the loss of substrate adhesion through a mechanism called anoikis that has been observed in tumor cells as well as in lung epithelial cells [112,113]. The LPA1 receptor has been shown to be important in the bone metastasis of breast carcinoma [114] and this is discussed in details in reviews in this volume [115]. Activation of LPA1 has also been reported to enhance the survival of cancer cells by increasing histone deacetylase activity and reducing histone acetylation [116].
Ovarian cancers predominantly express LPA2 receptors [117]. LPA2 is likely to play an important role in the aggressiveness of ovarian cancer in at least two ways. First, LPA2 promotes production of vascular endothelial growth factor (VEGF), urokinase (uPA), and matrix metalloproteinases [118–122]. LPA increases VEGF production, and in turn upregulates ATX production, which increases LPA levels [123] — a potential feed-forward loop that also promotes angiogenesis. Second, LPA-induced uPA production promotes invasiveness of several ovarian cancer cell lines [124,125]. High uPA levels indicate poor prognosis in ovarian cancer [126,127].
LPA3 is the dominant receptor subtype in some human melanomas and might be inhibitory to melanoma growth and viability [128]. Interestingly, overexpression of LPA3 in rat hepatoma cells enhanced cell migration and tumorigenicity [129,130]. Furthermore, RH7777 cells overexpressing LPA3 showed increased survival when treated with cisplatin or doxorubicin compared to the parental RH7777 cells. The enhanced survival correlated with the elevated expression level of multidrug-resistance related genes, Mdr1a, Mdr1b, and Gstp1. Hayashi and colleagues [131] examined the effect of the individual LPA receptors in neuroblastoma cells. They found in cell motility and invasion assays that LPA2 and LPA3 overexpressing cells showed significantly higher intrinsic activity without LPA treatment than B103 cells lacking these LPA receptor subtypes. These reports suggest that LPA receptor subtypes might exert distinct effects depending on the type and cellular origin of the individual carcinoma. If this is indeed the case, the therapeutic application of LPA receptor antagonists will be rather complicated.
A second cluster of LPAR is found within the purinergic cluster of receptors. The better-characterized members of the purinergic P2Y gene cluster are LPA4 (P2Y9) and LPA5 (GPR92). (LPA4) knockout (KO) mice generated using 129/Sv embryonic stem cells implanted into C57BL/6 mothers do not show a phenotype, although mouse embryonic fibroblasts isolated from these KOs display increased migration in response to LPA. This suggests that LPA4 inhibits cell motility [132]. However, the effects of LPA4 KO appear to be dependent on the genetic background of the stem cells because KO of this receptor subtype using C57BL/6 embryonic stem cells on the C57BL/6 genetic background revealed deficits in the development of lymphatic vessels [133].
Okabe and colleagues [130] examined the expression and methylation state of the Lpa5 gene and found that four out of six rat hepatocellular carcinomas and five out of six lung adenocarcinomas had elevated expression of Lpa5 with unmethylated status. In the lung-derived adenocarcinoma RLCNR and RH7777 hepatoma, the Lpa5 gene was unmethylated and that correlated with increased expressions of LPA5 receptor protein in these cells. These authors proposed that LPA5 expression could positively regulate the growth of these carcinomas. By contrast, Jongsma and colleagues showed that LPA5 appears to mediate chemorepulsion (inhibition of invasion) of B16 melanoma cells through the non-canonical elevation of cAMP level along with reduced PIP3 signaling [134].
GPR87 is under the transcriptional regulation of p53. This receptor subtype shows very high expression in squamous cell carcinomas, lung adenocarcinomas, and transitional carcinomas of the urinary bladder; and is involved in promoting cell survival and proliferation [135–137]. An unbiased genetic search for the gene causing familial Hypotrichosis simplex and autosomal recessive wooly hair has identified mutations in the LPA6 receptor [138,139] underlying the disease. There are no reports presently available for the role LPA6 in cancer.
8. Role of LPA receptors in mediating resistance to genotoxic agents (chemo and radiation therapy)
There is evidence for the oncogenic transforming action of LPA GPCR. Taghavi et al. [10] have evaluated the role of LPA1/2/3/4 in malignant transformation and found that the rank order of transforming activity was LPA2>LPA1>LPA4. LPA3 did not promote the growth in soft-agar of mouse embryonic fibroblasts immortalized with the Tbx2 transcriptional suppressor and c-Myc. When these fibroblasts were injected into nude mice the rank order of survival was LPA1<LPA2-LPA4 whereas, LPA3 cells did not develop into tumors. Heterologous overexpression of autotaxin and LPA1/2/3 targeted to the mammary tissues of FVB/N mice using the MMTV-LTR promoter increases mammary tumorigenesis, invasion, and metastases [140]. Hayashi and colleagues using soft agar growth assays examined the effect of a constitutively active LPA1 mutant (LPA1-Delta-1) and compared it to expression of wild type LPA2 and LPA3 in AB2-1bf neuroblastoma cells lacking LPA receptors [131]. In soft agar assays, LPA3 and LPA1-Delta-1 cells showed colony formation, whereas wild type LPA1 and LPA2 transfected AB2-1bf neuroblastoma did not grow. The different findings for the oncogenic potential of LPA3 in these three reports by van Lohuizen, Tsujiuchi, and Mills groups [10,131,140] again point to a difference between the cell type targeted.
In a genome-wide siRNA screen, we identified ATX as a candidate drug-resistance gene in ovarian cancer and showed that an inhibitor of ATX increases paclitaxel and carboplatin sensitivity of resistant cancer cells [141]. LPA2 receptors play a very profound role in the aggressiveness of ovarian carcinomas as described in the preceding paragraphs. These two observations combined logically lead to the hypothesis that the LPA2 receptor mediates the chemoresistance due to the increased LPA production by the overexpressed ATX in ovarian carcinomas. Indeed, there is evidence that LPA2 enhances resistance to apoptosis induced by genotoxic chemotherapeutics or radiation injury [142–145].
9. Unique aspects of LPA2 receptor signaling underlying radiation and chemoresistance
Tumor cells upregulate lipid synthesis as a requirement for increased cell proliferation. Lipogenesis is controlled by the sterol regulatory element binding proteins (SREBP). Mukherjee and colleagues recently showed in carcinoma cells, but not in nontransformed cells, that LPA upregulates the transcriptional activity of SREBP [146], which is the rate-limiting step in lipogenesis. The 5′ adenosine monophosphate-activated protein kinase (AMPK) plays a role in cellular energy homeostasis. The net effect of AMPK activation is stimulation of hepatic fatty acid oxidation and ketogenesis, inhibition of cholesterol synthesis, lipogenesis, and triglyceride synthesis, inhibition of adipocyte lipolysis, stimulation of skeletal muscle fatty acid oxidation, muscle glucose uptake, and modulation of insulin secretion by pancreatic β-cells. These authors also showed that LPA stimulation of ovarian carcinoma cells decreases AMP, which in turn promotes the lipogenetic effect mediated by SREBP signaling. Interestingly, the inhibition of AMPK and the stimulation of SREBP are uniquely mediated by LPA2, but not LPA1 or LPA3. Furthermore, the inhibition of AMPK is linked to the activation of the Gq-PLC, whereas the stimulation of SREBP is coupled to the G12/13-Rho-ROCK pathways downstream of LPA2.
High-energy ionizing radiation has been shown to activate ceramide production via the activation of acid sphingomyelinase [147–149]. Radiation oncotherapy is also known to lead to the elevation of ceramide level in the tumor cells. As mentioned in the previous paragraphs many chemotherapeutics cause cell death via elevation of ceramide levels [89–95]. In this context, it is important to recognize that LPA activation of cells can protect against ceramide-induced apoptosis [16,117,150]. Deng et al. [145] have shown that LPA reduced apoptosis when administered 1 h before or 2 h after γ-irradiation of IEC-6 cells, which is a nontransformed intestinal crypt-derived epithelial cell line. These authors also showed that LPA had a similar effect when apoptosis was elicited by the topoisomerase inhibitor chemotherapeutic camptothecin, TNF-α, or serum withdrawal. They also showed that oral delivery of LPA to C57BL/6 mice exposed to high levels of γ-irradiation (>10 Gy) reduced the number of apoptotic cells in the intestinal crypt-villus units indicating that LPA has a radioprotective effect. LPA not only prevents apoptosis, but also rescues apoptotically condemned cells when administered in a time window that extends for several hours after the apoptosis-inducing cellular injury. This observation has led to the recognition that LPA or LPA derivatives could mitigate the efficacy of radiation and chemotherapy by reducing apoptotic cell killing. In follow up publications, Deng and colleagues showed that the antiapoptotic effect of LPA is mediated through the attenuation of the mitochondrial apoptosis cascade uniquely linked to the activation of the LPA2 receptor [151]. The antiapoptotic signaling does not require LPA-mediated transphosphorylation of the EGF, ErbB2, or PDGF receptors [152].
Octadecenyl thiophosphate (OTP) is a metabolically stabilized mimic of LPA and a full agonist of the LPA2 receptor [153,154]. The plasma half-life of OTP in mice and nonhuman primates is ~10–14 h [155]. LPA and OTP were administered to mice irradiated with lethal levels of γ-irradiation (6–12 Gy) to elicit the hematopoietic or the gastrointestinal acute radiation syndromes. LPA and OTP treatment reduced the number of apoptotic cells in the crypt-villus units and decreased mortality due to the acute radiation syndrome [156]. The crypt cells of wild type C57BL/6 mice and LPA1 KO mice were protected by LPA or OTP administration from radiation injury [156]. By contrast, neither LPA nor OTP showed any protection in LPA2 KO mice against radiation injury. Furthermore, the LPA2 KO mice showed higher level of radiation sensitivity and injury compared to wild type or LPA1 KO mice. In vitro, the radioprotective effect of OTP was present in cells that expressed the LPA2 receptor subtype and was partially sensitive to pertussis toxin, indicating that non Gi-coupled signaling mechanisms are also important for the antiapoptotic effect [156]. An example demonstrating that LPA2 receptor knockout cells are more prone to radiation-induced apoptosis and that knock-in of LPA2 alone into mouse embryonic fibroblasts increases radiation resistance in a ligand-dependent manner is shown in Fig. 4 [157]. What might be the unique antiapoptotic signals that are activated by LPA2 but not by the highly homologous LPA1 or LPA3 receptors? Further analysis of signaling activated by the LPA2 receptor subtype has turned the focus of studies to non-GPRC signaling. Li and colleagues [158] have shown earlier that LPA2 forms a macromolecular signaling complex via its C-terminal type 1 motif recognized by the PSD95/Dlg/ZO-1 (PDZ) binding protein Na+/H+ exchange regulatory factor 2 (NHERF2). This signaling complex allows the coupling of LPA2 to the inhibition of the cystic fibrosis transmembrane regulator and the ensuing inhibition of secretory diarrhea due to the over-activity of this channel. This observation, along with the lack of pertussis toxin sensitivity of the antiapoptotic response steered our interest to the unique ability of LPA2 to form macromolecular signaling complexes via its C-terminus.
Fig. 4.

LPA2 receptor conveys increased resistance to radiation-induced apoptosis. In the experiment shown, mouse embryonic fibroblast (MEF) cells were derived from LPA1 and LPA2 double knockout mice and transduced with empty lentivirus vector (LPA1&2−/− MEF) or with the human LPA2 receptor (LPA2 Add-Back MEF). These cells also do not endogenously express LPA3 [159]. Both types of MEF cells were exposed to 15 Gy of γ-irradiation and treated with increasing concentrations of the LPA receptor agonist OTP. Apoptosis was quantified 5 h later using caspase 3/7 activity. Note that radiation alone elicits higher level of apoptosis in LPA1&2−/− MEF compared to LPA2 Add-Back MEF suggesting that LPA2 expression without exogenous ligand increases the radiation resistance of these cells. Also note, that the LPA mimic ligand OTP showed a dose-dependent attenuation of radiation-induced caspase 3/7 activity only in LPA2 Add-Back MEF whereas, it was ineffective in LPA1&2−/− MEF cells. This indicates that activation of LPA2 alone can lead to increased resistance to radiation-induced apoptosis.
Lin and colleagues [159] have shown that LPA2, but not LPA1 or LPA3, can interact with a number of zinc finger proteins through a C-terminus-mediated interaction. Ligand-activated binding of LPA2 to the zinc finger protein Siva-1 is of particular importance in this regard. Siva-1, a target of p53, is an early response gene activated by DNA-damage that promotes apoptosis. Siva-1 binds up the antiapoptotic Bxl-XL protein making the mitochondrial outer membrane more prone to apoptosis [160,161]. Siva-1 recognizes a C311-x-x-C motif in the C terminus of LPA2, where x can be any amino acid. Mutation of the two cysteine residues will lead to the disruption of the binding of LPA2 to Siva-1. On the other hand, ligand activation of LPA2 promotes the complex formation between this receptor subtype and Siva-1. Once this complex is formed, it is withdrawn for the endocytotic receptor recycling and instead it is polyubiquitinated and targeted to the proteasome for degradation. The overall effect of LPA2 activation is the depletion of the cell for Siva-1, which in turn leads to the attenuation of apoptotic signaling.
The C-x-x-C motif is only found in LPA2 but not in LPA1 or LPA3. It provides an interaction surface to the LIM family protein, the thyroid hormone receptor interacting protein 6 (TRIP6). TRIP6 has also a PDZ-binding motif and can physically interact with NHERF2. This PDZ- and Zn-finger-mediated interactions form the foundation of a ternary complex between LPA2–TRIP6–NHERF2. The functional significance of the LPA-activated assembly of the complex leads to a robust upregulation of the ERK1/2, PI3K-Akt- and NF-κB prosurvival pathways and the ensuing inhibition of apoptosis [162]. The proof of this hypothesis resides in experiments using a mutant LPA2 in which the residues C311/314 and L351 have been replaced by alanine rendering this construct unable to bind to NHERF2 and TRIP6. Cells that express this triple mutant of LPA2 can no longer be rescued from Adriamycin-(doxorubicin) or radiation-induced apoptosis upon LPA-stimulation. Furthermore, the siRNA-mediated downregulation of TRIP6 and/or NHERF2 blocks the chemoresistance of SKOV-3 ovarian cancer cells to cisplatin [162]. Lai and colleagues [163] showed that LPA induces the association of TRIP6 with NF-kB p65 and promotes its nuclear translocation and transcriptional activity. TRIP6 regulates LPA-mediated protection against Fas/CD95-mediated apoptosis in HEK 293 cells. Additional support for the role of LPA2 mediating radiation resistance is presented by Kiss et al. [157]. These authors demonstrate that an LPA2-specific agonist GRI977143 is capable of rescuing apoptotically condemned cells from cell death induced by γ-radiation-induced apoptosis [157] or genotoxic chemotherapeutics [164].
The unique C-terminal macromolecular complex-mediated signals activated by the stimulation of LPA2 overexpressed in many invasive and therapy-resistant carcinoma cells is likely to emerge as a novel therapeutic target for the enhancement of radiation- and/or chemotherapeutic-sensitivity of cancers. In this context ATX inhibitors alone or in combination with LPA2-selective antagonists can offer a new avenue in the treatment of malignancies (Fig. 5).
Fig. 5.

Scheme of the ATX–LPA2 signaling axis in the regulation of chemo- and radiation resistance. Genotoxic chemotherapeutics and radiation activate E2F and p53, which promote the initiation of apoptosis. Siva-1 is an immediate early response gene product regulated by p53 and E2F. In the cytoplasm, Siva-1 binds to Bcl-XL and depletion of this Bcl-2 antiapoptotic protein promotes apoptosis. Siva-1 in the nucleus complexes with p53 and the ubiquitin ligase Mdm2. The polyubiquitinated complex can eventually be degraded by the proteasome attenuating p53 signaling. ATX generates LPA by cleaving LPC at the cell surface. LPA activates LPA2 that binds to Siva-1 and the complex becomes polyubiquitinated and degraded. This will prevent the depletion of Bcl-XL levels due to the degradation of Siva-1. LPA2 activation leads to the assembly of a ternary complex with TRIP6 and NHEF2. This complex enhances the activation of NFkB- and ERK1/2-mediated prosurvival signaling downstream of LPA2.
10. Concluding remarks
The treatment of various cancers often fails due to the development of resistance to the actions of various chemotherapies and radiotherapy and patients die mainly because of the spread of metastases. This resistance depends largely on the effects of various growth factors including estrogens, EGF, PDGF, VEGF etc. that enable cancer cells to avoid cell death. We propose that another growth factor called LPA contributes to these problems of chemo-resistance. LPA is generated outside the cells mainly by ATX whose expression is associated with increased tumor progression, angiogenesis and metastasis. As discussed above, there is now a considerable body of evidence to show that increased expression of ATX and signaling by LPA is associated with the development of resistance to the actions of several chemotherapeutic agents and radiation-induced cell death. This is explained by the survival signals that are generated by several LPA receptors, but especially LPA2.
At present, there is no cancer treatment that is based on the inhibition of ATX, blocking LPA receptors, or signaling downstream of these receptors. We propose that improving our knowledge of the physiological and pathological consequences of signaling by the ATX/LPA axis will lead to the development of therapeutic agents that will enable us to target this signaling cascade. In particular, we predict that such treatments could be used as adjuvant therapies to improve the efficacies of established cancer treatments.
Acknowledgements
The work was supported by grants to DNB from the Canadian Institutes of Health Research, the Alberta Cancer Foundation and the Canadian Breast Cancer Foundation, by grants to GT from the National Cancer Institute, CA92160, AI80405 from the National Institute of Allergy Medical Countermeasures Radiological and Nuclear Threats Program, and by the Harriet Van Vleet Endowment for Basic Oncology Research.
Abbreviations
- AMPK
AMP-activated protein kinase
- ERK
extracellular signal-regulated kinases (p42/p44 mitogen-activated protein kinases)
- DG
diacylglycerol
- EGF
epidermal growth factor
- GPCR
G-protein coupled receptor
- KO
knockout
- LPA
lysophosphatidate
- LPA1–6
lysophosphatidate receptors 1–6
- LPC
lysophosphatidylcholine
- LPP
lipid phosphate phosphatase
- NHERF2
Na+/H+ exchange regulatory factor 2
- OTP
octadecenyl thiophosphate
- PA
phosphatidate
- PDGF
platelet-derived growth factor
- PDZ
PSD95/Dlg/ZO-1 binding protein
- PI3K
phosphatidylinositol 3-kinase
- PIP3
phosphatidylinositol 3,4,5-trisphosphate
- iPLA2β
Ca2+independent phospholipase A2β
- PLD
phospholipase D
- S1P
sphingosine 1-phosphate
- SREBP
sterol regulatory element-binding protein
- TRIP6
thyroid hormone receptor interacting protein 6
- VEGF
vascular endothelial growth factor
- uPA
urokinase
Footnotes
This article is part of a Special Issue entitled Advances in Lysophospholipid Research.
References
- [1].Umezu-Goto M. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002;158:227–233. doi: 10.1083/jcb.200204026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tokumura A, Majima E, Kariya Y, Tominaga K, Kogure K, Yasuda K, Fukuzawa K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 2002;277:39436–39442. doi: 10.1074/jbc.M205623200. [DOI] [PubMed] [Google Scholar]
- [3].van Meeteren LA, Moolenaar WH. Regulation and biological activities of the autotaxin–LPA axis. Prog. Lipid Res. 2007;46:145–160. doi: 10.1016/j.plipres.2007.02.001. [DOI] [PubMed] [Google Scholar]
- [4].Stracke ML, Krutzsch HC, Unsworth EJ, Arestad A, Cioce V, Schiffmann E, Liotta LA. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J. Biol. Chem. 1992;267:2524–2529. [PubMed] [Google Scholar]
- [5].Nam SW, Clair T, Kim YS, McMarlin A, Schiffmann E, Liotta LA, Stracke ML. Autotaxin (NPP-2), a metastasis-enhancing motogen, is an angiogenic factor. Cancer Res. 2001;61:6938–6944. [PubMed] [Google Scholar]
- [6].Umezu-Goto M, Kishi Y, Taira A, Hama K, Dohmae N, Takio K, Yamori T, Mills GB, Inoue K, Aoki J, Arai H. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002;158:227–233. doi: 10.1083/jcb.200204026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yang SY, Lee J, Park CG, Kim S, Hong S, Chung HC, Min SK, Han JW, Lee HW, Lee HY. Expression of autotaxin (NPP-2) is closely linked to invasiveness of breast cancer cells. Clin. Exp. Metastasis. 2002;19:603–608. doi: 10.1023/a:1020950420196. [DOI] [PubMed] [Google Scholar]
- [8].Samadi N, Bekele R, Capatos D, Venkatraman G, Sariahmetoglu M, Brindley DN. Regulation of lysophosphatidate signaling by autotaxin and lipid phosphate phosphatases with respect to tumor progression, angiogenesis, metastasis and chemo-resistance. Biochimie. 2011;93:61–70. doi: 10.1016/j.biochi.2010.08.002. [DOI] [PubMed] [Google Scholar]
- [9].Bekele R, Brindley DN. Role of autotaxin and lysophosphatidate in cancer progression and resistance to chemotherapy and radiotherapy. Clin. Lipidol. 2012;7:313–328. [Google Scholar]
- [10].Taghavi P, Verhoeven E, Jacobs JJ, Lambooij JP, Stortelers C, Tanger E, Moolenaar WH, van Lohuizen M. In vitro genetic screen identifies a cooperative role for LPA signaling and c-Myc in cell transformation. Oncogene. 2008;27:6806–6816. doi: 10.1038/onc.2008.294. [DOI] [PubMed] [Google Scholar]
- [11].Tigyi G. Aiming drug discovery at lysophosphatidic acid targets. Br. J. Pharmacol. 2010;161:241–270. doi: 10.1111/j.1476-5381.2010.00815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Moolenaar WH, van Meeteren LA, Giepmans BN. The ins and outs of lysophosphatidic acid signaling. Bioessays. 2004;26:870–881. doi: 10.1002/bies.20081. [DOI] [PubMed] [Google Scholar]
- [13].Brindley DN. Hepatic secretion of lysphosphatidylcholine: a novel transport system for polyunsaturated fatty acids and choline. J. Nutr. Biochem. 1993;4:442–449. [Google Scholar]
- [14].Aoki J, Taira A, Takanezawa Y, Kishi Y, Hama K, Kishimoto T, Mizuno K, Saku K, Taguchi R, Arai H. Serum lysophosphatidic acid is produced through diverse phospholipase pathways. J. Biol. Chem. 2002;277:48737–48744. doi: 10.1074/jbc.M206812200. [DOI] [PubMed] [Google Scholar]
- [15].Gaetano CG, Samadi N, Tomsig JL, Macdonald TL, Lynch KR, Brindley DN. Inhibition of autotaxin production or activity blocks lysophosphatidylcholine-induced migration of human breast cancer and melanoma cells. Mol. Carcinog. 2009;48:801–809. doi: 10.1002/mc.20524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Samadi N, Gaetano C, Goping IS, Brindley DN. Autotaxin protects MCF-7 breast cancer and MDA-MB-435 melanoma cells against Taxol-induced apoptosis. Oncogene. 2009;28:1028–1039. doi: 10.1038/onc.2008.442. [DOI] [PubMed] [Google Scholar]
- [17].Albers HM, Dong A, van Meeteren LA, Egan DA, Sunkara M, van Tilburg EW, Schuurman K, van Tellingen O, Morris AJ, Smyth SS, Moolenaar WH, Ovaa H. Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation. Proc. Natl. Acad. Sci. U. S. A. 2010;107:7257–7262. doi: 10.1073/pnas.1001529107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ferry G, Moulharat N, Pradere JP, Desos P, Try A, Genton A, Giganti A, Beucher-Gaudin M, Lonchampt M, Bertrand M, Saulnier-Blache JS, Tucker GC, Cordi A, Boutin JA. S32826, a nanomolar inhibitor of autotaxin: discovery, synthesis and applications as a pharmacological tool. J. Pharmacol. Exp. Ther. 2008;327:809–819. doi: 10.1124/jpet.108.141911. [DOI] [PubMed] [Google Scholar]
- [19].Gierse J, Thorarensen A, Beltey K, Bradshaw-Pierce E, Cortes-Burgos L, Hall T, Johnston A, Murphy M, Nemirovskiy O, Ogawa S, Pegg L, Pelc M, Prinsen M, Schnute M, Wendling J, Wene S, Weinberg R, Wittwer A, Zweifel B, Masferrer J. A novel autotaxin inhibitor reduces lysophosphatidic acid levels in plasma and the site of inflammation. J. Pharmacol. Exp. Ther. 2010;334:310–317. doi: 10.1124/jpet.110.165845. [DOI] [PubMed] [Google Scholar]
- [20].Parrill AL, Baker DL. Autotaxin inhibition: challenges and progress toward novel anti-cancer agents. Anticancer Agents Med Chem. 2008;8:917–923. doi: 10.2174/187152008786847765. [DOI] [PubMed] [Google Scholar]
- [21].Hausmann J, Kamtekar S, Christodoulou E, Day JE, Wu T, Fulkerson Z, Albers HM, van Meeteren LA, Houben AJ, van Zeijl L, Jansen S, Andries M, Hall T, Pegg LE, Benson TE, Kasiem M, Harlos K, Kooi CW, Smyth SS, Ovaa H, Bollen M, Morris AJ, Moolenaar WH, Perrakis A. Structural basis of substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Biol. 2011;18:198–204. doi: 10.1038/nsmb.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fulkerson Z, Wu T, Sunakura M, Vander Kooi C, Morris AJ, Smyth SS. Binding of autotaxin to integrins localizes lysophosphatidic acid production to platelets and mammalian cells. J. Biol. Chem. 2011;286:34654–34663. doi: 10.1074/jbc.M111.276725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Moolenaar WH, Perrakis A. Insights into autotaxin: how to produce and present a lipid mediator. Nat. Rev. Mol. Cell Biol. 2011;12:674–679. doi: 10.1038/nrm3188. [DOI] [PubMed] [Google Scholar]
- [24].Albelda SM, Mette SA, Elder DE, Stewart R, Damjanovich L, Herlyn M, Buck CA. Integrin distribution in malignant melanoma: association of the beta 3 subunit with tumor progression. Cancer Res. 1990;50:6757–6764. [PubMed] [Google Scholar]
- [25].Hsu MY, Shih DT, Meier FE, Van Belle P, Hsu JY, Elder DE, Buck CA, Herlyn M. Adenoviral gene transfer of beta3 integrin subunit induces conversion from radial to vertical growth phase in primary human melanoma. Am. J. Pathol. 1998;153:1435–1442. doi: 10.1016/s0002-9440(10)65730-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Desgrosellier JS, Barnes LA, Shields DJ, Huang M, Lau SK, Prevost N, Tarin D, Shattil SJ, Cheresh DA. An integrin alpha(v)beta(3)-c-Src oncogenic unit promotes anchorage-independence and tumor progression. Nat. Med. 2009;15:1163–1169. doi: 10.1038/nm.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yuelling LM, Fuss B. Autotaxin (ATX): a multi-functional and multi-modular protein possessing enzymatic lysoPLD activity and matricellular properties. Biochim. Biophys. Acta. 2008;1781:525–530. doi: 10.1016/j.bbalip.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jansen S, Andries M, Derua R, Waelkens E, Bollen M. Domain interplay mediated by an essential disulfide linkage is critical for the activity and secretion of the metastasis-promoting enzyme autotaxin. J. Biol. Chem. 2009;284:14296–14302. doi: 10.1074/jbc.M900790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fourcade O, Simon MF, Viode C, Rugani N, Leballe F, Ragab A, Fournie B, Sarda L, Chap H. Secretory phospholipase A2 generates the novel lipid mediator lysophosphatidic acid in membrane microvesicles shed from activated cells. Cell. 1995;80:919–927. doi: 10.1016/0092-8674(95)90295-3. [DOI] [PubMed] [Google Scholar]
- [30].Zhao X, Wang D, Zhao Z, Xiao Y, Sengupta S, Zhang R, Lauber K, Wesselborg S, Feng L, Rose TM, Shen Y, Zhang J, Prestwich G, Xu Y. Caspase-3-dependent activation of calcium-independent phospholipase A2 enhances cell migration in non-apoptotic ovarian cancer cells. J. Biol. Chem. 2006;281:29357–29368. doi: 10.1074/jbc.M513105200. [DOI] [PubMed] [Google Scholar]
- [31].Li H, Zhao Z, Wei G, Yan L, Wang D, Zhang H, Sandusky GE, Turk J, Xu Y. Group VIA phospholipase A2 in both host and tumor cells is involved in ovarian cancer development. FASEB J. 2010;24:4103–4116. doi: 10.1096/fj.10-161356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Moughal NA, Waters C, Sambi B, Pyne S, Pyne NJ. Nerve growth factor signaling involves interaction between the Trk A receptor and lysophosphatidate receptor 1 systems: nuclear translocation of the lysophosphatidate receptor 1 and Trk A receptors in pheochromocytoma 12 cells. Cell. Signal. 2004;16:127–136. doi: 10.1016/j.cellsig.2003.08.004. [DOI] [PubMed] [Google Scholar]
- [33].Gobeil F, Jr., Bernier SG, Vazquez-Tello A, Brault S, Beauchamp MH, Quiniou C, Marrache AM, Checchin D, Sennlaub F, Hou X, Nader M, Bkaily G, Ribeiro-da-Silva A, Goetzl EJ, Chemtob S. Modulation of pro-inflammatory gene expression by nuclear lysophosphatidic acid receptor type-1. J. Biol. Chem. 2003;278:38875–38883. doi: 10.1074/jbc.M212481200. [DOI] [PubMed] [Google Scholar]
- [34].McIntyre TM, Pontsler AV, Silva AR, St Hilaire A, Xu Y, Hinshaw JC, Zimmerman GA, Hama K, Aoki J, Arai H, Prestwich GD. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARgamma agonist. Proc. Natl. Acad. Sci. U. S. A. 2003;100:131–136. doi: 10.1073/pnas.0135855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Siess W, Tigyi G. Thrombogenic and atherogenic activities of lysophosphatidic acid. J. Cell. Biochem. 2004;92:1086–1094. doi: 10.1002/jcb.20108. [DOI] [PubMed] [Google Scholar]
- [36].Girnun GD, Chen L, Silvaggi J, Drapkin R, Chirieac LR, Padera RF, Upadhyay R, Vafai SB, Weissleder R, Mahmood U, Naseri E, Buckley S, Li D, Force J, McNamara K, Demetri G, Spiegelman BM, Wong KK. Regression of drug-resistant lung cancer by the combination of rosiglitazone and carboplatin. Clin. Cancer Res. 2008;14:6478–6486. doi: 10.1158/1078-0432.CCR-08-1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu. Rev. Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- [38].Girnun GD, Naseri E, Vafai SB, Qu L, Szwaya JD, Bronson R, Alberta JA, Spiegelman BM. Synergy between PPARgamma ligands and platinum-based drugs in cancer. Cancer Cell. 2007;11:395–406. doi: 10.1016/j.ccr.2007.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kok BP, Venkatraman G, Capatos D, Brindley DN. Unlike two peas in a pod: lipid phosphate phosphatases and phosphatidate phosphatases. Chem. Rev. Jun 28; doi: 10.1021/cr200433m. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- [40].Venkatraman G, Brindley DN. Lipid phosphate phosphatases: their role in lysolipid degradation and cell signaling. In: Hla T, Spiegel S, Moolenaar W, Chun J, editors. Lysolipid receptors: Signaling and Biochemistry. John Wiley and Sons Inc; NY, USA: (in press) [Google Scholar]
- [41].Zhang QX, Pilquil CS, Dewald J, Berthiaume LG, Brindley DN. Identification of structurally important domains of lipid phosphate phosphatase-1: implications for its sites of action. Biochem. J. 2000;345(Pt 2):181–184. [PMC free article] [PubMed] [Google Scholar]
- [42].Jasinska R, Zhang QX, Pilquil C, Singh I, Xu J, Dewald J, Dillon DA, Berthiaume LG, Carman GM, Waggoner DW, Brindley DN. Lipid phosphate phosphohydrolase-1 degrades exogenous glycerolipid and sphingolipid phosphate esters. Biochem. J. 1999;340:677–686. [PMC free article] [PubMed] [Google Scholar]
- [43].Tomsig JL, Snyder AH, Berdyshev EV, Skobeleva A, Mataya C, Natarajan V, Brindley DN, Lynch KR. Lipid phosphate phosphohydrolase type 1 (LPP1) degrades extracellular lysophosphatidic acid in vivo. Biochem. J. 2009;419:611–618. doi: 10.1042/BJ20081888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yue J, Yokoyama K, Balazs L, Baker DL, Smalley D, Pilquil C, Brindley DN, Tigyi G. Mice with transgenic overexpression of lipid phosphate phosphatase-1 display multiple organotypic deficits without alteration in circulating lysophosphatidate level. Cell. Signal. 2004;16:385–399. doi: 10.1016/j.cellsig.2003.08.012. [DOI] [PubMed] [Google Scholar]
- [45].Brindley D, Pilquil C, Sariahmetoglu M, Reue K. Phosphatidate degradation: phosphatidate phosphatases (lipins) and lipid phosphate phosphatases. Biochim. Biophys. Acta. 2009;1791:956–961. doi: 10.1016/j.bbalip.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Milstien S, Spiegel S. Targeting sphingosine-1-phosphate: a novel avenue for cancer therapeutics. Cancer Cell. 2006;9:148–150. doi: 10.1016/j.ccr.2006.02.025. [DOI] [PubMed] [Google Scholar]
- [47].Zhao Y, Kalari SK, Usatyuk PV, Gorshkova I, He D, Watkins T, Brindley DN, Sun C, Bittman R, Garcia JG, Berdyshev EV, Natarajan V. Intracellular generation of sphingosine 1-phosphate in human lung endothelial cells: role of lipid phosphate phosphatase-1 and sphingosine kinase 1. J. Biol. Chem. 2007;282:14165–14177. doi: 10.1074/jbc.M701279200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sukocheva O, Wang L, Verrier E, Vadas MA, Xia P. Restoring endocrine response in breast cancer cells by inhibition of the sphingosine kinase-1 signaling pathway. Endocrinology. 2009;150:4484–4492. doi: 10.1210/en.2009-0391. [DOI] [PubMed] [Google Scholar]
- [49].Martin A, Gomez-Munoz A, Waggoner DW, Stone JC, Brindley DN. Decreased activities of phosphatidate phosphohydrolase and phospholipase D in Ras and tyrosine kinase (fps) transformed fibroblasts. J. Biol. Chem. 1993;268:23924–23932. [PubMed] [Google Scholar]
- [50].Imai A, Furui T, Tamaya T, Mills GB. A gonadotropin-releasing hormoneresponsive phosphatase hydrolyses lysophosphatidic acid within the plasma membrane of ovarian cancer cells. J. Clin. Endocrinol. Metab. 2000;85:3370–3375. doi: 10.1210/jcem.85.9.6793. [DOI] [PubMed] [Google Scholar]
- [51].Tanyi JL, Hasegawa Y, Lapushin R, Morris AJ, Wolf JK, Berchuck A, Lu K, Smith DI, Kalli K, Hartmann LC, McCune K, Fishman D, Broaddus R, Cheng KW, Atkinson EN, Yamal JM, Bast RC, Felix EA, Newman RA, Mills GB. Role of decreased levels of lipid phosphate phosphatase-1 in accumulation of lysophosphatidic acid in ovarian cancer. Clin. Cancer Res. 2003;9:3534–3545. [PubMed] [Google Scholar]
- [52].Tanyi JL, Morris AJ, Wolf JK, Fang X, Hasegawa Y, Lapushin R, Auersperg N, Sigal YJ, Newman RA, Felix EA, Atkinson EN, Mills GB. The human lipid phosphate phosphatase-3 decreases the growth, survival, and tumorigenesis of ovarian cancer cells: validation of the lysophosphatidic acid signaling cascade as a target for therapy in ovarian cancer. Cancer Res. 2003;63:1073–1082. [PubMed] [Google Scholar]
- [53].Boucharaba A, Serre CM, Guglielmi J, Bordet JC, Clezardin P, Peyruchaud O. The type 1 lysophosphatidic acid receptor is a target for therapy in bone metastases. Proc. Natl. Acad. Sci. U. S. A. 2006;103:9643–9648. doi: 10.1073/pnas.0600979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Barila D, Plateroti M, Nobili F, Onetti Muda A, Xie Y, Morimoto T, Perozzi G. The Dri 42 gene, whose expression is up-regulated during epithelial differentiation, encodes a novel endoplasmic reticulum resident transmembrane protein. J. Biol. Chem. 1996;271:29928–29936. doi: 10.1074/jbc.271.47.29928. [DOI] [PubMed] [Google Scholar]
- [55].Kai M, Wada I, Imai S, Sakane F, Kanoh H. Cloning and characterization of two human isozymes of Mg2+−independent phosphatidic acid phosphatase. J. Biol. Chem. 1997;272:24572–24578. doi: 10.1074/jbc.272.39.24572. [DOI] [PubMed] [Google Scholar]
- [56].Brindley DN. Lipid phosphate phosphatases and related proteins: signaling functions in development, cell division, and cancer. J. Cell. Biochem. 2004;92:900–912. doi: 10.1002/jcb.20126. [DOI] [PubMed] [Google Scholar]
- [57].Pyne S, Kong KC, Darroch PI. Lysophosphatidic acid and sphingosine 1-phosphate biology: the role of lipid phosphate phosphatases. Semin. Cell Dev. Biol. 2004;15:491–501. doi: 10.1016/j.semcdb.2004.05.007. [DOI] [PubMed] [Google Scholar]
- [58].Alderton F, Darroch P, Sambi B, McKie A, Ahmed IS, Pyne N, Pyne S. G-protein-coupled receptor stimulation of the p42/p44 mitogen-activated protein kinase pathway is attenuated by lipid phosphate phosphatases 1, 1a, and 2 in human embryonic kidney 293 cells. J. Biol. Chem. 2001;276:13452–13460. doi: 10.1074/jbc.M006582200. [DOI] [PubMed] [Google Scholar]
- [59].Long JS, Yokoyama K, Tigyi G, Pyne NJ, Pyne S. Lipid phosphate phosphatase-1 regulates lysophosphatidic acidand platelet-derived-growth-factor-induced cell migration. Biochem. J. 2006;394:495–500. doi: 10.1042/BJ20051674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Long JS, Yokoyama K, Tigyi G, Pyne NJ, Pyne S. Lipid phosphate phosphatase-1 regulates lysophosphatidic acid- and platelet-derived-growth-factor-induced cell migration. Biochem. J. 2006;394:495–500. doi: 10.1042/BJ20051674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Long J, Darroch P, Wan KF, Kong KC, Ktistakis N, Pyne NJ, Pyne S. Regulation of cell survival by lipid phosphate phosphatases involves the modulation of intracellular phosphatidic acid and sphingosine 1-phosphate pools. Biochem. J. 2005;391:25–32. doi: 10.1042/BJ20050342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Pilquil C, Dewald J, Cherney A, Gorshkova I, Tigyi G, English D, Natarajan V, Brindley DN. Lipid phosphate phosphatase-1 regulates lysophosphatidate-induced fibroblast migration by controlling phospholipase D2-dependent phosphatidate generation. J. Biol. Chem. 2006;281:38418–38429. doi: 10.1074/jbc.M601670200. [DOI] [PubMed] [Google Scholar]
- [63].Leung DW, Tompkins CK, White T. Molecular cloning of two alternatively spliced forms of human phosphatidic acid phosphatase cDNAs that are differentially expressed in normal and tumor cells. DNA Cell Biol. 1998;17:377–385. doi: 10.1089/dna.1998.17.377. [DOI] [PubMed] [Google Scholar]
- [64].Garcia-Murillas I, Pettitt T, Macdonald E, Okkenhaug H, Georgiev P, Trivedi D, Hassan B, Wakelam M, Raghu P. Lazaro encodes a lipid phosphate phosphohydrolase that regulates phosphatidylinositol turnover during Drosophila phototransduction. Neuron. 2006;49:533–546. doi: 10.1016/j.neuron.2006.02.001. [DOI] [PubMed] [Google Scholar]
- [65].Sciorra VA, Morris AJ. Sequential actions of phospholipase D and phosphatidic acid phosphohydrolase 2b generate diglyceride in mammalian cells. Mol. Biol. Cell. 1999;10:3863–3876. doi: 10.1091/mbc.10.11.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Pyne S, Long JS, Ktistakis NT, Pyne NJ. Lipid phosphate phosphatases and lipid phosphate signalling. Biochem. Soc. Trans. 2005;33:1370–1374. doi: 10.1042/BST0331370. [DOI] [PubMed] [Google Scholar]
- [67].Martin A, Duffy PA, Liossis C, Gomez-Munoz A, O’Brien L, Stone JC, Brindley DN. Increased concentrations of phosphatidate, diacylglycerol and ceramide in ras- and tyrosine kinase (fps)-transformed fibroblasts. Oncogene. 1997;14:1571–1580. doi: 10.1038/sj.onc.1200987. [DOI] [PubMed] [Google Scholar]
- [68].Ghosh S, Strum JC, Sciorra VA, Daniel L, Bell RM. Raf-1 kinase possesses distinct binding domains for phosphatidylserine and phosphatidic acid. Phosphatidic acid regulates the translocation of Raf-1 in 12-O-tetradecanoylphorbol-13-acetate-stimulated Madin–Darby canine kidney cells. J. Biol. Chem. 1996;271:8472–8480. doi: 10.1074/jbc.271.14.8472. [DOI] [PubMed] [Google Scholar]
- [69].Rizzo MA, Shome K, Vasudevan C, Stolz DB, Sung TC, Frohman MA, Watkins SC, Romero G. Phospholipase D and its product, phosphatidic acid, mediate agonist-dependent raf-1 translocation to the plasma membrane and the activation of the mitogen-activated protein kinase pathway. J. Biol. Chem. 1999;274:1131–1139. doi: 10.1074/jbc.274.2.1131. [DOI] [PubMed] [Google Scholar]
- [70].Zhao C, Du G, Skowronek K, Frohman MA, Bar-Sagi D. Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat. Cell Biol. 2007;9:706–712. doi: 10.1038/ncb1594. [DOI] [PubMed] [Google Scholar]
- [71].Delon C, Manifava M, Wood E, Thompson D, Krugmann S, Pyne S, Ktistakis NT. Sphingosine kinase 1 is an intracellular effector of phosphatidic acid. J. Biol. Chem. 2004;279:44763–44774. doi: 10.1074/jbc.M405771200. [DOI] [PubMed] [Google Scholar]
- [72].Buchanan FG, McReynolds M, Couvillon A, Kam Y, Holla VR, Dubois RN, Exton JH. Requirement of phospholipase D1 activity in H-RasV12-induced transformation. Proc. Natl. Acad. Sci. U. S. A. 2005;102:1638–1642. doi: 10.1073/pnas.0406698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Uchida N, Okamura S, Nagamachi Y, Yamashita S. Increased phospholipase D activity in human breast cancer. J. Cancer Res. Clin. Oncol. 1997;123:280–285. doi: 10.1007/BF01208639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Noh DY, Ahn SJ, Lee RA, Park IA, Kim JH, Suh PG, Ryu SH, Lee KH, Han JS. Overexpression of phospholipase D1 in human breast cancer tissues. Cancer Lett. 2000;161:207–214. doi: 10.1016/s0304-3835(00)00612-1. [DOI] [PubMed] [Google Scholar]
- [75].Chen Y, Zheng Y, Foster DA. Phospholipase D confers rapamycin resistance in human breast cancer cells. Oncogene. 2003;22:3937–3942. doi: 10.1038/sj.onc.1206565. [DOI] [PubMed] [Google Scholar]
- [76].Hui L, Zheng Y, Yan Y, Bargonetti J, Foster DA. Mutant p53 in MDA-MB-231 breast cancer cells is stabilized by elevated phospholipase D activity and contributes to survival signals generated by phospholipase D. Oncogene. 2006;25:7305–7310. doi: 10.1038/sj.onc.1209735. [DOI] [PubMed] [Google Scholar]
- [77].Kang DW, Lee JY, Oh DH, Park SY, Woo TM, Kim MK, Park MH, Jang YH, Min do S. Triptolide-induced suppression of phospholipase D expression inhibits proliferation of MDA-MB-231 breast cancer cells. Exp. Mol. Med. 2009;41:678–685. doi: 10.3858/emm.2009.41.9.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Rodrik V, Zheng Y, Harrow F, Chen Y, Foster DA. Survival signals generated by estrogen and phospholipase D in MCF-7 breast cancer cells are dependent on Myc. Mol. Cell. Biol. 2005;25:7917–7925. doi: 10.1128/MCB.25.17.7917-7925.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Yamada Y, Hamajima N, Kato T, Iwata H, Yamamura Y, Shinoda M, Suyama M, Mitsudomi T, Tajima K, Kusakabe S, Yoshida H, Banno Y, Akao Y, Tanaka M, Nozawa Y. Association of a polymorphism of the phospholipase D2 gene with the prevalence of colorectal cancer. J. Mol. Med. (Berl) 2003;81:126–131. doi: 10.1007/s00109-002-0411-x. [DOI] [PubMed] [Google Scholar]
- [80].Merida I, Avila-Flores A. Tumor metabolism: new opportunities for cancer therapy. Clin. Transl. Oncol. 2006;8:711–716. doi: 10.1007/s12094-006-0117-6. [DOI] [PubMed] [Google Scholar]
- [81].Mandala SM. Sphingosine-1-phosphate phosphatases. Prostaglandins Other Lipid Mediat. 2001;64:143–156. doi: 10.1016/s0090-6980(01)00111-3. [DOI] [PubMed] [Google Scholar]
- [82].Pettus BJ, Kitatani K, Chalfant CE, Taha TA, Kawamori T, Bielawski J, Obeid LM, Hannun YA. The coordination of prostaglandin E2 production by sphingosine-1-phosphate and ceramide-1-phosphate. Mol. Pharmacol. 2005;68:330–335. doi: 10.1124/mol.104.008722. [DOI] [PubMed] [Google Scholar]
- [83].Zhao Y, Usatyuk PV, Cummings R, Saatian B, He D, Watkins T, Morris A, Spannhake EW, Brindley DN, Natarajan V. Lipid phosphate phosphatase-1 regulates lysophosphatidic acid-induced calcium release, NF-kappaB activation and interleukin-8 secretion in human bronchial epithelial cells. Biochem. J. 2005;385:493–502. doi: 10.1042/BJ20041160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Morris KE, Schang LM, Brindley DN. Lipid phosphate phosphatase-2 activity regulates S-phase entry of the cell cycle in Rat2 fibroblasts. J. Biol. Chem. 2006;281:9297–9306. doi: 10.1074/jbc.M511710200. [DOI] [PubMed] [Google Scholar]
- [85].Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- [86].Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- [87].Zhang N, Sundberg JP, Gridley T. Mice mutant for Ppap2c, a homolog of the germ cell migration regulator wunen, are viable and fertile. Genesis. 2000;27:137–140. doi: 10.1002/1526-968x(200008)27:4<137::aid-gene10>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- [88].Flanagan JM, Funes JM, Henderson S, Wild L, Carey N, Boshoff C. Genomics screen in transformed stem cells reveals RNASEH2A, PPAP2C, and ADARB1 as putative anticancer drug targets. Mol. Cancer Ther. 2009;8:249–260. doi: 10.1158/1535-7163.MCT-08-0636. [DOI] [PubMed] [Google Scholar]
- [89].Charles AG, Han TY, Liu YY, Hansen N, Giuliano AE, Cabot MC. Taxol-induced ceramide generation and apoptosis in human breast cancer cells. Cancer Chemother. Pharmacol. 2001;47:444–450. doi: 10.1007/s002800000265. [DOI] [PubMed] [Google Scholar]
- [90].Kolesnick R, Fuks Z. Radiation and ceramide-induced apoptosis. Oncogene. 2003;22:5897–5906. doi: 10.1038/sj.onc.1206702. [DOI] [PubMed] [Google Scholar]
- [91].Modrak DE, Gold DV, Goldenberg DM. Sphingolipid targets in cancer therapy. Mol. Cancer Ther. 2006;5:200–208. doi: 10.1158/1535-7163.MCT-05-0420. [DOI] [PubMed] [Google Scholar]
- [92].Martinez R, Navarro R, Lacort M, Ruiz-Sanz JI, Ruiz-Larrea MB. Doxorubicin induces ceramide and diacylglycerol accumulation in rat hepatocytes through independent routes. Toxicol. Lett. 2009;190:86–90. doi: 10.1016/j.toxlet.2009.07.010. [DOI] [PubMed] [Google Scholar]
- [93].Simstein R, Burow M, Parker A, Weldon C, Beckman B. Apoptosis, chemoresistance, and breast cancer: insights from the MCF-7 cell model system. Exp. Biol. Med. (Maywood) 2003;228:995–1003. doi: 10.1177/153537020322800903. [DOI] [PubMed] [Google Scholar]
- [94].Delpy E, Hatem SN, Andrieu N, de Vaumas C, Henaff M, Rucker-Martin C, Jaffrezou JP, Laurent G, Levade T, Mercadier JJ. Doxorubicin induces slow ceramide accumulation and late apoptosis in cultured adult rat ventricular myocytes. Cardiovasc. Res. 1999;43:398–407. doi: 10.1016/s0008-6363(99)00142-x. [DOI] [PubMed] [Google Scholar]
- [95].Zhang J, Alter N, Reed JC, Borner C, Obeid LM, Hannun YA. Bcl-2 interrupts the ceramide-mediated pathway of cell death. Proc. Natl. Acad. Sci. U. S. A. 1996;93:5325–5328. doi: 10.1073/pnas.93.11.5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Parra V, Eisner V, Chiong M, Criollo A, Moraga F, Garcia A, Hartel S, Jaimovich E, Zorzano A, Hidalgo C, Lavandero S. Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovasc. Res. 2008;77:387–397. doi: 10.1093/cvr/cvm029. [DOI] [PubMed] [Google Scholar]
- [97].Shida D, Takabe K, Kapitonov D, Milstien S, Spiegel S. Targeting SphK1 as a new strategy against cancer. Curr. Drug Targets. 2008;9:662–673. doi: 10.2174/138945008785132402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- [99].Huang WC, Chen CL, Lin YS, Lin CF. Apoptotic sphingolipid ceramide in cancer therapy. J Lipids. 2011;2011:565316. doi: 10.1155/2011/565316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Gomez-Munoz A, Martin A, O’Brien L, Brindley DN. Cell-permeable ceramides inhibit the stimulation of DNA synthesis and phospholipase D activity by phosphatidate and lysophosphatidate in rat fibroblasts. J. Biol. Chem. 1994;269:8937–8943. [PubMed] [Google Scholar]
- [101].Gómez-Muñoz A, Waggoner DW, O’Brien L, Brindley DN. Interaction of ceramides, sphingosine, and sphingosine 1-phosphate in regulating DNA synthesis and phospholipase D activity. J. Biol. Chem. 1995;270:26318–26325. doi: 10.1074/jbc.270.44.26318. [DOI] [PubMed] [Google Scholar]
- [102].Shida D, Fang X, Kordula T, Takabe K, Lepine S, Alvarez SE, Milstien S, Spiegel S. Cross-talk between LPA1 and epidermal growth factor receptors mediates up-regulation of sphingosine kinase 1 to promote gastric cancer cell motility and invasion. Cancer Res. 2008;68:6569–6577. doi: 10.1158/0008-5472.CAN-08-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Tabata K, Baba K, Shiraishi A, Ito M, Fujita N. The orphan GPCR GPR87 was deorphanized and shown to be a lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 2007;363:861–866. doi: 10.1016/j.bbrc.2007.09.063. [DOI] [PubMed] [Google Scholar]
- [104].Wetter JA, Revankar C, Hanson BJ. Utilization of the Tango beta-arrestin recruitment technology for cell-based EDG receptor assay development and interrogation. J. Biomol. Screen. 2009;14:1134–1141. doi: 10.1177/1087057109343809. [DOI] [PubMed] [Google Scholar]
- [105].Nonaka Y, Hiramoto T, Fujita N. Identification of endogenous surrogate ligands for human P2Y12 receptors by in silico and in vitro methods. Biochem. Biophys. Res. Commun. 2005;337:281–288. doi: 10.1016/j.bbrc.2005.09.052. [DOI] [PubMed] [Google Scholar]
- [106].Oka S, Ota R, Shima M, Yamashita A, Sugiura T. GPR35 is a novel lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 2010;395:232–237. doi: 10.1016/j.bbrc.2010.03.169. [DOI] [PubMed] [Google Scholar]
- [107].Zhao P, Abood ME. GPR55 and GPR35 and their relationship to cannabinoid and lysophospholipid receptors. Life Sci. 2012 doi: 10.1016/j.lfs.2012.06.039. (Epub ahead of print 24 July) [DOI] [PubMed] [Google Scholar]
- [108].Muller R, Berliner C, Leptin J, Portner D, Bialecki W, Kleuser B, Schumacher U, Milicevic NM. Expression of sphingosine-1-phosphate receptors and lysophosphatidic acid receptors on cultured and xenografted human colon, breast, melanoma, and lung tumor cells. Tumour Biol. 2010;31:341–349. doi: 10.1007/s13277-010-0043-7. [DOI] [PubMed] [Google Scholar]
- [109].Chen M, Towers LN, O’Connor KL. LPA2 (EDG4) mediates Rho-dependent chemotaxis with lower efficacy than LPA1 (EDG2) in breast carcinoma cells. Am. J. Physiol. Cell Physiol. 2007;292:C1927–C1933. doi: 10.1152/ajpcell.00400.2006. [DOI] [PubMed] [Google Scholar]
- [110].Horak CE, Mendoza A, Vega-Valle E, Albaugh M, Graff-Cherry C, McDermott WG, Hua E, Merino MJ, Steinberg SM, Khanna C, Steeg PS. Nm23-H1 suppresses metastasis by inhibiting expression of the lysophosphatidic acid receptor EDG2. Cancer Res. 2007;67:11751–11759. doi: 10.1158/0008-5472.CAN-07-3175. [DOI] [PubMed] [Google Scholar]
- [111].Park SY, Jeong KJ, Panupinthu N, Yu S, Lee J, Han JW, Kim JM, Lee JS, Kang J, Park CG, Mills GB, Lee HY. Lysophosphatidic acid augments human hepatocellular carcinoma cell invasion through LPA1 receptor and MMP-9 expression. Oncogene. 2011;30:1351–1359. doi: 10.1038/onc.2010.517. [DOI] [PubMed] [Google Scholar]
- [112].Funke M, Zhao Z, Xu Y, Chun J, Tager AM. The lysophosphatidic acid receptor LPA1 promotes epithelial cell apoptosis after lung injury. Am. J. Respir. Cell Mol. Biol. 2012;46:355–364. doi: 10.1165/rcmb.2010-0155OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Furui T, LaPushin R, Mao M, Khan H, Watt SR, Watt MA, Lu Y, Fang X, Tsutsui S, Siddik ZH, Bast RC, Mills GB. Overexpression of edg-2/vzg-1 induces apoptosis and anoikis in ovarian cancer cells in a lysophosphatidic acid-independent manner. Clin. Cancer Res. 1999;5:4308–4318. [PubMed] [Google Scholar]
- [114].David M, Wannecq E, Descotes F, Jansen S, Deux B, Ribeiro J, Serre CM, Gres S, Bendriss-Vermare N, Bollen M, Saez S, Aoki J, Saulnier-Blache JS, Clezardin P, Peyruchaud O. Cancer cell expression of autotaxin controls bone metastasis formation in mouse through lysophosphatidic acid-dependent activation of osteoclasts. PLoS One. 2010;5:e9741. doi: 10.1371/journal.pone.0009741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Peyruchaud O, Leblanc R, David M. Pleiotropic activity of lysophosphatidic acid in bone metastasis. Biochim. Biophys. Acta. doi: 10.1016/j.bbalip.2012.06.004. (in press) (Epub ahead of print 2012/07/24) [DOI] [PubMed] [Google Scholar]
- [116].Ishdorj G, Graham BA, Hu X, Chen J, Johnston JB, Fang X, Gibson SB. Lysophosphatidic acid protects cancer cells from histone deacetylase (HDAC) inhibitor-induced apoptosis through activation of HDAC. J. Biol. Chem. 2008;283:16818–16829. doi: 10.1074/jbc.M710177200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Goetzl EJ, Kong Y, Mei B. Lysophosphatidic acid and sphingosine 1-phosphate protection of T cells from apoptosis in association with suppression of Bax. J. Immunol. 1999;162:2049–2056. [PubMed] [Google Scholar]
- [118].Hu YL, Tee MK, Goetzl EJ, Auersperg N, Mills GB, Ferrara N, Jaffe RB. Lysophosphatidic acid induction of vascular endothelial growth factor expression in human ovarian cancer cells. J. Natl. Cancer Inst. 2001;93:762–768. doi: 10.1093/jnci/93.10.762. [DOI] [PubMed] [Google Scholar]
- [119].Huang MC, Lee HY, Yeh CC, Kong Y, Zaloudek CJ, Goetzl EJ. Induction of protein growth factor systems in the ovaries of transgenic mice overexpressing human type 2 lysophosphatidic acid G protein-coupled receptor (LPA2) Oncogene. 2004;23:122–129. doi: 10.1038/sj.onc.1206986. [DOI] [PubMed] [Google Scholar]
- [120].So J, Navari J, Wang FQ, Fishman DA. Lysophosphatidic acid enhances epithelial ovarian carcinoma invasion through the increased expression of interleukin-8. Gynecol. Oncol. 2004;95:314–322. doi: 10.1016/j.ygyno.2004.08.001. [DOI] [PubMed] [Google Scholar]
- [121].So J, Wang FQ, Navari J, Schreher J, Fishman DA. LPA-induced epithelial ovarian cancer (EOC) in vitro invasion and migration are mediated by VEGF receptor-2 (VEGF-R2) Gynecol. Oncol. 2005;97:870–878. doi: 10.1016/j.ygyno.2005.03.004. [DOI] [PubMed] [Google Scholar]
- [122].Zebrowski BK, Liu W, Ramirez K, Akagi Y, Mills GB, Ellis LM. Markedly elevated levels of vascular endothelial growth factor in malignant ascites. Ann. Surg. Oncol. 1999;6:373–378. doi: 10.1007/s10434-999-0373-0. [DOI] [PubMed] [Google Scholar]
- [123].Ptaszynska MM, Pendrak ML, Bandle RW, Stracke ML, Roberts DD. Positive feedback between vascular endothelial growth factor-A and autotaxin in ovarian cancer cells. Mol. Cancer Res. 2008;6:352–363. doi: 10.1158/1541-7786.MCR-07-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Gil OD, Lee C, Ariztia EV, Wang FQ, Smith PJ, Hope JM, Fishman DA. Lysophosphatidic acid (LPA) promotes E-cadherin ectodomain shedding and OVCA429 cell invasion in an uPA-dependent manner. Gynecol. Oncol. 2008;108:361–369. doi: 10.1016/j.ygyno.2007.10.027. [DOI] [PubMed] [Google Scholar]
- [125].Pustilnik TB, Estrella V, Wiener JR, Mao M, Eder A, Watt MA, Bast RC, Jr., Mills GB. Lysophosphatidic acid induces urokinase secretion by ovarian cancer cells. Clin. Cancer Res. 1999;5:3704–3710. [PubMed] [Google Scholar]
- [126].Murthi P, Barker G, Nowell CJ, Rice GE, Baker MS, Kalionis B, Quinn MA. Plasminogen fragmentation and increased production of extracellular matrix-degrading proteinases are associated with serous epithelial ovarian cancer progression. Gynecol. Oncol. 2004;92:80–88. doi: 10.1016/j.ygyno.2003.09.016. [DOI] [PubMed] [Google Scholar]
- [127].Schmalfeldt B, Prechtel D, Harting K, Spathe K, Rutke S, Konik E, Fridman R, Berger U, Schmitt M, Kuhn W, Lengyel E. Increased expression of matrix metalloproteinases (MMP)-2, MMP-9, and the urokinase-type plasminogen activator is associated with progression from benign to advanced ovarian cancer. Clin. Cancer Res. 2001;7:2396–2404. [PubMed] [Google Scholar]
- [128].Altman MK, Gopal V, Jia W, Yu S, Hall H, Mills GB, McGinnis AC, Bartlett MG, Jiang G, Madan D, Prestwich GD, Xu Y, Davies MA, Murph MM. Targeting melanoma growth and viability reveals dualistic functionality of the phosphonothionate analogue of carba cyclic phosphatidic acid. Mol. Cancer. 2010;9:140. doi: 10.1186/1476-4598-9-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Okabe K, Hayashi M, Kato K, Okumura M, Fukui R, Honoki K, Fukushima N, Tsujiuchi T. Lysophosphatidic acid receptor-3 increases tumorigenicity and aggressiveness of rat hepatoma RH7777 cells. Mol. Carcinog. 2011 doi: 10.1002/mc.21851. http://dx.doi.org/10.1002/mc.21851 (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- [130].Okabe K, Hayashi M, Yamawaki Y, Teranishi M, Honoki K, Mori T, Fukushima N, Tsujiuchi T. Possible involvement of lysophosphatidic acid receptor-5 gene in the acquisition of growth advantage of rat tumor cells. Mol. Carcinog. 2011;50:635–642. doi: 10.1002/mc.20750. [DOI] [PubMed] [Google Scholar]
- [131].Hayashi M, Okabe K, Kato K, Okumura M, Fukui R, Fukushima N, Tsujiuchi T. Differential function of lysophosphatidic acid receptors in cell proliferation and migration of neuroblastoma cells. Cancer Lett. 2012;316:91–96. doi: 10.1016/j.canlet.2011.10.030. [DOI] [PubMed] [Google Scholar]
- [132].Lee Z, Cheng CT, Zhang H, Subler MA, Wu J, Mukherjee A, Windle JJ, Chen CK, Fang X. Role of LPA4/p2y9/GPR23 in negative regulation of cell motility. Mol. Biol. Cell. 2008;19:5435–5445. doi: 10.1091/mbc.E08-03-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Sumida H, Noguchi K, Kihara Y, Abe M, Yanagida K, Hamano F, Sato S, Tamaki K, Morishita Y, Kano MR, Iwata C, Miyazono K, Sakimura K, Shimizu T, Ishii S. LPA4 regulates blood and lymphatic vessel formation during mouse embryogenesis. Blood. 2010;116:5060–5070. doi: 10.1182/blood-2010-03-272443. [DOI] [PubMed] [Google Scholar]
- [134].Jongsma M, Matas-Rico E, Rzadkowski A, Jalink K, Moolenaar WH. LPA is a chemorepellent for B16 melanoma cells: action through the cAMP-elevating LPA5 receptor. PLoS One. 2011;6:e29260. doi: 10.1371/journal.pone.0029260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Glatt S, Halbauer D, Heindl S, Wernitznig A, Kozina D, Su KC, Puri C, Garin-Chesa P, Sommergruber W. hGPR87 contributes to viability of human tumor cells. Int. J. Cancer. 2008;122:2008–2016. doi: 10.1002/ijc.23349. [DOI] [PubMed] [Google Scholar]
- [136].Gugger M, White R, Song S, Waser B, Cescato R, Riviere P, Reubi JC. GPR87 is an overexpressed G-protein coupled receptor in squamous cell carcinoma of the lung. Dis. Markers. 2008;24:41–50. doi: 10.1155/2008/857474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Zhang Y, Qian Y, Lu W, Chen X. The G protein-coupled receptor 87 is necessary for p53-dependent cell survival in response to genotoxic stress. Cancer Res. 2009;69:6049–6056. doi: 10.1158/0008-5472.CAN-09-0621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Pasternack SM, von Kugelgen I, Aboud KA, Lee YA, Ruschendorf F, Voss K, Hillmer AM, Molderings GJ, Franz T, Ramirez A, Nurnberg P, Nothen MM, Betz RC. G protein-coupled receptor P2Y5 and its ligand LPA are involved in maintenance of human hair growth. Nat. Genet. 2008;40:329–334. doi: 10.1038/ng.84. [DOI] [PubMed] [Google Scholar]
- [139].Shimomura Y, Wajid M, Ishii Y, Shapiro L, Petukhova L, Gordon D, Christiano AM. Disruption of P2RY5, an orphan G protein-coupled receptor, underlies autosomal recessive woolly hair. Nat. Genet. 2008;40:335–339. doi: 10.1038/ng.100. [DOI] [PubMed] [Google Scholar]
- [140].Liu S, Umezu-Goto M, Murph M, Lu Y, Liu W, Zhang F, Yu S, Stephens LC, Cui X, Murrow G, Coombes K, Muller W, Hung MC, Perou CM, Lee AV, Fang X, Mills GB. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell. 2009;15:539–550. doi: 10.1016/j.ccr.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Vidot S, Witham J, Agarwal R, Greenhough S, Bamrah HS, Tigyi GJ, Kaye SB, Richardson A. Autotaxin delays apoptosis induced by carboplatin in ovarian cancer cells. Cell. Signal. 2010;22:926–935. doi: 10.1016/j.cellsig.2010.01.017. [DOI] [PubMed] [Google Scholar]
- [142].Zhang R, Wang J, Ma S, Huang Z, Zhang G. Requirement of Osteopontin in the migration and protection against Taxol-induced apoptosis via the ATX–LPA axis in SGC7901 cells. BMC Cell Biol. 2011;12:11. doi: 10.1186/1471-2121-12-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Sun Y, Nam JS, Han DH, Kim NH, Choi HK, Lee JK, Rhee HJ, Huh SO. Lysophosphatidic acid induces upregulation of Mcl-1 and protects apoptosis in a PTX-dependent manner in H19-7 cells. Cell. Signal. 2010;22:484–494. doi: 10.1016/j.cellsig.2009.11.002. [DOI] [PubMed] [Google Scholar]
- [144].Yu S, Murph MM, Lu Y, Liu S, Hall HS, Liu J, Stephens C, Fang X, Mills GB. Lysophosphatidic acid receptors determine tumorigenicity and aggressiveness of ovarian cancer cells. J. Natl. Cancer Inst. 2008;100:1630–1642. doi: 10.1093/jnci/djn378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Deng W, Balazs L, Wang DA, Van Middlesworth L, Tigyi G, Johnson LR. Lysophosphatidic acid protects and rescues intestinal epithelial cells from radiation- and chemotherapy-induced apoptosis. Gastroenterology. 2002;123:206–216. doi: 10.1053/gast.2002.34209. [DOI] [PubMed] [Google Scholar]
- [146].Mukherjee A, Wu J, Barbour S, Fang X. Lysophosphatidic acid activates lipogenic pathways and de novo lipid synthesis in ovarian cancer cells. J. Biol. Chem. 2012;287(30):24990–5000. doi: 10.1074/jbc.M112.340083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Morita Y, Perez GI, Paris F, Miranda SR, Ehleiter D, Haimovitz-Friedman A, Fuks Z, Xie Z, Reed JC, Schuchman EH, Kolesnick RN, Tilly JL. Oocyte apoptosis is suppressed by disruption of the acid sphingomyelinase gene or by sphingosine-1-phosphate therapy. Nat. Med. 2000;6:1109–1114. doi: 10.1038/80442. [DOI] [PubMed] [Google Scholar]
- [148].Pena LA, Fuks Z, Kolesnick RN. Radiation-induced apoptosis of endothelial cells in the murine central nervous system: protection by fibroblast growth factor and sphingomyelinase deficiency. Cancer Res. 2000;60:321–327. [PubMed] [Google Scholar]
- [149].Santana P, Pena LA, Haimovitz-Friedman A, Martin S, Green D, McLoughlin M, Cordon-Cardo C, Schuchman EH, Fuks Z, Kolesnick R. Acid sphingomyelinase-deficient human lymphoblasts and mice are defective in radiation-induced apoptosis. Cell. 1996;86:189–199. doi: 10.1016/s0092-8674(00)80091-4. [DOI] [PubMed] [Google Scholar]
- [150].Gueguen G, Granci V, Rogalle P, Briand-Mesange F, Wilson M, Klaebe A, Terce F, Chap H, Salles JP, Simon MF, Gaits F. A lysophosphatidic acid analogue is revealed as a potent inhibitor of phosphatidylcholine synthesis, inducing apoptosis. Biochem. J. 2002;368:447–459. doi: 10.1042/BJ20020273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Deng W, Wang DA, Gosmanova E, Johnson LR, Tigyi G. LPA protects intestinal epithelial cells from apoptosis by inhibiting the mitochondrial pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2003;284:G821–G829. doi: 10.1152/ajpgi.00406.2002. [DOI] [PubMed] [Google Scholar]
- [152].Deng W, Poppleton H, Yasuda S, Makarova N, Shinozuka Y, Wang DA, Johnson LR, Patel TB, Tigyi G. Optimal lysophosphatidic acid-induced DNA synthesis and cell migration but not survival require intact autophosphorylation sites of the epidermal growth factor receptor. J. Biol. Chem. 2004;279:47871–47880. doi: 10.1074/jbc.M405443200. [DOI] [PubMed] [Google Scholar]
- [153].Durgam GG, Tsukahara R, Makarova N, Walker MD, Fujiwara Y, Pigg KR, Baker DL, Sardar VM, Parrill AL, Tigyi G, Miller DD. Synthesis and pharmacological evaluation of second-generation phosphatidic acid derivatives as lysophosphatidic acid receptor ligands. Bioorg. Med. Chem. Lett. 2006;16:633–640. doi: 10.1016/j.bmcl.2005.10.031. [DOI] [PubMed] [Google Scholar]
- [154].Virag T, Elrod DB, Liliom K, Sardar VM, Parrill AL, Yokoyama K, Durgam G, Deng W, Miller DD, Tigyi G. Fatty alcohol phosphates are subtype-selective agonists and antagonists of LPA receptors. Mol. Pharmacol. 2003;63:1032–1042. doi: 10.1124/mol.63.5.1032. [DOI] [PubMed] [Google Scholar]
- [155].Kosanam H, Ma F, He H, Ramagiri S, Gududuru V, Tigyi GJ, Van Rompay K, Miller DD, Yates CR. Development of an LC-MS/MS assay to determine plasma pharmacokinetics of the radioprotectant octadecyl thiophosphate (OTP) in monkeys. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2010;878:2379–2383. doi: 10.1016/j.jchromb.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Deng W, Shuyu E, Tsukahara R, Valentine WJ, Durgam G, Gududuru V, Balazs L, Manickam V, Arsura M, VanMiddlesworth L, Johnson LR, Parrill AL, Miller DD, Tigyi G. The lysophosphatidic acid type 2 receptor is required for protection against radiation-induced intestinal injury. Gastroenterology. 2007;132:1834–1851. doi: 10.1053/j.gastro.2007.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Kiss GN, Lee S-C, Fells JI, Liu J, Valentine WJ, Fujiwara Y, Emmons-Thompson K, Yates CR, Sümegi B, Tigyi G. Mitigation of radiation injury by selective stimulation of the LPA2 receptor. Biochim. Biophys. Acta. 2013;1831:117–125. doi: 10.1016/j.bbalip.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Li C, Dandridge KS, Di A, Marrs KL, Harris EL, Roy K, Jackson JS, Makarova NV, Fujiwara Y, Farrar PL, Nelson DJ, Tigyi GJ, Naren AP. Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhea through CFTR-dependent protein interactions. J. Exp. Med. 2005;202:975–986. doi: 10.1084/jem.20050421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Lin FT, Lai YJ, Makarova N, Tigyi G, Lin WC. The lysophosphatidic acid 2 receptor mediates down-regulation of Siva-1 to promote cell survival. J. Biol. Chem. 2007;282:37759–37769. doi: 10.1074/jbc.M705025200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Xue L, Chu F, Cheng Y, Sun X, Borthakur A, Ramarao M, Pandey P, Wu M, Schlossman SF, Prasad KV. Siva-1 binds to and inhibits BCL-X(L)-mediated protection against UV radiation-induced apoptosis. Proc. Natl. Acad. Sci. U. S. A. 2002;99:6925–6930. doi: 10.1073/pnas.102182299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Chu F, Barkinge J, Hawkins S, Gudi R, Salgia R, Kanteti PV. Expression of Siva-1 protein or its putative amphipathic helical region enhances cisplatin-induced apoptosis in breast cancer cells: effect of elevated levels of BCL-2. Cancer Res. 2005;65:5301–5309. doi: 10.1158/0008-5472.CAN-04-3270. [DOI] [PubMed] [Google Scholar]
- [162].Shuyu E, Lai YJ, Tsukahara R, Chen CS, Fujiwara Y, Yue J, Yu JH, Guo H, Kihara A, Tigyi G, Lin FT. Lysophosphatidic acid 2 receptor-mediated supramolecular complex formation regulates its antiapoptotic effect. J. Biol. Chem. 2009;284:14558–14571. doi: 10.1074/jbc.M900185200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Lai YJ, Lin VT, Zheng Y, Benveniste EN, Lin FT. The adaptor protein TRIP6 antagonizes Fas-induced apoptosis but promotes its effect on cell migration. Mol. Cell. Biol. 2010;30:5582–5596. doi: 10.1128/MCB.00134-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Kiss GN, J.I. F, Gupte R, Lee S-C, Liu J, Nusser N, Ray RM, Lin F-T, Parrill AL, Sümegi B, Miller DD, Tigyi GJ. Virtual screening for LPA2-specific agonists reveals nonlipid compounds with antiapoptotic actions. Mol. Pharm. doi: 10.1124/mol.112.079699. (submitted for publication) [DOI] [PMC free article] [PubMed] [Google Scholar]