

Abstract
Disruption of the blood-brain barrier (BBB) and edema formation play a key role in the development of neurological dysfunction in acute and chronic cerebral ischemia. Animal studies have revealed the molecular cascades that are initiated with hypoxia/ischemia in the cells forming the neurovascular unit (NVU) and that contribute to cell death. Matrix metalloproteinases (MMPs) cause reversible degradation of tight junction proteins (TJPs) early after the onset of ischemia and a delayed secondary opening during a neuroinflammatory response occurring from 24 to 72 hrs. Cyclooxgyenases (COX) are important in the delayed opening as the neuroinflammatory response progresses. An early opening of the BBB within the three-hour therapeutic window for tissue plasminogen activator (tPA) can allow it to enter the brain and increase the risk of hemorrhage. Chronic hypoxic hypoperfusion opens the BBB, which contributes to the cognitive changes seen with lacunar strokes and white matter injury in subcortical ischemic vascular disease (SIVD). This review will describe the molecular and cellular events associated with BBB disruption and potential therapies directed toward restoring the integrity of the NVU.
Introduction
Molecular biology methods and advances in magnetic resonance imaging (MRI) have provided novel insights into the cerebral vasculature in acute and chronic stroke. Protection of the neuronal microenvironment is provided by the neuro(glio)vascular unit (NVU) that includes endothelial cells, astrocytes, pericytes, neurons and extracellular matrix around the vessels.1 Tight junction proteins (TJPs) form the first defence in the endothelial barrier disruption of which leads to vasogenic edema and cell death. In stroke the ischemic injury induces a molecular cascade that culminates in the formation of toxic proteases and free radicals, which participate in the damage to the tissue and the removal of dead cells. However, the same molecules that are damaging in the early stages of the injury perform essential functions in the recovery phase, drastically complicating the therapeutic use of these agents. The best studied of these dual functioning molecules are the matrix metalloproteinases (MMPs), cyclooxygenases (COX), and free radicals, nitric oxide (NO) and reactive oxygen species (ROS).
Biology of Tight Junctions and the Neurovascular Unit
Tight junction proteins form the initial barrier at the endothelial cells between blood and brain cells. Major TJPs are occludin and claudins close to the blood and junctional adhesion molecules (JAM) deeper in the endothelial cell clefts.2 Surrounding the abluminal surface of the endothelial cell is a basal lamina composed mainly of type IV collagen, fibronectin, heparan sulfate, and laminin, which functions as a charge and molecular weight barrier and interacts in complex ways with integrins to regulate permeability and cellular transport across the BBB.3 Embedded in the basal lamina are the pericytes, which are hybrid cells with both macrophage and smooth muscle properties.4 Astrocytes are generally thought to be an essential cell for tight junction formation. However, a recent study in embryos challenges this concept by showing that the BBB qualities of the capillary form when the endothelial cells invade the central nervous system and pericytes are recruited to the developing vessels; this occurs over a week before astrocyte generation, suggesting that pericytes are critical for tight junction formation.5 Another interesting study of pericytes showed that they decrease with age, paralleling an increase in BBB permeability.6
Disruption of the BBB in Ischemia/Hypoxia and Hemorrhage
Proteases, which are the final common pathway for disruption of the NVU, are normally present in a latent form. Some proteases are constitutively expressed and participate in normal processes. This includes 72-kDa gelatinase A (MMP-2), which is normally found in cerebrospinal fluid and in astrocytes. Activation of the latent MMP-2 to the 62-64kDa forms requires the formation of a trimolecular complex with MMP-2, tissue inhibitor of metalloproteinases-2 (TIMP-2) and membrane-type 1 metalloproteinase (MT1-MMP), which constrains the activity of MMP-2 to the vicinity of the membrane.
Another group of proteases are induced during an injury. The major inducible MMPs are stromelysin-1 (MMP-3) and 92-kDa gelatinase B (MMP-9). When these are released they are unrestrained and act on multiple substrates in the extracellular matrix (Figure 1). Cytokines induce the expression of MMP-3 and -9 through the action of nuclear-factor-κB (NF-κB) and the activator protein-1 (AP-1) gene transcription sites. Another source of MMPs is through the influx of white blood cells, such as neutrophils, which can release activated 84-kDa MMP-9. In reperfusion injury, proteases participate in the biphasic opening of the BBB. An initial reversible phase related to activation of latent MMP-2 precedes a later phase at 24 to 48 hrs associated with the induction of MMP-3 and MMP-9 (Figure 2).
Fig. 1.

Mechanism for MMP-mediated BBB disruption in hypoxia/ischemia. Induction of hypoxia inducible factor-α (HIF-α) by loss of oxygen and ATP leads to activation of MMP-2. The constitutive enzyme, proMT1-MMP, is activated by the convertase, Furin, and it activates proMMP-2. Secondary neuroinflammation with the formation of cytokines (TNF-α and IL-1β) and induction of MMP-9 and -3 occurs. ProMMP-9 is activated by MMP-3 and free radicals. Active MMPs degrade the basal lamina and tight junctions of endothelial cells, thereby opening the blood–brain barrier (BBB). Opening of the BBB leads to vasogenic edema.
Fig. 2.

Schematic drawing to show the theoretical mechanisms leading to the initial reversible opening of the BBB and the later more slowly irreversible opening. Reperfusion injury leads to a biphasic opening of the BBB. The early opening occurs several hours after the onset of reperfusion due to activation of the constitutive enzyme gelatinase A (MMP-2). This initial opening is transient and reversible. At 24 to 72 hours later, the inflammatory response leads to the induction of MMP-3 and MMP-9, which induce more intense and irreversible damage to the blood vessel.
Cyclooxygenases (COX) are another important family of inflammatory enzymes. COX-1 is a constitutive enzyme, while COX-2 is inducible and contributes to BBB damage as part of a secondary inflammatory response from 24 to 72 hrs after the initial insult.7 Both MMP and COX-2 inhibitors protect the BBB after an ischemic injury; MMP inhibitors act during the early phase, and COX-2 inhibitors block the secondary opening of the BBB, suggesting that a combination of both agents may be able to better control BBB disruption.
Tissue plasminogen activator (tPA) breaks fibrin clots and restores blood flow. In a small percentage of patients the tPA leads to a life-threatening hemorrhage. Since the BBB is disrupted early in the injury cascade, a possible mechanism of bleeding is that the tPA crosses the BBB. When tPA enters the brain rather than remaining in the circulation, it induces cytokines and activates MMP-9.8 MMP inhibitors prevent the early opening of the BBB and can reduce the risk of hemorrhage and death when tPA is given in stroke.9 Low-density lipoprotein receptor-related protein (LRP) is a serine protease that is a member of the low-density lipoprotein (LDL) receptor gene family. LRP binds tPA and participates in the action of tPA.10, 11
Aquaporins are pore-forming molecules that facilitate the passage of water molecules across the BBB. Deletion of AQP4 reduces edema in models in which cytotoxic edema is the pathophysiological mechanism. However, in conditions in which vasogenic edema is significant, AQP4 deletion exacerbated brain edema, since AQP4 functions as a passive pore, allowing water to follow pressure gradients to remove extracellular fluid and resolve vasogenic edema.12
Oxidative stress damages endothelial cells of the BBB and contributes to vasogenic edema. The superoxide radical (O2-.) has been identified as the primary reactive oxygen species (ROS) involved in increased vascular permeability and edema formation in global and focal cerebral ischemia, cold brain injury and brain tumors. Scavenging O2-. radicals using recombinant superoxide dismutase (SOD) or polyethylene glycol-SOD reduces ischemia-induced BBB injury and vasogenic edema. SOD1-overexpressing mice also have reduced activation of MMP-9 by ROS, which may be involved in early BBB disruption and progressive striatal damage induced by the mitochondrial excitotoxin, 3-nitropropionic acid.13
Normobaric hyperoxia in the early stages of stroke reduces the disruption of the BBB in reperfusion injury.14 The increased oxygen acts on the cells in the penumbra. When hyperoxia is used with tPA, there is reduction in the hemorrhagic complications from tPA, probably due to the protection of the BBB. Rats treated with combined normobaric hyperoxia and tPA showed significantly reduced tPA-associated mortality, brain edema, hemorrhage, and MMP-9 induction as compared with tPA alone.15
Blood-Brain Barrier Studies in Humans
In the early stages of an infarct in humans, blood vessels have increased permeability, which can be seen in the meninges over the area of the stroke. Gadolinium enhancement on fluid-attenuated inversion recovery (FLAIR) images obtained several hours after injection showed disruption of the BBB, which appeared as enhancement in sulci over the infarct area. This was termed, hyperintense acute reperfusion marker (HARM), and was found in one-third of ischemic stroke patients; those with the sign had a higher risk of hemorrhagic transformation and worse clinical outcome (Figure 3).16
Fig. 3.
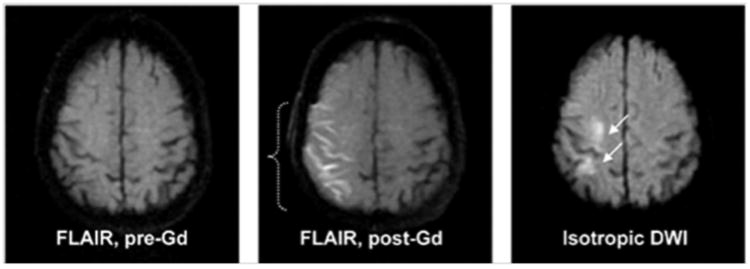
Evidence of HARM for a representative acute stroke patient with early blood–brain barrier (BBB) disruption (woman, 81 years old, baseline NIHSS 17, no tPA). Left: FLAIR pre-Gd contrast, Middle: FLAIR post-Gd contrast, Left: isotropic DWI (b =1000). FLAIR enhancement was never seen before Gd contrast. After Gd contrast, FLAIR images were positive for HARM. (permission pending)16
Growth of intracerebral hemorrhage (ICH) occurs in the first 24 hours.17 Treatment with factor VII, which promotes clotting, in an early study reduced the growth, but a second study failed to show an effect, and there is no proven treatment to reduce lesion growth in ICH.18 The extent of hemorrhagic transformation after stroke correlated with MMP-9 elevation in the blood.19
Blood-Brain Barrier Dysfunction in Chronic Cerebrovascular Disease
Blood-brain barrier abnormalities have been described in patients with small vessel disease secondary to hypertension and diabetes. CSF studies have documented increased albumin, which is an indicator of a disrupted BBB.20 Dynamic contrastenhanced MRI (DCEMRI) provides a quantitative measurement of permeability.21 Patients with large white matter hyperintensities and symptoms suggestive of subcortical ischemic vascular disease (SIVD) had increased BBB permeability as measured by DCEMRI, which paralleled changes in CSF albumin index (Figure 4).22
Fig. 4.

Two subcortical ischemic vascular disease (SIVD) patients with white matter hyperintensities (WMHs). A) FLAIR MRI shows WMHs in the centrum semiovale (arrowhead) without involvement of the cortex. B) The corresponding permeability map has regions of moderately increased permeability (light blue) and high permeability (red). C) FLAIR image from another SIVD patient with larger white matter lesions (arrowhead). D) Permeability map shows increased permeability is limited to two small regions within the WMHs.
Blood vessels undergo profound changes with aging. Tortuosity of the vessels and thickening of the vessel walls is found at autopsy in elderly patients, which results in reduced vascular reactivity in the regions of the white matter with hyperintensities on MRI.23 Hypertension, diabetes and hyperlipidemia are the major factors besides age that cause changes in the blood vessels responsible for the impairments in cerebral blood flow and oxygenation along with increase in permeability.24, 25 The vulnerability of the white matter is due to multiple factors since it is the end of the arterial circulation to the brain.26 Large epidemiological studies utilizing MRI and cognitive testing show a high incidence of changes in the white matter after age 65 associated with cognitive decline.27 There is a gradual growth in the white matter lesions over time.28
Autopsy studies show the presence of hypoxia inducible factor-1α in the affected white matter supporting a role of hypoxia in the process that leads to death of the oligodendrocytes and extensive gliosis.29 Several etiologies are proposed to explain the changes in the white matter. Silent strokes are suspected in many cases, and may be the initiating event particularly when hypertension is damaging the blood vessels.30 An alternative mechanism is disruption of the NVU with vasogenic edema. Support of a disturbed BBB comes from autopsy studies of patients with Binswanger's disease that show the presence of serum proteins in the brain.31 A possible cause of the BBB damage is the presence of MMPs in the white matter at autopsy in patients with vascular dementia.32 Reactive astrocytes that are positive for glial fibrillary acidic protein (GFAP) are immunoreative for MMP-2, while microglia/macrophages stain for MMP-3.
Mechanisms to explain the inflammatory process have been revealed by animal studies. Rats with hypoxic hypoperfusion induced by bilateral carotid artery occlusion (BCAO) have increased MMP-2 in the white matter and have chronic gliosis; mmp-2 knockout mice had less disruption of the BBB and damage to the white matter.33
Translational Aspects of Blood-Brain Barrier Disruption in Stroke
MMP inhibitors (MMPIs) block the early opening of the BBB and COX-2 inhibitors are effective in the delayed phase. Several MMPIs have been tested and shown to have efficacy in animals, including BB-94, BB-1101, and GM6001 along with selective COX-2 inhibitors.34, 35 Joint problems occurred when MMPIs were given chronically to cancer patient,36 but short-term use of MMPIs may avoid side-effects.37
Minocycline is an inhibitor of MMPs with multiple actions, including antiinflammatory efects, and is well tolerated in low doses over long periods of time.38 Treatment with minocycline benefits patients with multiple sclerosis, most likely through protection of the BBB.39 Another potential treatment is the use of inhibitors of COX-2, which block the secondary opening of the BBB in experimental stroke. Concern about the cardiac side effects, which were seen with long-term use of these agents in arthritis, is less likely to be present in the short-term use.
Contrary to their deleterious actions in the early stages of an acute stroke, MMPs may aid in the recovery process. Post-stroke angiogenesis is the key step for recovery after ischemia and provide the critical neurovascular substrates for neuronal remodeling after stroke.40 In the process of angiogenesis, loss of vascular integrity and degradation of cell matrix are crucial initiating steps. MMPs degrade the extracellular matrix and prepare the stage for growth factors and guidance molecules. MMPs can also cleave many signaling molecules, such as vascular endothelial growth factor (VEGF), that induces the proliferation of endothelial cells, and promotes their release from matrixbound compartments or cell surface.41 Recent studies have shown that long-term inhibition of MMPs decreased migration of neuroblast cells from the subventricular zone, reduced neuronal plasticity and the number of newly formed vessels, leading to an increase in tissue damage in peri-infarct cortex.42
An important aspect of the design of treatment protocols for acute and chronic stroke is the dual nature of the molecules targeted for treatment. Many of those molecules that participate in the death of cells in the early stages of the injury also play a critical role in the recovery period. An example is the benefit derived from treatment with MMPIs in the early stages of injury may be lost if the same enzymes are blocked in the later stages when they are used in angiogensis and neurogenesis.43 Understanding the timing of the expression of each of the agents to be blocked and their actions at each point of the injury cycle is necessary in planning the use of inhibitors. In the future, as the multiple cascades are better understood, treatments will be tailored to start when the damaging effects of that agent are maximal and to be stopped as the beneficial effects are beginning.
In conclusion, we have described some of the major advances in understanding the function of the NVU at the molecular level. Unraveling the proteins that comprise the tight junctions provided tools to observe the effects of proteases, such as the MMPs, on the tight junctions following acute stroke. Agents that block the BBB disruption can protect the brain from the adverse effects of tPA, extending the therapeutic window. The combined effect of agents that act early, including MMPIs and those that protect the delayed opening of the BBB, such as the COX-2 inhibitors, need to be tested. The recent studies of chronic effects of hypoperfusion in humans and animals demonstrate a role for MMPs in both the disruption of the BBB and the breakdown of myelin, which may contribute to the death of oligodendrocytes. Defining the molecular mechanisms underlying damage to the vasculature provides important information on which to base further trials of novel therapies to protect the BBB.
Acknowledgments
Funding: Studies supported by grants from the American Heart Association Beginning Grant-in-Aids (0765473Z and 10BGIA4310034) to YY, and from the NIH (R01NS052305, R01NS045847, R21NS066418) to GAR.
Footnotes
Disclosures: No conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- 2.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. PharmacolRev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 3.Milner R, Hung S, Wang X, Berg GI, Spatz M, del Zoppo GJ. Responses of endothelial cell and astrocyte matrix-integrin receptors to ischemia mimic those observed in the neurovascular unit. Stroke. 2008;39:191–197. doi: 10.1161/STROKEAHA.107.486134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dore-Duffy P. Pericytes: Pluripotent cells of the blood brain barrier. Curr Pharm Des. 2008;14:1581–1593. doi: 10.2174/138161208784705469. [DOI] [PubMed] [Google Scholar]
- 5.Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Candelario-Jalil E, Gonzalez-Falcon A, Garcia-Cabrera M, Leon OS, Fiebich BL. Post-ischaemic treatment with the cyclooxygenase-2 inhibitor nimesulide reduces blood-brain barrier disruption and leukocyte infiltration following transient focal cerebral ischaemia in rats. J Neurochem. 2007;100:1108–1120. doi: 10.1111/j.1471-4159.2006.04280.x. [DOI] [PubMed] [Google Scholar]
- 8.Tsuji K, Aoki T, Tejima E, Arai K, Lee SR, Atochin DN, et al. Tissue plasminogen activator promotes matrix metalloproteinase-9 upregulation after focal cerebral ischemia. Stroke. 2005;36:1954–1959. doi: 10.1161/01.STR.0000177517.01203.eb. [DOI] [PubMed] [Google Scholar]
- 9.Lapchak PA, Chapman DF, Zivin JA. Metalloproteinase inhibition reduces thrombolytic (tissue plasminogen activator)-induced hemorrhage after thromboembolic stroke. Stroke. 2000;31:3034–3040. doi: 10.1161/01.str.31.12.3034. [DOI] [PubMed] [Google Scholar]
- 10.Wang X, Lee SR, Arai K, Lee SR, Tsuji K, Rebeck GW, et al. Lipoprotein receptor-mediated induction of matrix metalloproteinase by tissue plasminogen activator. NatMed. 2003;9:1313–1317. doi: 10.1038/nm926. [DOI] [PubMed] [Google Scholar]
- 11.Yepes M, Sandkvist M, Moore EG, Bugge TH, Strickland DK, Lawrence DA. Tissue-type plasminogen activator induces opening of the blood-brain barrier via the ldl receptor-related protein. JClinInvest. 2003;112:1533–1540. doi: 10.1172/JCI19212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zador Z, Bloch O, Yao X, Manley GT. Aquaporins: Role in cerebral edema and brain water balance. Prog Brain Res. 2007;161:185–194. doi: 10.1016/S0079-6123(06)61012-1. [DOI] [PubMed] [Google Scholar]
- 13.Kim GW, Gasche Y, Grzeschik S, Copin JC, Maier CM, Chan PH. Neurodegeneration in striatum induced by the mitochondrial toxin 3-nitropropionic acid: Role of matrix metalloproteinase-9 in early blood-brain barrier disruption? J Neurosci. 2003;23:8733–8742. doi: 10.1523/JNEUROSCI.23-25-08733.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim HY, Singhal AB, Lo EH. Normobaric hyperoxia extends the reperfusion window in focal cerebral ischemia. AnnNeurol. 2005;57:571–575. doi: 10.1002/ana.20430. [DOI] [PubMed] [Google Scholar]
- 15.Liu W, Hendren J, Qin XJ, Liu KJ. Normobaric hyperoxia reduces the neurovascular complications associated with delayed tissue plasminogen activator treatment in a rat model of focal cerebral ischemia. Stroke. 2009;40:2526–2531. doi: 10.1161/STROKEAHA.108.545483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henning EC, Latour LL, Warach S. Verification of enhancement of the csf space, not parenchyma, in acute stroke patients with early blood-brain barrier disruption. J Cereb Blood Flow Metab. 2008;28:882–886. doi: 10.1038/sj.jcbfm.9600598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brott T, Broderick J, Kothari R, Barsan W, Tomsick T, Sauerbeck L, et al. Early hemorrhage growth in patients with intracerebral hemorrhage. Stroke. 1997;28:1–5. doi: 10.1161/01.str.28.1.1. [DOI] [PubMed] [Google Scholar]
- 18.Mayer SA, Brun NC, Begtrup K, Broderick J, Davis S, Diringer MN, et al. Efficacy and safety of recombinant activated factor vii for acute intracerebral hemorrhage. N Engl J Med. 2008;358:2127–2137. doi: 10.1056/NEJMoa0707534. [DOI] [PubMed] [Google Scholar]
- 19.Montaner J, Alvarez-Sabin J, Molina CA, Angles A, Abilleira S, Arenillas J, et al. Matrix metalloproteinase expression is related to hemorrhagic transformation after cardioembolic stroke. Stroke. 2001;32:2762–2767. doi: 10.1161/hs1201.99512. [DOI] [PubMed] [Google Scholar]
- 20.Skoog I, Wallin A, Fredman P, Hesse C, Aevarsson O, Karlsson I, et al. A population study on blood-brain barrier function in 85-year- olds: Relation to alzheimer's disease and vascular dementia. Neurology. 1998;50:966–971. doi: 10.1212/wnl.50.4.966. [DOI] [PubMed] [Google Scholar]
- 21.Ewing JR, Knight RA, Nagaraja TN, Yee JS, Nagesh V, Whitton PA, et al. Patlak plots of gd-dtpa mri data yield blood-brain transfer constants concordant with those of 14C-sucrose in areas of blood-brain opening. Magn ResonMed. 2003;50:283–292. doi: 10.1002/mrm.10524. [DOI] [PubMed] [Google Scholar]
- 22.Taheri S, Gasparovic C, Huisa BN, Adair JC, Edmonds C, Prestopnik J, Grossetete M, Shah NJ, Wills J, Qualls C, Rosenberg GA. Blood-brain barrier permeability abnormalities in vascular cognitive impairment. Stroke. 2011 doi: 10.1161/STROKEAHA.110.611731. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marstrand JR, Garde E, Rostrup E, Ring P, Rosenbaum S, Mortensen EL. Cerebral perfusion and cerebrovascular reactivity are reduced in white matter hyperintensities. Stroke. 2002;33:972–976. doi: 10.1161/01.str.0000012808.81667.4b. [DOI] [PubMed] [Google Scholar]
- 24.Topakian R, Barrick TR, Howe FA, Markus HS. Blood-brain barrier permeability is increased in normal-appearing white matter in patients with lacunar stroke and leucoaraiosis. J Neurol Neurosurg Psychiatry. 2010;81:192–197. doi: 10.1136/jnnp.2009.172072. [DOI] [PubMed] [Google Scholar]
- 25.Wardlaw JM, Doubal F, Armitage P, Chappell F, Carpenter T, Munoz Maniega S, et al. Lacunar stroke is associated with diffuse blood-brain barrier dysfunction. Ann Neurol. 2009;65:194–202. doi: 10.1002/ana.21549. [DOI] [PubMed] [Google Scholar]
- 26.De Reuck J, Crevits L, De Coster W, Sieben G, vander Eecken H. Pathogenesis of binswanger chronic progressive subcortical encephalopathy. Neurology. 1980;30:920–928. doi: 10.1212/wnl.30.9.920. [DOI] [PubMed] [Google Scholar]
- 27.de Groot JC, de Leeuw FE, Oudkerk M, van Gijn J, Hofman A, Jolles J, et al. Periventricular cerebral white matter lesions predict rate of cognitive decline. AnnNeurol. 2002;52:335–341. doi: 10.1002/ana.10294. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt R, Scheltens P, Erkinjuntti T, Pantoni L, Markus HS, Wallin A, et al. White matter lesion progression: A surrogate endpoint for trials in cerebral small-vessel disease. Neurology. 2004;63:139–144. doi: 10.1212/01.wnl.0000132635.75819.e5. [DOI] [PubMed] [Google Scholar]
- 29.Fernando MS, Simpson JE, Matthews F, Brayne C, Lewis CE, Barber R, et al. White matter lesions in an unselected cohort of the elderly: Molecular pathology suggests origin from chronic hypoperfusion injury. Stroke. 2006;37:1391–1398. doi: 10.1161/01.STR.0000221308.94473.14. [DOI] [PubMed] [Google Scholar]
- 30.Vermeer SE, Longstreth WT, Jr, Koudstaal PJ. Silent brain infarcts: A systematic review. Lancet Neurol. 2007;6:611–619. doi: 10.1016/S1474-4422(07)70170-9. [DOI] [PubMed] [Google Scholar]
- 31.Akiguchi I, Tomimoto H, Suenaga T, Wakita H, Budka H. Blood-brain barrier dysfunction in binswanger's disease; an immunohistochemical study. Acta Neuropathologica. 1998;95:78–84. doi: 10.1007/s004010050768. [DOI] [PubMed] [Google Scholar]
- 32.Rosenberg GA, Sullivan N, Esiri MM. White matter damage is assoiciated with matrix metalloproteinases in vascular dementia. Stroke. 2001;32:1162–1168. doi: 10.1161/01.str.32.5.1162. [DOI] [PubMed] [Google Scholar]
- 33.Nakaji K, Ihara M, Takahashi C, Itohara S, Noda M, Takahashi R, et al. Matrix metalloproteinase-2 plays a critical role in the pathogenesis of white matter lesions after chronic cerebral hypoperfusion in rodents. Stroke. 2006;37:2816–2823. doi: 10.1161/01.STR.0000244808.17972.55. [DOI] [PubMed] [Google Scholar]
- 34.Rosenberg GA. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 2009;8:205–216. doi: 10.1016/S1474-4422(09)70016-X. [DOI] [PubMed] [Google Scholar]
- 35.Candelario-Jalil E, Yang Y, Rosenberg GA. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience. 2009;158:983–994. doi: 10.1016/j.neuroscience.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 37.Hu J, Van den Steen PE, Sang QX, Opdenakker G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat Rev Drug Discov. 2007;6:480–498. doi: 10.1038/nrd2308. [DOI] [PubMed] [Google Scholar]
- 38.Matsukawa N, Yasuhara T, Hara K, Xu L, Maki M, Yu G, et al. Therapeutic targets and limits of minocycline neuroprotection in experimental ischemic stroke. BMC Neurosci. 2009;10:126. doi: 10.1186/1471-2202-10-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zabad RK, Metz LM, Todoruk TR, Zhang Y, Mitchell JR, Yeung M. The clinical response to minocycline in multiple sclerosis is accompanied by beneficial immune changes: A pilot study. Mult Scler. 2007;13:517–526. doi: 10.1177/1352458506070319. [DOI] [PubMed] [Google Scholar]
- 40.Snapyan M, Lemasson M, Brill MS, Blais M, Massouh M, Ninkovic J, et al. Vasculature guides migrating neuronal precursors in the adult mammalian forebrain via brain-derived neurotrophic factor signaling. J Neurosci. 2009;29:4172–4188. doi: 10.1523/JNEUROSCI.4956-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hermann DM, Zechariah A. Implications of vascular endothelial growth factor for postischemic neurovascular remodeling. J Cereb Blood Flow Metab. 2009;29:1620–1643. doi: 10.1038/jcbfm.2009.100. [DOI] [PubMed] [Google Scholar]
- 42.Lee SR, Kim HY, Rogowska J, Zhao BQ, Bhide P, Parent JM, et al. Involvement of matrix metalloproteinase in neuroblast cell migration from the subventricular zone after stroke. J Neurosci. 2006;26:3491–3495. doi: 10.1523/JNEUROSCI.4085-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao BQ, Wang S, Kim HY, Storrie H, Rosen BR, Mooney DJ, et al. Role of matrix metalloproteinases in delayed cortical responses after stroke. NatMed. 2006;12:441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]