

Abstract
Regulation of the extracellular matrix by proteases and protease inhibitors is a fundamental biological process for normal growth, development and repair in the central nervous system. Matrix metalloproteinases (MMPs) and the tissue inhibitors of metalloproteinases (TIMPs) are the major extracellular-degrading enzymes. Two other enzyme families, a disintegrin and metalloproteinase (ADAM), and the serine proteases, plasminogen/plasminogen activator (P/PA) system, are also involved in extracellular matrix degradation. Normally, the highly integrated action of these enzyme families remodel all of the components of the matrix and perform essential functions at the cell surface involved in signaling, cell survival, and cell death. During the inflammatory response induced in infection, autoimmune reactions and hypoxia/ischemia, abnormal expression and activation of these proteases lead to breakdown of the extracellular matrix, resulting in the opening of the blood-brain barrier (BBB), preventing normal cell signaling, and eventually leading to cell death. There are several key MMPs and ADAMs that have been implicated in neuroinflammation: gelatinases A and B (MMP-2 and -9), stromelysin-1 (MMP-3), membrane-type MMP (MT1-MMP or MMP-14), and tumor necrosis factor-α converting enzyme (TACE). In addition, TIMP-3, which is bound to the cell surface, promotes cell death and impedes angiogenesis. Inhibitors of metalloproteinases are available, but balancing the beneficial and detrimental effects of these agents remains a challenge.
Keywords: blood-brain barrier, stroke, gelatinases, stromelysin-1, extracellular matrix, MMP inhibitors
Introduction
During neuroinflammation and ischemia, molecular cascades are initiated with the purpose of removing damaged cells and preparing the brain for repair (Dirnagl et al., 1999). This dual role necessitates a full understanding of the timing of the injury phases and of those involved in repair. Early after the injury, constitutive proteases are activated and begin the process of disassembling the extracellular matrix, opening the blood-brain barrier (BBB), and initiating cell death by apoptosis (Rosenberg et al., 1998; Heo et al., 1999; Yang et al., 2007). The second stage of injury involves MMPs in processes of angiogenesis and neurogenesis (Lee et al., 2006; Wang et al., 2006; Zhao et al., 2006). In this second phase, treatment with MMP inhibitors may interfere with repair (Rosell and Lo, 2008; Sood et al., 2008). Remodeling of the extracellular matrix characterizes the third phase when gliosis forms impenetrable scar tissues that block the regrowth and re-projection of axons. The action of the MMPs on the basal lamina and tight junction proteins (TJPs) in endothelial cells is the final common pathway for opening of the BBB, which allows cells to enter the central nervous system and attack invading organisms. This is probably a protective mechanism during CNS infection, but when no infection exists, this inflammatory response contributes to tissue damage. When matrix proteins around neurons are degraded, there is loss of contact and cell death by anoikis (Gu et al., 2002). While a great deal has been learned in the past decade as information on the MMPs and TIMPs has emerged from many laboratories, the multiple functions of the MMPs and TIMPs have made the search of clinically useful MMP inhibitors a challenge.
The first enzyme in the large gene family now known as the MMPs was discovered in the regenerating frog tail (see for discussion (Brinckerhoff and Matrisian, 2002)). The next major breakthrough in MMP biology was the discovery that metastatic melanoma cells secrete a 72-kDa type IV collagenase (MMP-2) to facilitate tumor cells’ passage from the blood into the tissues (Liotta et al., 1980). This important discovery lead to a great deal of interest by the pharmaceutical industry to identify MMP inhibitors for the treatment of cancer (Overall and Lopez-Otin, 2002). Initially the emphasis of drug discovery and clinical trials was on the role of the MMPs in cancer with many agents entering clinical trials (Coussens et al., 2002). The results were disappointing because long-term use of these agents resulted in overgrowth of extracellular matrix in joints, which was painful. Subsequently, a shift has occurred with the realization that short-term use of MMP inhibitors may be possible in neurological disorders, particularly for treatment of cerebrovascular and cardiovascular diseases (Hu et al., 2007). This selective review will focus on the role of the MMPs in the neuroinflammatory response to injury with special emphasis on cerebral ischemia. This article will not cover the role of MMPs in other neurodegenerative diseases (e.g., multiple sclerosis, inflammatory myopathies) where the inflammation is the underlying cause of the disease. Several recent reviews have been published detailing the basic biology and role in the central nervous system of the MMPs (Lo et al., 2003; Cunningham et al., 2005; Liu and Rosenberg, 2005; Yong, 2005).
Biology of the MMPs: expression and mechanisms of activation
The MMPs are zinc- and calcium-dependent endopeptidases, identified as matrix-degrading enzymes. MMPs cleave most components of the extracellular matrix including fibronectin, laminin, proteoglycans and type IV collagen (Sternlicht and Werb, 2001; Rosenberg, 2002). MMPs are also capable of processing other proMMPs and a number of bioactive molecules, including proforms of cytokines such as TNF-α and IL-1β (Schonbeck et al., 1998; Cauwe et al., 2007), and pro-neurotrophins such as proNGF and proBDNF (Schonbeck et al., 1998; Lee et al., 2001; Sternlicht and Werb, 2001). Regulation of MMP expression and activation is very complex and tightly controlled. MMPs are synthesized as zymogens and are secreted into the extracellular space as inactive zymogens. ProMMPs are activated by disruption of the zinc-thiol interaction between the catalytic site and the pro-domain. The pro-peptide of the zymogen has to be proteolytically cleaved by other MMPs or proteases for an MMP to be active (Sternlicht and Werb, 2001). The proteases plasmin, tissue plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA) are important physiological activators of the MMPs (Fig. 1) (Gasche et al., 2006). Protease-independent activation of the MMPs by S-nitrosylation or oxidation can unmask the catalytic domain producing activation of MMPs without pro-domain cleavage (Gu et al., 2002; Meli et al., 2003; Pei et al., 2006).
Fig. 1.
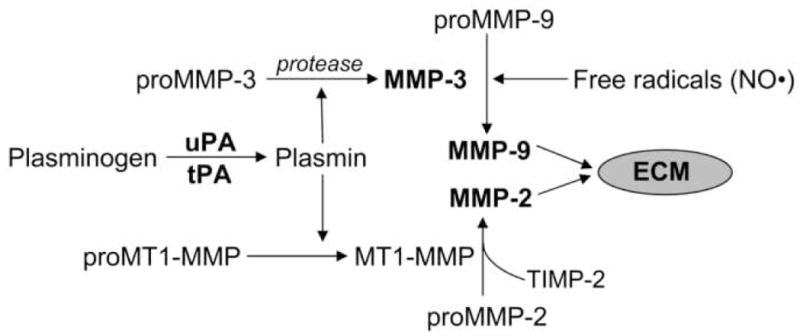
Mechanisms of activation of MMPs. Plasmin plays an active role in the activation of MMPs. MT1-MMP (MMP-14) binds to TIMP-2 and proMMP-2 leading to the formation of catalytically active MMP-2. ProMMP-9 is activated by MMP-3 and free radicals (nitric oxide, NO•). Plasmin has been shown to activate proMMP-3. Recently, it was shown that there is an intracellular mechanism of activation of MMP-3 which involves a serine protease other than furin (Choi et al., 2008). Although proMMP-3 possesses the furin recognition sequence, the cleavage would leave 9 amino acids belonging to the prodomain resulting in a form which is not catalytically active (Choi et al., 2008).
The activities of MMPs are regulated by tissue inhibitors of MMPs (TIMPs). Four member of this family have been characterized (Brew et al., 2000). TIMP-2 inhibits MMP-2, TIMP-1 inhibits MMP-9 and TIMP-3 acts on MMPs and TACE. Although individual TIMPs have preferences for one or another of the MMPs, they can inhibit all of the MMPs (Brew et al., 2000). TIMP-3 is unique because it is membrane bound. The main substrates inhibited by TIMP-3 include MT1-MMP, MMP-3 and TACE (Cunningham et al., 2005).
Constitutive expression of MMP-2 provides an on-going, well-controlled remodeling of the extracellular matrix. MMP-2 remains in the pro or latent form until activated by a molecular cascade that involves a trimolecular complex made up of MMP-2, TIMP-2, and MT1-MMP (Fig. 2). This reaction occurs close to the cell surface where it provides local proteolysis without involvement of the surrounding tissues (Zucker et al., 2003). TIMP-3 reduces the activation of MT1-MMP, which affects the activation of MMP-2.
Fig. 2.
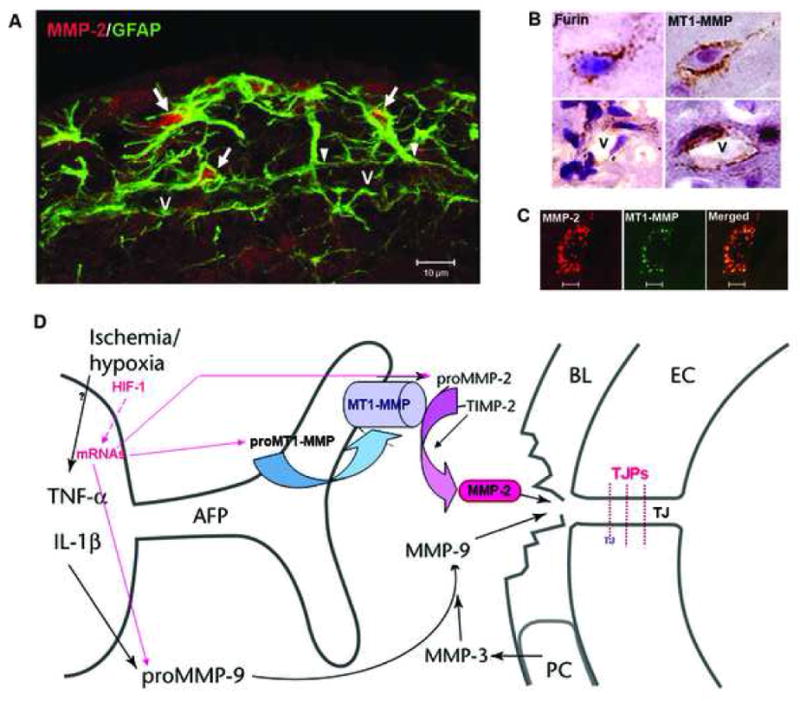
(A) Confocal immunohistochemistry shows GFAP-positive astrocytes around a vessel (V) that express MMP-2 (arrows) in intact rat brain tissue. The arrowheads indicate the astrocyte endfeet around the vessel. (B) Expression of furin and MT1-MMP in brain cells and around vessels (V). (C) Confocal images show the co-localization of MMP-2 and MT1-MMP immunohistochemistry in brain cells. (D) Schematic drawing to show that the activation of MMP-2 occurs through the action of the trimolecular complex during the early opening of the BBB in 3h-reperfusion after 90 min MCAO. In the astrocytic foot processes (AFP), the membrane-type 1 matrix metalloproteinase (MT1-MMP) joins with tissue inhibitor of metalloproteinases-2 (TIMP-2) to activate proMMP-2 in a spatially constrained manner close to the basal lamina (BL). In the BL are the pericytes (PC). The endothelial cells (EC) have tight junctions (TJ). The activated MMP-2 has direct access to the portion of the BL beneath the AFP and components of the BL are degraded. The manner in which this disruption of the BL leads to increased permeability is unclear since the role of the BL in maintaining the integrity of the blood vessel is uncertain.
The early events in the molecular cascade of hypoxia/ischemia probably involve the induction of the proconvertase, furin, by hypoxia inducible factor-1α (HIF-1α) (McMahon et al., 2005). Furin is an activator of MT1-MMP (MMP-14), which is required for the activation of MMP-2 (McMahon et al., 2005). Activation of MT1-MMP by furin has been established for tumorigenesis and can only be proposed for cerebral ischemia. Unlike other MMPs, MMP-2 is constitutively present in large quantities in the normal brain and is found in astrocytes and CSF (Fig. 2). The rate-limiting step is activation, making MT1-MMP critical in the process (Fig. 1). Abnormal increase in MMP-2 expression and activity in hypoxia/ischemia may disrupt basal lamina and tight junctions between endothelial cells leading to BBB disruption. On the other hand, latent MMP-9 is activated by free radicals and MMP-3 during neuroinflammatory and ischemic conditions (Hahn-Dantona et al., 1999; Gasche et al., 2001; Liu and Rosenberg, 2005) (Fig. 1). The link between hypoxia, HIF-1α, furin and MT1-MMP activation has been demonstrated in peripheral tissues, and remains to be studied in the brain.
MMPs and neuroinflammatory responses
The two major inducible MMPs that have been identified in the neuroinflammatory response are MMP-3 and MMP-9. In brain, a number of cell types express MMP-9, including neurons, endothelial cells, reactive astrocytes and microglia. Studies to identify the factors involved in induction of MMPs in neuroinflammation have used lipopolysaccharide (LPS), which is a potent stimulus for MMP-3 and -9 in cultures of brain astrocytes and microglia (Gottschall and Deb, 1996; Rosenberg et al., 2001; Lee et al., 2003; Kim et al., 2008). It has been suggested that the effects of LPS on the expression of MMPs are due in part to the formation of proinflammatory cytokines. TNF-α and IL-1β produce a significant increase in the production of MMP-3 and -9 in cultured astrocytes and microglia (Gottschall and Yu, 1995; Kauppinen and Swanson, 2005; Crocker et al., 2006). A recent study found that endothelin-1, which is increased in cerebral ischemia, is a potent inducer of MMP-3 production in primary rat astrocytes (Koyama and Tanaka, 2008).
Intracerebral injection of LPS disrupts the BBB through the action of MMP-9 (Mun-Bryce and Rosenberg, 1998). Furthermore, intracerebral injection of LPS significantly elevates mRNA levels of MMP-2 and -3, and TNF-α mRNA levels (Mun-Bryce et al., 2002). Since MMP-9 protein was seen after LPS injection, but mRNA was not, the source of MMP-9 was most likely the invading neutrophils. Active MMP-9 is prepackaged in neutrophils and released under neuroinflammatory conditions (Justicia et al., 2003; Gidday et al., 2005; Rosell et al., 2008).
Studies in the MMP-3 knockout mouse, using the LPS intracerebral injection model, showed induction of MMP-3 and -9 with disruption of the BBB; MMP-3 knockout mice have significantly reduced BBB opening and neutrophil infiltration (Gurney et al., 2006). The MMP-3 co-localized with the macrophages/microglia cells (Fig. 3).
Fig. 3.
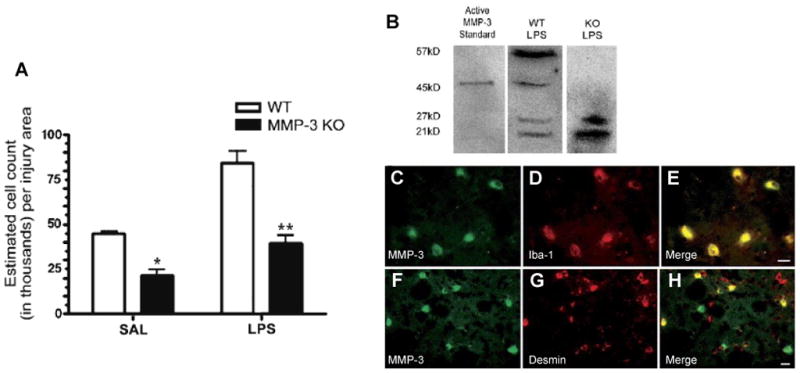
(A) Stereology for neutrophil counts 24 h after LPS or saline injection. Counts of MPO-immunoreactive neutrophils were greater in the LPS-injected caudate for both WT and MMP-3 KO. However, in the MMP-3 KO, there were significantly fewer neutrophils in the caudate than in WT in both saline and LPS-injected hemispheres (*P < 0.01, **P < 0.001; n = 4 for both WT and KO). Panels B–H: Western blotting and immunohistochemical staining for MMP-3, pericytes, and microglia. (B) Western blotting for MMP-3 showed the proform at 57 kDa and the active form at 45 kDa along with two lower, unidentified bands. The MMP-3 KO mouse did not show the 57 or 45-kDa bands of MMP-3. (C–E) MMP-3 colocalized with Iba-1-immunoreactive microglia/macrophages. (C) MMP-3 staining in LPS-injected WT; (D) Iba-1 immunostaining; (E) merged image of C and D. (F–H) MMP-3 was also seen in pericytes stained for desmin in LPS-injected WT. (F) MMP-3 staining; (G) Pericyte staining; (H) merged composite of panels F and G. Scale bars are 10 μm. Adapted from Gurney et al., 2006.
Direct injection of tumor necrosis factor-α (TNF-α) into the rat brain results in a dramatic increase in the expression and activation of MMP-9 and MMP-3 (Rosenberg et al., 1995; Candelario-Jalil et al., 2007b), which is associated with a significant opening of the BBB. Microglia/macrophages and neurons surrounding the injection site are the major cellular sources of MMP-3 and -9 following intracerebral TNF-α administration. In this model of neuroinflammation, cyclooxygenase (COX)-derived products are involved in the molecular mechanisms responsible for the expression and activation of both MMP-3 and -9. Indomethacin, an inhibitor of COX-1 and COX-2, significantly reduced the expression and activity of MMP-9 as assessed by immunoblotting and gelatin-substrate zymography. Similarly, indomethacin treatment reduced MMP-3 expression and activity. Indomethacin and the COX-1 inhibitor valeroyl salicylate significantly attenuated TNF-α-induced BBB breakdown and free radical formation (Candelario-Jalil et al., 2007b), indicating that MMP-mediated BBB disruption during neuroinflammation can be significantly reduced by administration of COX inhibitors. Furthermore, COX-2 inhibition reduced BBB damage, vasogenic edema and leukocyte infiltration following transient focal cerebral ischemia (Candelario-Jalil et al., 2007a). These studies suggest that enhanced COX activity during neuroinflammatory and/or ischemic conditions is an important mechanism involved in BBB injury, possibly by increasing MMP production and inducing oxidative damage.
Novel roles of MMPs in cellular signaling
Data from recent studies indicate that MMP-3 plays a critical role as an intercellular signaling molecule that modulates neuroinflammatory responses (Kim et al., 2005; Kim et al., 2007; Woo et al., 2008). Neurons that are under cellular stress and undergoing apoptosis release the active form of MMP-3, and this catalytically active MMP-3 in turn triggers microglial activation and production of pro-inflammatory cytokines such as TNF-α, IL-6 and IL-1β (Kim et al., 2005). Inhibition of MMP-3 activity by NNGH [N-isobutyl-N-(4-methoxyphenylsulfonyl)-glycylhydroxamic acid] completely prevented microglial activation in terms of reduction of proinflammatory cytokines release (Kim et al., 2005) and formation of reactive oxygen species (Kim et al., 2007; Woo et al., 2008). In line with these studies, it has been recently reported that inhibition of MMP-3 or -9 significantly suppressed the expression of inducible nitric oxide synthase, IL-1β and IL-6 at the transcriptional level through a mechanism involving suppression of NF-κB, AP-1 and mitogen-activated protein kinases (MAPKs) in LPS-stimulated microglial cells (Woo et al., 2008). Furthermore, treatment with BB-94, a broad spectrum MMP inhibitor, suppressed TNF-α production in LPS-treated human microglia (Nuttall et al., 2007).
MMP-3 deficient mice displayed a significant reduction in microglial activation following in vivo administration of the neurotoxin MPTP (Kim et al., 2007). MMPs are secreted as the pro-form and processed to the active form extracellularly (Sternlicht and Werb, 2001). However, neurons undergoing apoptosis release the active form of MMP-3 (Kim et al., 2005; Kim et al., 2007), which is cleaved inside the cell by a serine protease other than furin (Choi et al., 2008), suggesting for the first time that there is a mechanism of activation of MMP-3 inside the cell.
The biological significance of the release of active MMP-3 by neurons undergoing apoptosis is not completely clear. It has been suggested that it may disrupt physical connections between apoptotic cells and the extracellular matrix, which may facilitate engulfment by phagocytes (Kim et al., 2005). Active MMP-3 may also induce microglial activation near the site at which apoptosis occurs to promote clearance of apoptotic cells (Kim et al., 2005). It should be noted that the exact molecular mechanisms through which active MMP-3 activates microglial cells remain to be clarified.
The Neurovascular Unit, Tight Junction Proteins and MMPs
Brain capillaries, which are the major interface between blood and the brain tissues, play a major role in controlling the neuronal microenvironment. The neurovascular unit includes the endothelial cells (ECs), astrocytes, pericytes, basal lamina and the neurons. Around the capillary is a basal lamina with pericytes and surrounding them are the astrocyte endfeet. MMPs are found in all of the elements of the neurovascular unit, but different MMPs have predilection for certain cell types. Endothelial cells have mainly MMP-9, pericytes express MMP-3 and -9, while astrocytes have MMP-2 and MT1-MMP in the endfeet that surround the endothelial cells. This pattern of MMPs facilitates the opening of the BBB in inflammation, but also allows for the gradual changes in the extracellular matrix that are most likely on going and involve the action of the MMP-2/MT1-MMP complex remodeling the matrix to prevent excessive build-up.
Cerebrovascular endothelial tight junctions restrict molecules from moving between the blood and the brain. Tight junction proteins join ECs together, forming an interface between blood and brain (Brightman and Reese, 1969; Liebner et al., 2000; Ballabh et al., 2004). Trans-membrane tight-junction proteins consist of three integral proteins, claudins, occludin, and junctional adhesion molecules (JAM) (Furuse et al., 1993; Papadopoulos et al., 2001). Zona occludens (ZO)-1, -3, and cingulin are considered to be cytoplasmic tight junctional accessory proteins, which connect tight junctions to the actin cytoskeleton (Itoh et al., 1997; Haskins et al., 1998). The extracellular loops of occludin, claudin, and JAM originating from neighboring cells form the paracellular barrier of the TJ, which selectively exclude most blood-born substances from entering the brain. In rodents and adult human brains, claudin-1, claudin-5, and occludin have been found to be present in brain endothelial tight junctions forming the BBB. Tight junctions are dynamic structures and proteins forming the TJ are subject to changes in expression, subcellular location, post-translational modification, and protein-protein interactions under both physiological and pathophysiological conditions (Hawkins and Davis, 2005). Occludin, claudin-5 and ZO-1, which are the main structural barrier proteins, are considered sensitive indicators of normal and disturbed functional state of the BBB (Hirase et al., 1997; Hirase et al., 2001; Dobrogowska and Vorbrodt, 2004; Kanda et al., 2004; Wen et al., 2004). The claudins have been proven to be one of the essential proteins for TJ strands and the composition of the claudin species directly determines the barrier function (Hawkins and Davis, 2005). Taking together, the regulation of TJPs is essential for the maintenance of the BBB permeability.
The role of the basal lamina molecules may be to provide a charge barrier, and the astrocytes and neurons may secrete various vasoactive substances to modulate the structure and physical state of the endothelial cells. Elegant studies of astrocytes with confocal microscopy shows a large cellular domain with extensive connections to the capillaries and rapid activation of calcium signals that travel over large regions affecting capillary function at a distance from the stimulus (Simard et al., 2003).
Involvement of MMPs in Bacterial Meningitis
Acute inflammation of the meninges triggers the release of MMPs, which can be detected in the CSF, opening the BBB at the brain surface. Elevated levels of MMPs are found in viral, bacterial, and fungal meningitis (Leppert et al., 2001). Matrix metalloproteinases and TACE contribute synergistically to the pathophysiology of bacterial meningitis; TACE proteolytically releases several cell-surface proteins, including the proinflammatory cytokine, TNF-α and its receptors, which in turn stimulate cells to produce active MMPs, facilitating leukocyte extravasation and brain edema by degradation of extracellular matrix components (Leib et al., 2001). Treatment with BB1101, a hydroxamic acid-based inhibitor of MMP and TACE, reduced the CSF concentration of TNF-α and decreased the incidences of seizures and mortality. Several types of meningitis cause an increase in MMPs in the CSF, including Lyme disease, viral infections, and tuberculosis (Kolb et al., 1998; Perides et al., 1998; Sporer et al., 1998; Lee et al., 2004). A water-soluble MMP inhibitor, TNF484, with actions against both MMPs and TACE was shown to be effective in an experimental model of bacterial meningitis (Meli et al., 2004; Echchannaoui et al., 2007). Doxycycline, a tetracycline derivative that inhibits MMPs, was also shown to effectively treat meningitis (Meli et al., 2006).
MMPs in Cerebral Ischemia
Emerging evidence from many laboratories indicates a complex pattern of MMP expression in hypoxia/ischemia (Rosenberg et al., 1996; Gasche et al., 1999; Heo et al., 1999; Wang et al., 2000; Planas et al., 2001; Magnoni et al., 2004; Sole et al., 2004; Yang et al., 2007). In the reperfusion model, there is an early increase in MMP-2, which is transient, but results in the early reversible opening of the BBB. An elevation in MMP-2 in the early stages of the injury has been observed in rodents and nonhuman primates (Chang et al., 2003; Yang et al., 2007). Claudin-5 is degraded by MMP-2, but remains within the vessels after 3 hour of reperfusion (Fig. 4). However, by 24 hours the tight junction proteins are no longer seen in the vessels (Yang et al., 2007). Following the initial opening of the BBB, there is a second opening between 24 and 48 hours, depending on the time of occlusion (Rosenberg et al., 1998). During this phase there is marked increase in MMP-9, which leads to more extensive damage to the blood vessels. Treatment with MMP inhibitors or MMP neutralizing antibodies decreases infarct size and prevents BBB breakdown after focal ischemic stroke (Romanic et al., 1998; Rosenberg et al., 1998; Asahi et al., 2000; Asahi et al., 2001b). When rats were treated with the MMP inhibitor, BB1101, immediately after the onset of reperfusion, the early opening of the BBB seen at 3 h was blocked (Fig. 5). However, MMP inhibition failed to reduce infarct size at 48 h and interfered with recovery, as demonstrated by a worse neurological deficit score at 3–4 weeks after stroke (Sood et al., 2008).
Fig. 4.
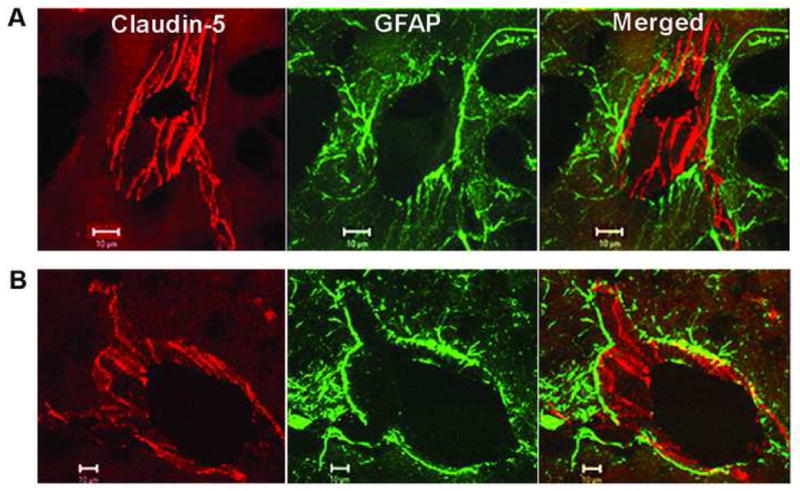
Confocal micrographs showing claudin-5 immunoreactivity after 3 h of reperfusion following 90 min of MCAO in the rat. (A) The nonischemic side show that the claudin-5 (Cy-3) in blood vessels is separated from the astrocytes (GFAP-FITC) surrounding them. The merged images show that the claudin-5 and astrocytes are separate. (B) In the ischemic hemisphere, there is fragmentation and degeneration of the claudin-5 immunoreactivity. Co-localization of claudin-5 and GFAP was seen in the ischemic hemisphere. Adapted from Yang et al., 2007.
Fig. 5.
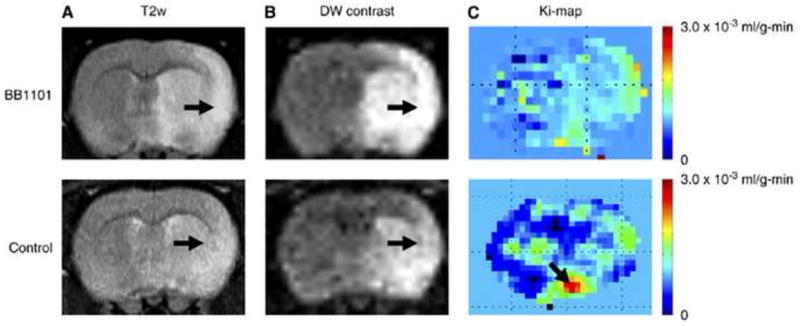
(A) T2 weighted image (B) diffusion-weighted (DW) contrast image, and (C) color coded permeability coefficient map for BB1101 treated (top row) and control (bottom row) rats. The ischemia can be seen as a hyperintense lesion (arrow) on T2 weighted images. A 53.1 and 48.4% reduction in the ADC value on the ischemic side (region of interest on lesion) compared with that in the matching contralateral side was observed in BB1101-treated and control rats, respectively, after 3 h of reperfusion. The arrows on diffusion-weighted contrast image point at ischemic regions with diffusion changes. Color-coded permeability maps show clearly the regions of high (arrow) and low permeability in treated and control rats. Taken from Sood et al., 2008.
A number of other factors are involved in the later disruption of the blood vessels since there are cytokines, proteases, and free radicals released at this stage of the injury. Another major difference between the first wave of MMP-induced injury and the second is that MMP-2 is tethered to the cell surface by MT1-MMP and requires the presence of TIMP-2 in order to undergo activation. This restricts the proteolytic action of MMP-2 to the immediate vicinity of the protease. On the other hand, MMP-9 is released into the extracellular space where it is not constrained and degrades multiple proteins in the extracellular matrix, including those in the matrix around neurons.
MMP-9, but not MMP-2 gene knockout is associated with a reduction in infarction and attenuation of BBB opening after focal cerebral ischemia (Asahi et al., 2000; Asahi et al., 2001a; Asahi et al., 2001b).
In human ischemic stroke, active MMP-2 is increased first on days 2–5 compared to active MMP-9, which is elevated up to months after the ischemic episode (Clark et al., 1997). In stroke patients, there is a correlation between levels of MMP-9 in plasma and the final NIHSS score (Montaner et al., 2001).
Apoptosis by injury to the matrix around the neuron has been proposed as a mechanism of cell death by anoikis (Gu et al., 2002). Furthermore, treatment with a selective inhibitor of MMP-2 and MMP-9 blocks cell death in transient focal ischemia (Gu et al., 2005). Mice overexpressing superoxide dismutase (SOD) are protected from ischemic injury and have reduced MMP production (Morita-Fujimura et al., 2000). Oxidative stress leads to the induction of MMP-9 in transient focal cerebral ischemia (Gasche et al., 2001). Hyperglycemia increases oxidative stress and MMP-9 activity, exacerbating BBB disruption following temporary focal cerebral ischemia (Kamada et al., 2007).
The early increase in gelatinase activity at 2 h of reperfusion following focal ischemia is involved in the maturation of IL-1β in the ischemic brain (Amantea et al., 2007). IL-1β is synthesized as a precursor molecule, pro-IL-1β (31 kDa), which is cleaved and converted into the mature, biologically active, form of the cytokine (17 kDa) by caspase-1 (Thornberry and Molineaux, 1995). After 2 h of recirculation following ischemia, the increase in MMP-2 and -9 in the ischemic cortex coincides with elevation of mature IL-1β. This early increase in IL-1β does not implicate the caspase-1-dependent processing of pro-IL-1β to yield mature IL-1β. More importantly, administration of the MMP inhibitor GM6001 abolished the increase of IL-1β and reduced the cleavage of the pro-IL-1β form, indicating that the early increase in MMPs following ischemia may initiate IL-1β processing. GM6001 treatment also produced a significant reduction in infarct size at 24 h after focal ischemia (Amantea et al., 2007).
MMPs and tPA-induced hemorrhagic transformation
Magnetic resonance imaging (MRI) in human cardioembolic stroke showed hemorrhagic transformation in 68.6% of infarcts, suggesting that hemorrhagic transformation is a regular finding in medium and large cardioembolic infarcts (Hornig et al., 1993). Plasma levels of MMP-9 correlate with hemorrhagic transformation and intracerebral hemorrhage (Montaner et al., 2003; Rosell et al., 2006). Treatment with tissue plasminogen activator (tPA) is the only approved effective therapy in stroke. tPA, which is given for acute strokes within 3 hours of onset, increases the risk of hemorrhage approximately 10-fold (NINDS, 1995; Kidwell et al., 2008). Therefore, therapeutic strategies that reduce these side effects of tPA are highly desirable.
Treatment of rats with tPA increases mortality by opening the BBB and inducing hemorrhage; treatment with the broad-spectrum MMP inhibitor, BB94, blocks the opening of the BBB and dramatically reduces hemorrhage (Fig. 6) (Lapchak et al., 2000; Pfefferkorn and Rosenberg, 2003). Opening of the BBB after tPA leads to increased mortality; when the BBB is closed with a MMP inhibitor, the death rate is dramatically reduced (Fig. 6). Tissue plasminogen activator increased the expression and activation of MMP-9; at 12 hours, tPA-treated rats showed significantly higher levels of proMMP-9 and cleaved MMP-9 than untreated controls and by 24 hours, all rats showed evidence of hemorrhagic transformation in the ischemic territory. Rats treated with BB-94 and tPA showed significantly reduced hemorrhage volumes compared with those that received tPA alone (Sumii and Lo, 2002). When the BBB remains intact the fibrinolytic agent acts on fibrin within the blood vessels, however, if the BBB is compromised, the tPA escapes into the brain and acts on the MMPs. Agents that maintain the integrity of the BBB may therefore extend the therapeutic window for treatment. A small percentage of stroke patients arrive at a hospital early enough to qualify for tPA treatment. Therefore, extending the time window for thrombolytic therapy by reducing the hemorrhagic risk would be beneficial.
Fig. 6.
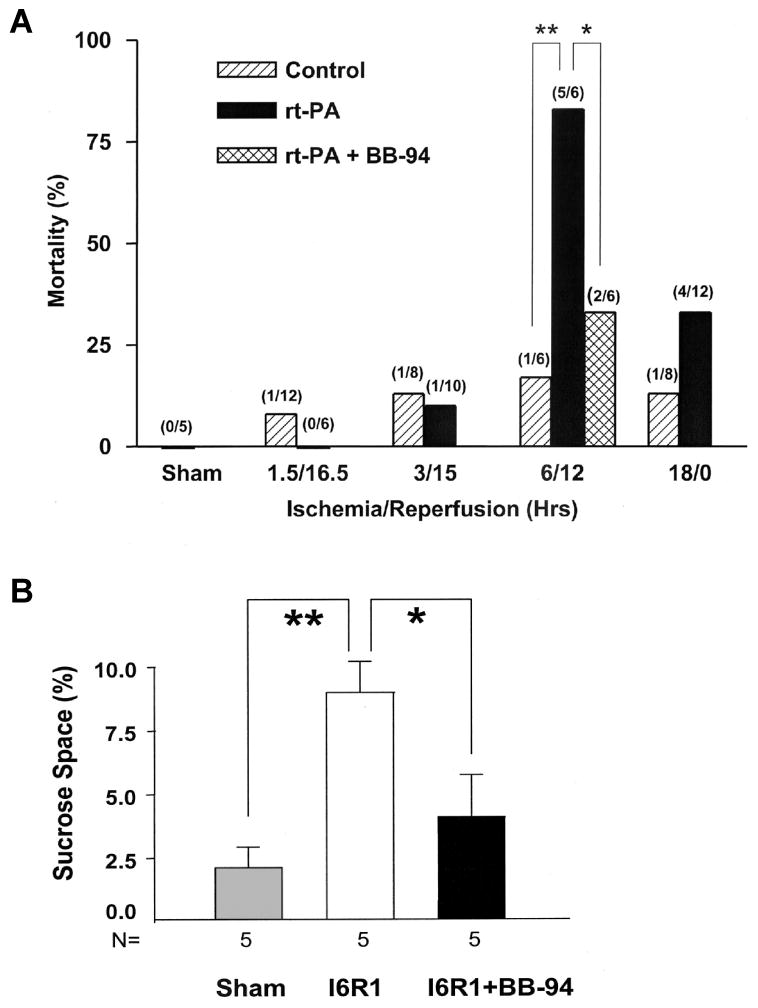
(A) Effect of rtPA on mortality in animals with different intervals of ischemia and reperfusion: Mortality is shown in percent with the numbers of animals dying and the number of animals studied shown in parentheses above the bars. When reperfusion was delayed to 6 hours, mortality was increased markedly in rtPA-treated animals (**P<0.01). Treatment with BB-94 reduced rtPA-associated mortality significantly (*P<0.05). Control animals had MCAO without rtPA treatment. (B) Opening of the BBB as measured by sucrose space in rats with 6 hours of ischemia and 1 hour of reperfusion (I6R1). Compared with sham-operated animals, BBB permeability was markedly increased in untreated rats (**P<0.01). BB-94 given 2 and 5 hours after MCAO markedly decreased BBB opening (*P<0.01). Adapted from Pfefferkorn and Rosenberg, 2003.
It has been shown that the serine proteases, urokinase plasminogen activator (uPA) and tPA induced a dose-dependent upregulation of MMP-2 and MMP-9 in rat cortical astrocytes (Lee et al., 2007). Furthermore, tPA promotes neutrophil degranulation and MMP-9 release, suggesting that neutrophils are good candidates to be the main source of MMP-9 following tPA stroke treatment and thus, partially responsible for thrombolysis-related hemorrhagic transformation (Cuadrado et al., 2008).
Emerging evidence indicates that MMP-3 is important in intracerebral hemorrhage induced by tPA treatment of ischemic stroke (Suzuki et al., 2007). Mice with genetic deficiency of plasminogen (Plg−/−), stromelysin-1 (MMP-3−/−) or gelatinase B (MMP-9−/−) were treated with either vehicle or tPA at 4 h after MCAO, and intracerebral hemorrhage was quantified 20 h later. Interestingly, tPA enhanced intracerebral bleeding in MMP-9−/− mice, but not in Plg−/− or MMP-3−/− animals. With tPA treatment, Plg−/− and MMP-3−/− mice showed significantly less hemorrhage than the corresponding wild-type animals, whereas this was not the case for MMP-9−/− mice, indicating that plasminogen and MMP-3, more than MMP-9, appear to play a critical role in the tPA-enhanced intracerebral bleeding after stroke, whereas MMP-9, more than plasminogen or MMP-3, contributes to hemorrhagic transformation in the absence of tPA treatment (Suzuki et al., 2007).
TIMP-3, MMP-3 and Apoptosis
Intrinsic and extrinsic pathways mediate apoptosis. The death receptors, Fas/CD95 and TNF receptors (TNFR1 and TNFR2) are cleaved from the cell surface by the sheddase activity of MMP-3 and TACE, respectively. Freeing a death receptor from the surface reduces apoptosis (Bond et al., 2002; Wetzel et al., 2003; Liu et al., 2008; Wetzel et al., 2008). TIMP-3 blocks the action of MMP-3, which enhances the apoptosis by preventing the release of Fas from the cell surface (Wetzel et al., 2008). In a rat model of focal cerebral ischemia, TIMP-3 immunostaining was increased in neurons on the ischemic side, and by 24 hours the majority of the ischemic neurons were TIMP-3-positive (Wallace et al., 2002). Knockout of the TIMP-3 gene reduces cell death and infarct size after cerebral ischemia in the reperfusion injury model in the mouse (Fig. 7), suggesting that TIMP-3 facilitates cell death in ischemic neurons (Wetzel et al., 2008). Similarly, data from a recent study showed that TIMP-3 is upregulated in cultured neurons undergoing apoptosis induced by serum deprivation. In this model, TIMP-3 mediated neuronal apoptosis through inhibition of MMP-3 and subsequent activation of the Fas/FasL pathway (Lee et al., 2008).
Fig. 7.
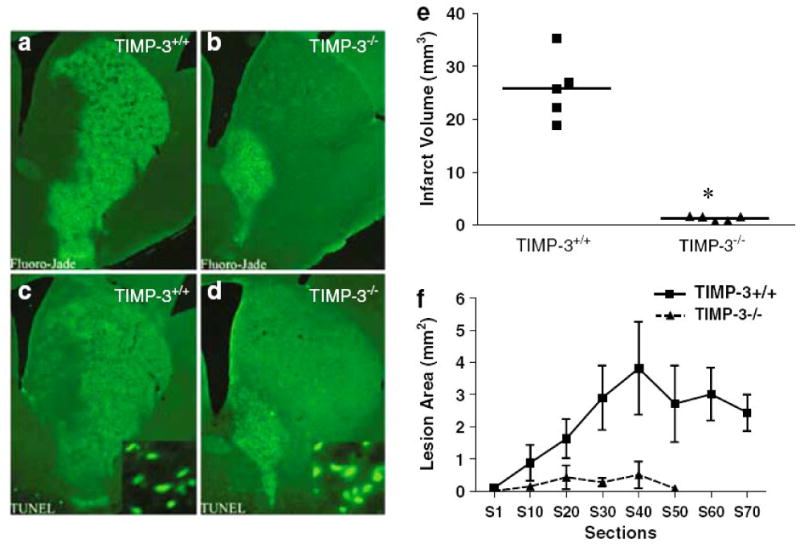
Lesion size in timp-3 +/+ versus timp-3 −/− at 3 days of reperfusion following 30 min transient MCAO. Timp-3 +/+ and timp-3 −/− mice were subjected to 30 min transient MCAO followed by 3 days of reperfusion. Coronal sections were stained for degenerating neurons using Fluoro-Jade (a, b) or stained for apoptotic nuclei using TUNEL (c, d). Infarct volume in timp-3 +/+ versus timp-3 −/− mice as assessed by morphometric analysis of Fluoro-Jade-stained histological sections, (e) n=5 mice per strain, *P<0.001. (f) Average lesion area per histological section in rostrocaudal direction in timp-3 +/+ versus timp-3 −/− mice. Taken from Wetzel et al., 2008.
A new role for intracellular active MMP-3 in neuronal apoptosis has been described (Choi et al., 2008). Suppression of MMP-3 activity using different approaches, including pharmacological inhibition, gene deletion or siRNA, resulted in protection of neuronal cells from apoptosis. Inhibition of MMP-3 activity reduced the activation of caspase-3 and the increase in DNA fragmentation (Choi et al., 2008). This supports a previous study showing that MMP-3 can induce apoptosis in hepatic cells via its catalytic activity. Interestingly, MMP-3 was present in the cell nucleus, and increased apoptosis was abolished by site-directed mutagenesis of the catalytic site of MMP-3 or by using the MMP inhibitor GM6001 (Si-Tayeb et al., 2006).
Implications for therapy and conclusions
MMP inhibitors have been used in a number of animal studies to block BBB injury, reduce infarct size and cell death (Rosenberg et al., 1998; Asahi et al., 2000; Gu et al., 2005). However, the same inhibitors blocked neurogenesis and neurovascular remodeling during delayed phases after stroke (Lee et al., 2006; Zhao et al., 2006; Rosell and Lo, 2008). These results underscore the complexity of the effects of MMPs during ischemic brain injury, ranging from detrimental effects during the early phases after stroke to beneficial roles at later stages. The major effects of the MMP inhibitors have been in the early stages of stroke when closing of the BBB is desirable in order to extend the treatment window for tPA. MRI provides a unique method to follow the opening of the BBB with Gadolinium-enhanced contrast agents. Combining Gd with fast T1-weighted imaging allows for a dynamic view of the BBB from which graphical data can be extracted for quantification of BBB permeability (Ewing et al., 2003). The Patlak Plot graphical method can be used to study the effect of drugs on the BBB (Sood et al., 2008). When a broad-based MMP inhibitor was used, the early opening of the BBB was blocked, but the later recovery was slowed (Sood et al., 2008). The major obstacles for use of these agents in clinical trials are the low specificity and poor solubility of the most commonly used MMP inhibitors; those with a hydroxymate base. The other obstacle is poor understanding of the time to initiate and the time to stop the use of the agents. Improvements in drug design and increased understanding of the timing of MMP expression should improve our therapeutic options with MMP inhibitors.
Acknowledgments
This work was supported by grants from the National Institutes of Health (NIH) to GAR (R01 NS045847 and R01 NS052305). ECJ and YY were supported by research grants from the American Heart Association (AHA). Confocal images were generated in the University of New Mexico Cancer Center Fluorescence Microscopy Facility, supported as detailed on the webpage: http://hsc.unm.edu/crtc/microscopy
List of abbreviations
- BBB
blood-brain barrier
- COX
cyclooxygenase
- CSF
cerebrospinal fluid
- ECs
endothelial cells
- ECM
extracellular matrix
- IL-1β
interleukin-1β
- LPS
lipopolysaccharide
- MCAO
middle cerebral artery occlusion
- MMP
matrix metalloproteinase
- MT1-MMP
membrane-type 1 matrix metalloproteinase
- TACE
tumor necrosis factor-α converting enzyme
- TIMP
tissue inhibitor of metalloproteinase
- TJPs
tight junction proteins
- TNF-α
tumor necrosis factor-α
- tPA
tissue plasminogen activator
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amantea D, Russo R, Gliozzi M, Fratto V, Berliocchi L, Bagetta G, Bernardi G, Corasaniti MT. Early upregulation of matrix metalloproteinases following reperfusion triggers neuroinflammatory mediators in brain ischemia in rat. Int Rev Neurobiol. 2007;82:149–169. doi: 10.1016/S0074-7742(07)82008-3. [DOI] [PubMed] [Google Scholar]
- Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000;20:1681–1689. doi: 10.1097/00004647-200012000-00007. [DOI] [PubMed] [Google Scholar]
- Asahi M, Sumii T, Fini ME, Itohara S, Lo EH. Matrix metalloproteinase 2 gene knockout has no effect on acute brain injury after focal ischemia. Neuroreport. 2001a;12:3003–3007. doi: 10.1097/00001756-200109170-00050. [DOI] [PubMed] [Google Scholar]
- Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001b;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16:1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- Bond M, Murphy G, Bennett MR, Newby AC, Baker AH. Tissue inhibitor of metalloproteinase-3 induces a Fas-associated death domain-dependent type II apoptotic pathway. J Biol Chem. 2002;277:13787–13795. doi: 10.1074/jbc.M111507200. [DOI] [PubMed] [Google Scholar]
- Brew K, Dinakarpandian D, Nagase H. Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim Biophys Acta. 2000;1477:267–283. doi: 10.1016/s0167-4838(99)00279-4. [DOI] [PubMed] [Google Scholar]
- Brightman MW, Reese TS. Junctions between intimately apposed cell membranes in the vertebrate brain. J Cell Biol. 1969;40:648–677. doi: 10.1083/jcb.40.3.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinckerhoff CE, Matrisian LM. Matrix metalloproteinases: a tail of a frog that became a prince. Nat Rev Mol Cell Biol. 2002;3:207–214. doi: 10.1038/nrm763. [DOI] [PubMed] [Google Scholar]
- Candelario-Jalil E, Gonzalez-Falcon A, Garcia-Cabrera M, Leon OS, Fiebich BL. Post-ischaemic treatment with the cyclooxygenase-2 inhibitor nimesulide reduces blood-brain barrier disruption and leukocyte infiltration following transient focal cerebral ischaemia in rats. J Neurochem. 2007a;100:1108–1120. doi: 10.1111/j.1471-4159.2006.04280.x. [DOI] [PubMed] [Google Scholar]
- Candelario-Jalil E, Taheri S, Yang Y, Sood R, Grossetete M, Estrada EY, Fiebich BL, Rosenberg GA. Cyclooxygenase inhibition limits blood-brain barrier disruption following intracerebral injection of tumor necrosis factor-alpha in the rat. J Pharmacol Exp Ther. 2007b;323:488–498. doi: 10.1124/jpet.107.127035. [DOI] [PubMed] [Google Scholar]
- Cauwe B, Van den Steen PE, Opdenakker G. The biochemical, biological, and pathological kaleidoscope of cell surface substrates processed by matrix metalloproteinases. Crit Rev Biochem Mol Biol. 2007;42:113–185. doi: 10.1080/10409230701340019. [DOI] [PubMed] [Google Scholar]
- Chang DI, Hosomi N, Lucero J, Heo JH, Abumiya T, Mazar AP, del Zoppo GJ. Activation systems for latent matrix metalloproteinase-2 are upregulated immediately after focal cerebral ischemia. J Cereb Blood Flow Metab. 2003;23:1408–1419. doi: 10.1097/01.WCB.0000091765.61714.30. [DOI] [PubMed] [Google Scholar]
- Choi DH, Kim EM, Son HJ, Joh TH, Kim YS, Kim D, Beal MF, Hwang O. A novel intracellular role of matrix metalloproteinase-3 during apoptosis of dopaminergic cells. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05399.x. (in press) [DOI] [PubMed] [Google Scholar]
- Clark AW, Krekoski CA, Bou SS, Chapman KR, Edwards DR. Increased gelatinase A (MMP-2) and gelatinase B (MMP-9) activities in human brain after focal ischemia. Neurosci Lett. 1997;238:53–56. doi: 10.1016/s0304-3940(97)00859-8. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- Crocker SJ, Milner R, Pham-Mitchell N, Campbell IL. Cell and agonist-specific regulation of genes for matrix metalloproteinases and their tissue inhibitors by primary glial cells. J Neurochem. 2006;98:812–823. doi: 10.1111/j.1471-4159.2006.03927.x. [DOI] [PubMed] [Google Scholar]
- Cuadrado E, Ortega L, Hernandez-Guillamon M, Penalba A, Fernandez-Cadenas I, Rosell A, Montaner J. Tissue plasminogen activator (t-PA) promotes neutrophil degranulation and MMP-9 release. J Leukoc Biol. 2008 doi: 10.1189/jlb.0907606. (in press) [DOI] [PubMed] [Google Scholar]
- Cunningham LA, Wetzel M, Rosenberg GA. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia. 2005;50:329–339. doi: 10.1002/glia.20169. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Dobrogowska DH, Vorbrodt AW. Immunogold localization of tight junctional proteins in normal and osmotically-affected rat blood-brain barrier. J Mol Histol. 2004;35:529–539. doi: 10.1007/10.1007/s10735-004-1318-3. [DOI] [PubMed] [Google Scholar]
- Echchannaoui H, Leib SL, Neumann U, Landmann RM. Adjuvant TACE inhibitor treatment improves the outcome of TLR2−/− mice with experimental pneumococcal meningitis. BMC Infect Dis. 2007;7:25. doi: 10.1186/1471-2334-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing JR, Knight RA, Nagaraja TN, Yee JS, Nagesh V, Whitton PA, Li L, Fenstermacher JD. Patlak plots of Gd-DTPA MRI data yield blood-brain transfer constants concordant with those of 14C-sucrose in areas of blood-brain opening. Magn Reson Med. 2003;50:283–292. doi: 10.1002/mrm.10524. [DOI] [PubMed] [Google Scholar]
- Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123:1777–1788. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasche Y, Copin JC, Sugawara T, Fujimura M, Chan PH. Matrix metalloproteinase inhibition prevents oxidative stress-associated blood-brain barrier disruption after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:1393–1400. doi: 10.1097/00004647-200112000-00003. [DOI] [PubMed] [Google Scholar]
- Gasche Y, Fujimura M, Morita-Fujimura Y, Copin JC, Kawase M, Massengale J, Chan PH. Early appearance of activated matrix metalloproteinase-9 after focal cerebral ischemia in mice: a possible role in blood-brain barrier dysfunction. J Cereb Blood Flow Metab. 1999;19:1020–1028. doi: 10.1097/00004647-199909000-00010. [DOI] [PubMed] [Google Scholar]
- Gasche Y, Soccal PM, Kanemitsu M, Copin JC. Matrix metalloproteinases and diseases of the central nervous system with a special emphasis on ischemic brain. Front Biosci. 2006;11:1289–1301. doi: 10.2741/1883. [DOI] [PubMed] [Google Scholar]
- Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, Chan PH, Park TS. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol. 2005;289:H558–568. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- Gottschall PE, Deb S. Regulation of matrix metalloproteinase expressions in astrocytes, microglia and neurons. Neuroimmunomodulation. 1996;3:69–75. doi: 10.1159/000097229. [DOI] [PubMed] [Google Scholar]
- Gottschall PE, Yu X. Cytokines regulate gelatinase A and B (matrix metalloproteinase 2 and 9) activity in cultured rat astrocytes. J Neurochem. 1995;64:1513–1520. doi: 10.1046/j.1471-4159.1995.64041513.x. [DOI] [PubMed] [Google Scholar]
- Gu Z, Cui J, Brown S, Fridman R, Mobashery S, Strongin AY, Lipton SA. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci. 2005;25:6401–6408. doi: 10.1523/JNEUROSCI.1563-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- Gurney KJ, Estrada EY, Rosenberg GA. Blood-brain barrier disruption by stromelysin-1 facilitates neutrophil infiltration in neuroinflammation. Neurobiol Dis. 2006;23:87–96. doi: 10.1016/j.nbd.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Hahn-Dantona E, Ramos-DeSimone N, Sipley J, Nagase H, French DL, Quigley JP. Activation of proMMP-9 by a plasmin/MMP-3 cascade in a tumor cell model. Regulation by tissue inhibitors of metalloproteinases. Ann N Y Acad Sci. 1999;878:372–387. doi: 10.1111/j.1749-6632.1999.tb07696.x. [DOI] [PubMed] [Google Scholar]
- Haskins J, Gu L, Wittchen ES, Hibbard J, Stevenson BR. ZO-3, a novel member of the MAGUK protein family found at the tight junction, interacts with ZO-1 and occludin. J Cell Biol. 1998;141:199–208. doi: 10.1083/jcb.141.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- Heo JH, Lucero J, Abumiya T, Koziol JA, Copeland BR, del Zoppo GJ. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:624–633. doi: 10.1097/00004647-199906000-00005. [DOI] [PubMed] [Google Scholar]
- Hirase T, Kawashima S, Wong EY, Ueyama T, Rikitake Y, Tsukita S, Yokoyama M, Staddon JM. Regulation of tight junction permeability and occludin phosphorylation by Rhoa-p160ROCK-dependent and -independent mechanisms. J Biol Chem. 2001;276:10423–10431. doi: 10.1074/jbc.M007136200. [DOI] [PubMed] [Google Scholar]
- Hirase T, Staddon JM, Saitou M, Ando-Akatsuka Y, Itoh M, Furuse M, Fujimoto K, Tsukita S, Rubin LL. Occludin as a possible determinant of tight junction permeability in endothelial cells. J Cell Sci. 1997;110 (Pt 14):1603–1613. doi: 10.1242/jcs.110.14.1603. [DOI] [PubMed] [Google Scholar]
- Hornig CR, Bauer T, Simon C, Trittmacher S, Dorndorf W. Hemorrhagic transformation in cardioembolic cerebral infarction. Stroke. 1993;24:465–468. doi: 10.1161/01.str.24.3.465. [DOI] [PubMed] [Google Scholar]
- Hu J, Van den Steen PE, Sang QX, Opdenakker G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat Rev Drug Discov. 2007;6:480–498. doi: 10.1038/nrd2308. [DOI] [PubMed] [Google Scholar]
- Itoh M, Nagafuchi A, Moroi S, Tsukita S. Involvement of ZO-1 in cadherin-based cell adhesion through its direct binding to alpha catenin and actin filaments. J Cell Biol. 1997;138:181–192. doi: 10.1083/jcb.138.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justicia C, Panes J, Sole S, Cervera A, Deulofeu R, Chamorro A, Planas AM. Neutrophil infiltration increases matrix metalloproteinase-9 in the ischemic brain after occlusion/reperfusion of the middle cerebral artery in rats. J Cereb Blood Flow Metab. 2003;23:1430–1440. doi: 10.1097/01.WCB.0000090680.07515.C8. [DOI] [PubMed] [Google Scholar]
- Kamada H, Yu F, Nito C, Chan PH. Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in rats: relation to blood-brain barrier dysfunction. Stroke. 2007;38:1044–1049. doi: 10.1161/01.STR.0000258041.75739.cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda T, Numata Y, Mizusawa H. Chronic inflammatory demyelinating polyneuropathy: decreased claudin-5 and relocated ZO-1. J Neurol Neurosurg Psychiatry. 2004;75:765–769. doi: 10.1136/jnnp.2003.025692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen TM, Swanson RA. Poly(ADP-ribose) polymerase-1 promotes microglial activation, proliferation, and matrix metalloproteinase-9-mediated neuron death. J Immunol. 2005;174:2288–2296. doi: 10.4049/jimmunol.174.4.2288. [DOI] [PubMed] [Google Scholar]
- Kidwell CS, Latour L, Saver JL, Alger JR, Starkman S, Duckwiler G, Jahan R, Vinuela F, Kang DW, Warach S. Thrombolytic toxicity: blood brain barrier disruption in human ischemic stroke. Cerebrovasc Dis. 2008;25:338–343. doi: 10.1159/000118379. [DOI] [PubMed] [Google Scholar]
- Kim KS, Kim HY, Joe EH, Jou I. Matrix metalloproteinase-3 induction in rat brain astrocytes: focus on the role of two AP-1 elements. Biochem J. 2008;410:605–611. doi: 10.1042/BJ20071207. [DOI] [PubMed] [Google Scholar]
- Kim YS, Choi DH, Block ML, Lorenzl S, Yang L, Kim YJ, Sugama S, Cho BP, Hwang O, Browne SE, Kim SY, Hong JS, Beal MF, Joh TH. A pivotal role of matrix metalloproteinase-3 activity in dopaminergic neuronal degeneration via microglial activation. FASEB J. 2007;21:179–187. doi: 10.1096/fj.06-5865com. [DOI] [PubMed] [Google Scholar]
- Kim YS, Kim SS, Cho JJ, Choi DH, Hwang O, Shin DH, Chun HS, Beal MF, Joh TH. Matrix metalloproteinase-3: a novel signaling proteinase from apoptotic neuronal cells that activates microglia. J Neurosci. 2005;25:3701–3711. doi: 10.1523/JNEUROSCI.4346-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb SA, Lahrtz F, Paul R, Leppert D, Nadal D, Pfister HW, Fontana A. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in viral meningitis: upregulation of MMP-9 and TIMP-1 in cerebrospinal fluid. J Neuroimmunol. 1998;84:143–150. doi: 10.1016/s0165-5728(97)00247-6. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Tanaka K. Endothelins stimulate the production of stromelysin-1 in cultured rat astrocytes. Biochem Biophys Res Commun. 2008 doi: 10.1016/j.bbrc.2008.04.064. (in press) [DOI] [PubMed] [Google Scholar]
- Lapchak PA, Chapman DF, Zivin JA. Metalloproteinase inhibition reduces thrombolytic (tissue plasminogen activator)-induced hemorrhage after thromboembolic stroke. Stroke. 2000;31:3034–3040. doi: 10.1161/01.str.31.12.3034. [DOI] [PubMed] [Google Scholar]
- Lee JK, Shin JH, Suh J, Choi IS, Ryu KS, Gwag BJ. Tissue inhibitor of metalloproteinases-3 (TIMP-3) expression is increased during serum deprivation-induced neuronal apoptosis in vitro and in the G93A mouse model of amyotrophic lateral sclerosis: a potential modulator of Fas-mediated apoptosis. Neurobiol Dis. 2008;30:174–185. doi: 10.1016/j.nbd.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Lee KY, Kim EH, Yang WS, Ryu H, Cho SN, Lee BI, Heo JH. Persistent increase of matrix metalloproteinases in cerebrospinal fluid of tuberculous meningitis. J Neurol Sci. 2004;220:73–78. doi: 10.1016/j.jns.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Lee R, Kermani P, Teng KK, Hempstead BL. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945–1948. doi: 10.1126/science.1065057. [DOI] [PubMed] [Google Scholar]
- Lee SR, Guo SZ, Scannevin RH, Magliaro BC, Rhodes KJ, Wang X, Lo EH. Induction of matrix metalloproteinase, cytokines and chemokines in rat cortical astrocytes exposed to plasminogen activators. Neurosci Lett. 2007;417:1–5. doi: 10.1016/j.neulet.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Lee SR, Kim HY, Rogowska J, Zhao BQ, Bhide P, Parent JM, Lo EH. Involvement of matrix metalloproteinase in neuroblast cell migration from the subventricular zone after stroke. J Neurosci. 2006;26:3491–3495. doi: 10.1523/JNEUROSCI.4085-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WJ, Shin CY, Yoo BK, Ryu JR, Choi EY, Cheong JH, Ryu JH, Ko KH. Induction of matrix metalloproteinase-9 (MMP-9) in lipopolysaccharide-stimulated primary astrocytes is mediated by extracellular signal-regulated protein kinase 1/2 (Erk1/2) Glia. 2003;41:15–24. doi: 10.1002/glia.10131. [DOI] [PubMed] [Google Scholar]
- Leib SL, Clements JM, Lindberg RL, Heimgartner C, Loeffler JM, Pfister LA, Tauber MG, Leppert D. Inhibition of matrix metalloproteinases and tumour necrosis factor alpha converting enzyme as adjuvant therapy in pneumococcal meningitis. Brain. 2001;124:1734–1742. doi: 10.1093/brain/124.9.1734. [DOI] [PubMed] [Google Scholar]
- Leppert D, Lindberg RL, Kappos L, Leib SL. Matrix metalloproteinases: multifunctional effectors of inflammation in multiple sclerosis and bacterial meningitis. Brain Res Brain Res Rev. 2001;36:249–257. doi: 10.1016/s0165-0173(01)00101-1. [DOI] [PubMed] [Google Scholar]
- Liebner S, Kniesel U, Kalbacher H, Wolburg H. Correlation of tight junction morphology with the expression of tight junction proteins in blood-brain barrier endothelial cells. Eur J Cell Biol. 2000;79:707–717. doi: 10.1078/0171-9335-00101. [DOI] [PubMed] [Google Scholar]
- Liotta LA, Tryggvason K, Garbisa S, Hart I, Foltz CM, Shafie S. Metastatic potential correlates with enzymatic degradation of basement membrane collagen. Nature. 1980;284:67–68. doi: 10.1038/284067a0. [DOI] [PubMed] [Google Scholar]
- Liu KJ, Rosenberg GA. Matrix metalloproteinases and free radicals in cerebral ischemia. Free Radic Biol Med. 2005;39:71–80. doi: 10.1016/j.freeradbiomed.2005.03.033. [DOI] [PubMed] [Google Scholar]
- Liu L, Kim JY, Koike MA, Yoon YJ, Tang XN, Ma H, Lee H, Steinberg GK, Lee JE, Yenari MA. FasL shedding is reduced by hypothermia in experimental stroke. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05411.x. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- Magnoni S, Baker A, George SJ, Duncan WC, Kerr LE, McCulloch J, Horsburgh K. Differential alterations in the expression and activity of matrix metalloproteinases 2 and 9 after transient cerebral ischemia in mice. Neurobiol Dis. 2004;17:188–197. doi: 10.1016/j.nbd.2004.07.020. [DOI] [PubMed] [Google Scholar]
- McMahon S, Grondin F, McDonald PP, Richard DE, Dubois CM. Hypoxia-enhanced expression of the proprotein convertase furin is mediated by hypoxia-inducible factor-1: impact on the bioactivation of proproteins. J Biol Chem. 2005;280:6561–6569. doi: 10.1074/jbc.M413248200. [DOI] [PubMed] [Google Scholar]
- Meli DN, Christen S, Leib SL. Matrix metalloproteinase-9 in pneumococcal meningitis: activation via an oxidative pathway. J Infect Dis. 2003;187:1411–1415. doi: 10.1086/374644. [DOI] [PubMed] [Google Scholar]
- Meli DN, Coimbra RS, Erhart DG, Loquet G, Bellac CL, Tauber MG, Neumann U, Leib SL. Doxycycline reduces mortality and injury to the brain and cochlea in experimental pneumococcal meningitis. Infect Immun. 2006;74:3890–3896. doi: 10.1128/IAI.01949-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meli DN, Loeffler JM, Baumann P, Neumann U, Buhl T, Leppert D, Leib SL. In pneumococcal meningitis a novel water-soluble inhibitor of matrix metalloproteinases and TNF-alpha converting enzyme attenuates seizures and injury of the cerebral cortex. J Neuroimmunol. 2004;151:6–11. doi: 10.1016/j.jneuroim.2004.01.026. [DOI] [PubMed] [Google Scholar]
- Montaner J, Alvarez-Sabin J, Molina C, Angles A, Abilleira S, Arenillas J, Gonzalez MA, Monasterio J. Matrix metalloproteinase expression after human cardioembolic stroke: temporal profile and relation to neurological impairment. Stroke. 2001;32:1759–1766. doi: 10.1161/01.str.32.8.1759. [DOI] [PubMed] [Google Scholar]
- Montaner J, Molina CA, Monasterio J, Abilleira S, Arenillas JF, Ribo M, Quintana M, Alvarez-Sabin J. Matrix metalloproteinase-9 pretreatment level predicts intracranial hemorrhagic complications after thrombolysis in human stroke. Circulation. 2003;107:598–603. doi: 10.1161/01.cir.0000046451.38849.90. [DOI] [PubMed] [Google Scholar]
- Morita-Fujimura Y, Fujimura M, Gasche Y, Copin JC, Chan PH. Overexpression of copper and zinc superoxide dismutase in transgenic mice prevents the induction and activation of matrix metalloproteinases after cold injury-induced brain trauma. J Cereb Blood Flow Metab. 2000;20:130–138. doi: 10.1097/00004647-200001000-00017. [DOI] [PubMed] [Google Scholar]
- Mun-Bryce S, Lukes A, Wallace J, Lukes-Marx M, Rosenberg GA. Stromelysin-1 and gelatinase A are upregulated before TNF-alpha in LPS-stimulated neuroinflammation. Brain Res. 2002;933:42–49. doi: 10.1016/s0006-8993(02)02303-x. [DOI] [PubMed] [Google Scholar]
- Mun-Bryce S, Rosenberg GA. Gelatinase B modulates selective opening of the blood-brain barrier during inflammation. Am J Physiol. 1998;274:R1203–R1211. doi: 10.1152/ajpregu.1998.274.5.R1203. [DOI] [PubMed] [Google Scholar]
- NINDS . Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med. 1995;333:1581–1587. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- Nuttall RK, Silva C, Hader W, Bar-Or A, Patel KD, Edwards DR, Yong VW. Metalloproteinases are enriched in microglia compared with leukocytes and they regulate cytokine levels in activated microglia. Glia. 2007;55:516–526. doi: 10.1002/glia.20478. [DOI] [PubMed] [Google Scholar]
- Overall CM, Lopez-Otin C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer. 2002;2:657–672. doi: 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Saadoun S, Woodrow CJ, Davies DC, Costa-Martins P, Moss RF, Krishna S, Bell BA. Occludin expression in microvessels of neoplastic and non-neoplastic human brain. Neuropathol Appl Neurobiol. 2001;27:384–395. doi: 10.1046/j.0305-1846.2001.00341.x. [DOI] [PubMed] [Google Scholar]
- Pei P, Horan MP, Hille R, Hemann CF, Schwendeman SP, Mallery SR. Reduced nonprotein thiols inhibit activation and function of MMP-9: implications for chemoprevention. Free Radic Biol Med. 2006;41:1315–1324. doi: 10.1016/j.freeradbiomed.2006.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perides G, Charness ME, Tanner LM, Peter O, Satz N, Steere AC, Klempner MS. Matrix metalloproteinases in the cerebrospinal fluid of patients with Lyme neuroborreliosis. J Infect Dis. 1998;177:401–408. doi: 10.1086/514198. [DOI] [PubMed] [Google Scholar]
- Pfefferkorn T, Rosenberg GA. Closure of the blood-brain barrier by matrix metalloproteinase inhibition reduces rtPA-mediated mortality in cerebral ischemia with delayed reperfusion. Stroke. 2003;34:2025–2030. doi: 10.1161/01.STR.0000083051.93319.28. [DOI] [PubMed] [Google Scholar]
- Planas AM, Sole S, Justicia C. Expression and activation of matrix metalloproteinase-2 and -9 in rat brain after transient focal cerebral ischemia. Neurobiol Dis. 2001;8:834–846. doi: 10.1006/nbdi.2001.0435. [DOI] [PubMed] [Google Scholar]
- Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 1998;29:1020–1030. doi: 10.1161/01.str.29.5.1020. [DOI] [PubMed] [Google Scholar]
- Rosell A, Cuadrado E, Ortega-Aznar A, Hernandez-Guillamon M, Lo EH, Montaner J. MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke. 2008;39:1121–1126. doi: 10.1161/STROKEAHA.107.500868. [DOI] [PubMed] [Google Scholar]
- Rosell A, Lo EH. Multiphasic roles for matrix metalloproteinases after stroke. Curr Opin Pharmacol. 2008;8:82–89. doi: 10.1016/j.coph.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Rosell A, Ortega-Aznar A, Alvarez-Sabin J, Fernandez-Cadenas I, Ribo M, Molina CA, Lo EH, Montaner J. Increased brain expression of matrix metalloproteinase-9 after ischemic and hemorrhagic human stroke. Stroke. 2006;37:1399–1406. doi: 10.1161/01.STR.0000223001.06264.af. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2002;39:279–291. doi: 10.1002/glia.10108. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Cunningham LA, Wallace J, Alexander S, Estrada EY, Grossetete M, Razhagi A, Miller K, Gearing A. Immunohistochemistry of matrix metalloproteinases in reperfusion injury to rat brain: activation of MMP-9 linked to stromelysin-1 and microglia in cell cultures. Brain Res. 2001;893:104–112. doi: 10.1016/s0006-8993(00)03294-7. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29:2189–2195. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Estrada EY, Dencoff JE, Stetler-Stevenson WG. Tumor necrosis factor-alpha-induced gelatinase B causes delayed opening of the blood-brain barrier: an expanded therapeutic window. Brain Res. 1995;703:151–155. doi: 10.1016/0006-8993(95)01089-0. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Navratil M, Barone F, Feuerstein G. Proteolytic cascade enzymes increase in focal cerebral ischemia in rat. J Cereb Blood Flow Metab. 1996;16:360–366. doi: 10.1097/00004647-199605000-00002. [DOI] [PubMed] [Google Scholar]
- Schonbeck U, Mach F, Libby P. Generation of biologically active IL-1 beta by matrix metalloproteinases: a novel caspase-1-independent pathway of IL-1 beta processing. J Immunol. 1998;161:3340–3346. [PubMed] [Google Scholar]
- Si-Tayeb K, Monvoisin A, Mazzocco C, Lepreux S, Decossas M, Cubel G, Taras D, Blanc JF, Robinson DR, Rosenbaum J. Matrix metalloproteinase 3 is present in the cell nucleus and is involved in apoptosis. Am J Pathol. 2006;169:1390–1401. doi: 10.2353/ajpath.2006.060005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M. Signaling at the gliovascular interface. J Neurosci. 2003;23:9254–9262. doi: 10.1523/JNEUROSCI.23-27-09254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sole S, Petegnief V, Gorina R, Chamorro A, Planas AM. Activation of matrix metalloproteinase-3 and agrin cleavage in cerebral ischemia/reperfusion. J Neuropathol Exp Neurol. 2004;63:338–349. doi: 10.1093/jnen/63.4.338. [DOI] [PubMed] [Google Scholar]
- Sood RR, Taheri S, Candelario-Jalil E, Estrada EY, Rosenberg GA. Early beneficial effect of matrix metalloproteinase inhibition on blood-brain barrier permeability as measured by magnetic resonance imaging countered by impaired long-term recovery after stroke in rat brain. J Cereb Blood Flow Metab. 2008;28:431–438. doi: 10.1038/sj.jcbfm.9600534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporer B, Paul R, Koedel U, Grimm R, Wick M, Goebel FD, Pfister HW. Presence of matrix metalloproteinase-9 activity in the cerebrospinal fluid of human immunodeficiency virus-infected patients. J Infect Dis. 1998;178:854–857. doi: 10.1086/515342. [DOI] [PubMed] [Google Scholar]
- Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumii T, Lo EH. Involvement of matrix metalloproteinase in thrombolysis-associated hemorrhagic transformation after embolic focal ischemia in rats. Stroke. 2002;33:831–836. doi: 10.1161/hs0302.104542. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Nagai N, Umemura K, Collen D, Lijnen HR. Stromelysin-1 (MMP-3) is critical for intracranial bleeding after t-PA treatment of stroke in mice. J Thromb Haemost. 2007;5:1732–1739. doi: 10.1111/j.1538-7836.2007.02628.x. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Molineaux SM. Interleukin-1 beta converting enzyme: a novel cysteine protease required for IL-1 beta production and implicated in programmed cell death. Protein Sci. 1995;4:3–12. doi: 10.1002/pro.5560040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JA, Alexander S, Estrada EY, Hines C, Cunningham LA, Rosenberg GA. Tissue inhibitor of metalloproteinase-3 is associated with neuronal death in reperfusion injury. J Cereb Blood Flow Metab. 2002;22:1303–1310. doi: 10.1097/01.WCB.0000040943.89393.c1. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhang ZG, Zhang RL, Gregg SR, Hozeska-Solgot A, LeTourneau Y, Wang Y, Chopp M. Matrix metalloproteinase 2 (MMP2) and MMP9 secreted by erythropoietin-activated endothelial cells promote neural progenitor cell migration. J Neurosci. 2006;26:5996–6003. doi: 10.1523/JNEUROSCI.5380-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Jung J, Asahi M, Chwang W, Russo L, Moskowitz MA, Dixon CE, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. J Neurosci. 2000;20:7037–7042. doi: 10.1523/JNEUROSCI.20-18-07037.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Watry DD, Marcondes MC, Fox HS. Selective decrease in paracellular conductance of tight junctions: role of the first extracellular domain of claudin-5. Mol Cell Biol. 2004;24:8408–8417. doi: 10.1128/MCB.24.19.8408-8417.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel M, Li L, Harms KM, Roitbak T, Ventura PB, Rosenberg GA, Khokha R, Cunningham LA. Tissue inhibitor of metalloproteinases-3 facilitates Fas-mediated neuronal cell death following mild ischemia. Cell Death Differ. 2008;15:143–151. doi: 10.1038/sj.cdd.4402246. [DOI] [PubMed] [Google Scholar]
- Wetzel M, Rosenberg GA, Cunningham LA. Tissue inhibitor of metalloproteinases-3 and matrix metalloproteinase-3 regulate neuronal sensitivity to doxorubicin-induced apoptosis. Eur J Neurosci. 2003;18:1050–1060. doi: 10.1046/j.1460-9568.2003.02838.x. [DOI] [PubMed] [Google Scholar]
- Woo MS, Park JS, Choi IY, Kim WK, Kim HS. Inhibition of MMP-3 or -9 suppresses lipopolysaccharide-induced expression of proinflammatory cytokines and iNOS in microglia. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05430.x. (in press) [DOI] [PubMed] [Google Scholar]
- Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- Yong VW. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci. 2005;6:931–944. doi: 10.1038/nrn1807. [DOI] [PubMed] [Google Scholar]
- Zhao BQ, Wang S, Kim HY, Storrie H, Rosen BR, Mooney DJ, Wang X, Lo EH. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med. 2006;12:441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]
- Zucker S, Pei D, Cao J, Lopez-Otin C. Membrane type-matrix metalloproteinases (MT-MMP) Curr Top Dev Biol. 2003;54:1–74. doi: 10.1016/s0070-2153(03)54004-2. [DOI] [PubMed] [Google Scholar]