

Abstract
PFKFB3is a target gene of peroxisome proliferator-activated receptor gamma (PPARγ) and encodes for inducible 6-phosphofructo-2-kinase (iPFK2). As a key regulatory enzyme that stimulates glycolysis, PFKFB3/iPFK2 links adipocyte metabolic and inflammatory responses. Additionally, PFKFB3/iPFK2 is involved in the effect of active PPARγ on suppressing overnutrition-induced adipose tissue inflammatory response, which accounts for the insulin-sensitizing and antidiabetic effects of PPARγ activation. Using PFKFB3/iPFK2-disrupted mice, the present study investigated the role of PFKFB3/iPFK2 in regulating overnutrition-associated intestine inflammatory response and in mediating the effects of PPARγ activation. In wild-type mice, intestine PFKFB3/iPFK2 was increased in response to high-fat diet (HFD) feeding compared with that in mice fed a low-fat diet. However, intestine PFKFB3/iPFK2 was decreased in PFKFB3/iPFK2-disrupted mice and did not respond to HFD feeding. Furthermore, on an HFD, PFKFB3/iPFK2-disrupted mice displayed a significant increase in major intestine proinflammatory indicators such as toll-like receptor 4 expression, c-Jun N-terminal kinase 1 and nuclear factor kappa B phosphorylation, and proinflammatory cytokine expression compared with wild-type littermates. Upon treatment with rosiglitazone, an agonist of PPARγ, intestine proinflammatory indicators were markedly decreased in wild-type mice, but to a much lesser degree in PFKFB3/iPFK2-disrupted mice. Overall, the status of HFD-induced intestine inflammatory response in all treated mice correlated inversely with systemic insulin sensitivity, indicated by the homeostasis model assessment of insulin resistance data. Together, these results suggest that PFKFB3/iPFK2 is critically involved in the effect of PPARγ activation on suppressing diet-induced intestine inflammatory response.
Keywords: Inducible 6-phosphofructo-2-kinase, PPARγ, Overnutrition, Intestine, Inflammatory response
1. Introduction
Chronic low-grade inflammation in adipose tissue is a feature of overnutrition and critically contributes to the pathogenesis of insulin resistance and a wide variety of metabolic diseases [1–4]. On the other hand, suppressing adipose tissue inflammatory response by active peroxisome proliferator-activated receptor gamma (PPARγ), a nuclear receptor whose activation accounts for the insulin-sensitizing and antidiabetic effects of thiazolidinediones (TZDs) [5–12], is considered as one of the two adipose tissue-based mechanisms that underlie the beneficial effects of PPARγ activation [10,13]. However, the regulation of overnutrition-associated inflammatory response in intestine, where nutrients interact with host cells at a stage earlier than with adipocytes, is poorly understood. Additionally, the mechanisms underlying PPARγ suppression of intestine inflammation in relation to systemic insulin sensitivity remain to be elucidated.
Intestine is an organ for digestion, absorption and assimilation of nutrients. Physiologically, nutrients absorbed by intestine, along with a number of nutritional and hormonal signals, are delivered to both central and peripheral tissues in response to feeding. This leads to appropriate regulation of nutrient metabolism in key metabolic tissues such as liver, adipose tissue and skeletal muscle to maintain systemic metabolic homeostasis and insulin sensitivity [14,15]. Pathologically, nutrient overload disturbs glucose and lipid metabolism and triggers the inflammatory response in intestine. For example, feeding a high-fat diet (HFD) to mice activates nuclear factor kappa B (NF-κB) activity in intestine cells including epithelial cells, immune cells and endothelial cells of small intestine [16], which appears to contribute to HFD-induced insulin resistance and adiposity. Intestine also hosts microbes, whose composition, when altered by overnutrition, contributes to increased intestine inflammatory response and the development of systemic insulin resistance [16–19]. At this point, little is known about how glucose and lipid metabolism is orchestrated to regulate intestine inflammatory response. In terms of managing intestine inflammatory response, adequate expression of PPARγ in epithelial cells appears to be required for prevention of inflammatory bowel disease [20,21]. Given this, addressing the link between intestine nutrient metabolism and the anti-intestine-inflammatory effect of active PPARγ is of particular importance to a better understanding of the pathophysiology of overnutrition-associated insulin resistance and inflammatory intestine diseases.
PFKFB3 is the gene that encodes for the inducible 6-phosphofructo-2-kinase (iPFK2). Additionally, PFKFB3 is a target gene of PPARγ [22] and is stimulated by TZDs [23]. At the cellular level, iPFK2 generates fructose-2,6-bisphosphate, which is the most powerful activator of 6-phosphofructo-1-kinase to enhance glycolysis [24,25]. Using PFKFB3/iPFK2-disrupted (PFKFB3+/−) mice, it has been previously shown that PFKFB3/iPFK2 protects against diet-induced insulin resistance and adipose tissue inflammatory response [26]. Additionally, PFKFB3/iPFK2 is involved in the antidiabetic effect of PPARγ activation, at least, by suppressing excessive fatty acid oxidation-related reactive oxygen species (ROS) production and inflammatory responses in adipose tissue/adipocytes. In the present study, the effect of PFKFB3/iPFK2 disruption on HFD-induced intestine inflammatory response was determined, and the involvement of PFKFB3/iPFK2 in the effect of PPARγ activation on suppressing HFD-induced intestine inflammatory response was addressed.
2. Methods and materials
2.1. Animal experiments
All mice were maintained on a 12:12-h light–dark cycle (lights on at 06:00). For PFKFB3/iPFK2 distribution study, male wild-type C57BL/6J mice were fed ad libitum. At 12–14 weeks of age, mice were euthanized for collection of tissue samples. To examine dietary effects on intestine PFKFB3/iPFK2 expression and the inflammatory response, male wild-type C57BL/6J mice, at 5–6 weeks of age, were fed an HFD (60% fat calories, 20% protein calories and 20 carbohydrate calories) or a low-fat diet (LFD) (10% fat calories, 20% protein calories and 70 carbohydrate calories) for 12 weeks. Both diets are products of Research Diets, Inc. (New Brunswick, NJ, USA). Details of diet composition are provided in Table 1. To determine the role of PFKFB3/iPFK2 in regulating intestine inflammatory response, PFKFB3/iPFK2-disrupted mice were included. Homozygous disruption of PFKFB3/iPFK2 is embryonically lethal [27]. Thus, PFKFB3+/− mice, generated as previously described [27], were used in the present study. Given that rosiglitazone lowers the levels of plasma glucose and improves insulin sensitivity only in diabetic mice, male PFKFB3+/− and wild-type littermates (C57BL/6J background), at 5–6 weeks of age, were fed an HFD for 12 weeks [26]. During the last 4 weeks of the feeding regimen, HFD-fed mice were treated with rosiglitazone [10 mg/kg/day in phosphate-buffered saline (PBS); Avandia tablets] or vehicle (PBS) via oral gavages as previously described [28]. Before and during rosiglitazone or PBS treatment regimen, fecal samples of the treated mice were collected and used for microbiota analysis. At the end of the feeding/treatment regimen, all mice were fasted for 4 h before collection of blood and tissue samples [29–31]. Frozen intestine samples were subjected to further analyses. All study protocols were approved by the Institutional Animal Care and Use Committees of Texas A&M University.
Table 1.
Composition of diets
| LFD | HFD | |||
|---|---|---|---|---|
| g% | kcal% | g% | kcal% | |
| Casein | 18.96 | 19.72 | 25.84 | 19.72 |
| L-Cystine | 0.28 | 0.30 | 0.39 | 0.30 |
| Corn starch | 29.86 | 31.06 | 0.00 | 0.00 |
| Maltodextrin | 3.32 | 3.45 | 16.15 | 12.32 |
| Sucrose | 33.17 | 34.51 | 8.89 | 6.78 |
| Cellulose | 4.74 | 0.00 | 6.46 | 0.00 |
| Soybean oil | 2.37 | 5.55 | 3.23 | 5.55 |
| Lard | 1.90 | 4.44 | 31.66 | 54.35 |
2.2. Determination of PFKFB3 mRNA and iPFK2 amount
Intestine PFKFB3 mRNA and iPFK2 amount were determined using real-time reverse transcriptase polymerase chain reaction (RT-PCR) and Western blot analyses, respectively, as described below.
2.3. RNA isolation, reverse transcription and real-time PCR
The total RNA was isolated from frozen intestine (jejunum and ileum) samples. RNA isolation and real-time RT-PCR were performed as previously described [31]. The mRNA levels were analyzed for toll-like receptor 4 (TLR4), tumor necrosis factor alpha (TNFα) and interleukin 6 (IL-6).
2.4. Western blot
Lysates were prepared from frozen intestine (jejunum and ileum) samples. Western blots were conducted as previously described [30,31]. The levels of iPFK2, c-Jun N-terminal kinase (JNK), phospho-JNK (Thr183), NF-κB p65 and phospho-p65 (Ser468) were analyzed.
2.5. Measurement of microbiota composition
Before and during treatment with rosiglitazone or PBS, fecal samples of HFD-fed mice were collected, pooled and homogenized. Total genomic DNA was isolated and subjected to real-time PCR using primers specific to Bifidobacterium [32] and Lactobacillus [33].
2.6. Evaluation of systemic insulin sensitivity
Plasma levels of glucose and insulin were measured as previously described [26] and used to calculate homeostasis model assessment of insulin resistance (HOMA-IR), an indicator of systemic insulin resistance, using the following equation: HOMA-IR=basal glucose (mmol/L)×basal insulin (mU/L)/22.5.
2.7. Statistical methods
Numeric data are presented as means±standard error (S.E.). Statistical significance was assessed by unpaired, two-tailed analysis of variance or Student’s t test. Differences were considered significant at the two-tailed P<.05.
3. Results
3.1. PFKFB3/iPFK2 is expressed at high abundance in intestine
The amount of iPFK2 was determined in various tissues in wild-type mice. Among the key tissues that are involved in the regulation of systemic insulin sensitivity and metabolic homeostasis, iPFK2 was at high abundance in intestine and white adipose tissue but at very low abundance in the liver, skeletal muscle and brown adipose tissue (Fig. 1).
Fig. 1.
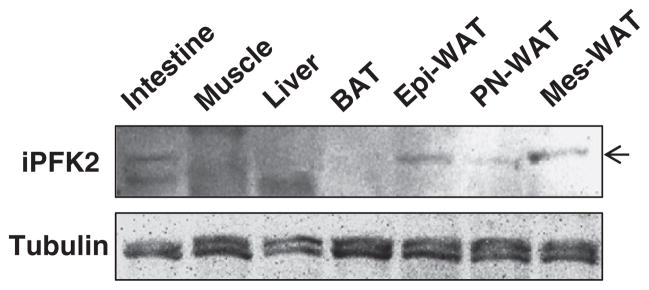
PFKFB3/iPFK2 expression in key metabolic tissues. Male wild-type C57BL/6J mice were fed ad libitum. At 12–14 weeks of age, mice were euthanized for collection of tissue samples. Tissue lysates were prepared to determine the amount of iPFK2 using Western blot analyses. BAT, brown adipose tissue; Epi-WAT, epididymal white adipose tissue; PN, perinephric; Mes, mesenteric.
3.2. HFD feeding stimulates intestine PFKFB3/iPFK2 expression and induces intestine inflammatory response
HFD feeding induces inflammation in intestine [16]. To address dietary responses of intestine PFKFB3/iPFK2 expression in relation to inflammatory responses in intestine, the amount of intestine iPFK2 in LFD- and/or HFD-fed wild-type C57BL/6J mice was examined. Compared with LFD-fed mice, HFD-fed mice displayed an increase in intestine iPFK2 amount (Fig. 2A). This increase in intestine PFKFB3/iPFK2 expression appears to be a defensive response, given a critical role for PFKFB3/iPFK2 in protecting against diet-induced intestine inflammatory response (see below in Fig. 4). Next, intestine inflammatory response was examined. Compared with controls, the phosphorylation of JNK1 in intestine of HFD-fed mice was increased (Fig. 2B), although the phosphorylation of NF-κB was undetectable in mice on either an HFD or LFD (data not shown). Additionally, in HFD-fed mice, intestine mRNA levels of TNFα and IL-6 were significantly higher than their respective levels in controls (Fig. 2C). These results demonstrate an increase in intestine inflammatory response in HFD-fed mice.
Fig. 2.

Effects of HFD feeding on intestine PFKFB3/iPFK2 and inflammatory responses. Male wild-type C57BL/6J mice, at 5–6 weeks of age, were fed an LFD or HFD for 12 weeks. For panels (A) and (B), intestine lysates were subjected to Western blot analyses. (A) Intestine amount of iPFK2. (B) Intestine amount of JNK and phospho-JNK. (C) The total RNA of intestine was prepared to determine the expression of proinflammatory cytokines using real-time RT-PCR. Data are means±S.E., n=4. *P<.05 HFD vs. LFD for the same gene.
Fig. 4.

Involvement of PFKFB3/iPFK2 in the effect of PPARγ activation on diet-induced intestine inflammatory response. Male PFKFB3+/− mice and wild-type littermates, at 5–6 weeks of age, were fed an HFD for 12 weeks and treated with rosiglitazone (Rosi, 10 mg/kg/day in PBS) or vehicle (PBS) orally for the last 4 weeks of the feeding regimen. (A) Intestine mRNA levels of TLR4 were quantified using real-time RT-PCR. (B) Intestine inflammatory signaling. Intestine lysates were prepared to determine the amount and phosphorylation states of JNK and NF-κB p65 using Western blot analyses. (C) Intestine mRNA levels of TNFα and IL-6 were quantified using real-time RT-PCR. Left panel, representative PCR products; and right two panels, relative intestine mRNA levels. For panels (A) and (C), numeric data are means±S.E.; n=4–6. *P<.05 and **P<.01, rosiglitazone vs. vehicle for the same genotype; †P<.05 and ††P<.01, PFKFB3+/− vs. wild type for the same treatment (rosiglitazone or vehicle).
3.3. PFKFB3/iPFK2 disruption blunts dietary response of intestine iPFK2
The response of intestine PFKFB3/iPFK2 to HFD feeding was examined in PFKFB3+/− mice. Heterozygous PFKFB3 disruption was confirmed using PCR analyses of genomic DNA (Fig. 3A). On an LFD, intestine iPFK2 amount in PFKFB3+/− mice was lower than in wild-type littermates (C57BL/6J background) (Fig. 3B), further demonstrating PFKFB3/iPFK2 disruption. Upon feeding an HFD, PFKFB3+/− mice did not exhibit an increase in intestine iPFK2 amount as did wild-type mice. Thus, intact PFKFB3/iPFK2 appears to be required for a defensive increase in intestine PFKFB3/iPFK2 in response to HFD feeding.
Fig. 3.
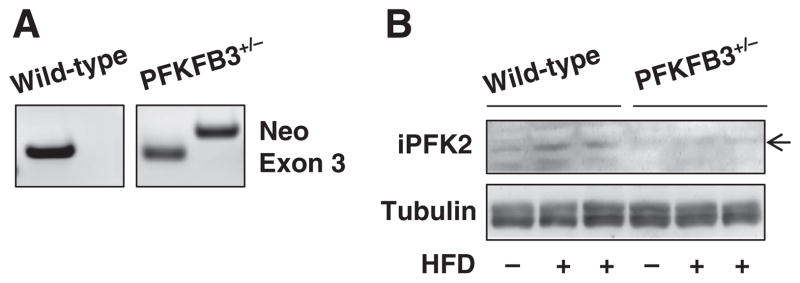
Effects of PFKFB3/iPFK2 disruption on dietary response of intestine iPFK2. (A) Validation of heterozygous PFKFB3/iPFK2 disruption. Genomic DNA of PFKFB3+/− mice and wild-type littermates was subjected to PCR analyses using an exon 2-specific primer with an exon 3-specific primer (WT) or a neomycin-specific primer (Neo). (B) Male PFKFB3+/− mice and wild-type littermates, at 5–6 weeks of age, were fed an LFD or HFD for 12 weeks. Intestine lysates were prepared to determine the amount of iPFK2 using Western blot analyses.
3.4. PFKFB3/iPFK2 disruption exacerbates HFD-induced intestine inflammatory response and partially blunts the effects of PPARγ activation
The effect of PFKFB3/iPFK2 disruption on diet-induced intestine inflammatory response was examined. Compared with HFD-fed wild-type littermates, HFD-fed PFKFB3+/− mice exhibited an increase in the mRNA levels of intestine TLR4 (Fig. 4A), a receptor whose activation leads to increased proinflammatory responses. In addition, the phosphorylation of JNK1 and NF-κB p65, two key signaling pathways that mediate proinflammatory responses, in intestine of HFD-fed PFKFB3+/− mice was much greater than that in controls (Fig. 4B). Consistent with increased signaling through proinflammatory pathways, the mRNA levels of intestine TNFα and IL-6 in HFD-fed PFKFB3+/− mice were higher than their respective levels in controls (Fig. 4C). These results, in combination, suggest a protective role for PFKFB3/iPFK2 in diet-induced intestine inflammatory response.
PPARγ has a protective role in intestine inflammation [20,21]. The effects of PPARγ activation on diet-induced intestine inflammatory response were examined. Upon treatment with rosiglitazone, the mRNA levels of TLR4 (Fig. 4A), the phosphorylation of JNK1 (Fig. 4B), and the mRNA levels of TNFα and IL-6 (Fig. 4C) were decreased in intestine of HFD-fed wild-type littermates and, to a much lesser degree, in intestine of HFD-fed PFKFB3+/− mice. Additionally, intestine NF-κB p65 phosphorylation remained high in HFD-fed PFKFB3+/− mice compared with that in HFD-fed wild-type littermates after treatment with rosiglitazone (Fig. 4B). Together, these results suggest that PFKFB3/iPFK2 disruption partially blunts the effect of PPARγ activation on suppressing HFD-induced intestine inflammatory response.
3.5. PFKFB3/iPFK2 disruption decreases intestine proliferation of lactobacilli in HFD-fed mice in response to PBS and/or rosiglitazone treatment
Intestine microbiotas not only control the inflammatory response in intestine but also critically regulate systemic insulin sensitivity [17–19]. The composition of Lactobacillus and Bifidobacterium in fecal samples of HFD-fed mice was analyzed and used to reflect changes in intestine proliferation of Lactobacillus and Bifidobacterium. Compared with controls, the proliferation of Lactobacillus in HFD-fed PFKFB3+/− mice was decreased upon treatment with either rosiglitazone or PBS (Fig. 5A). However, the proliferation of Bifidobacterium did not show significant differences among all four groups of mice (Fig. 5B). These results suggest that oral dosing decreases intestine Lactobacillus proliferation when PFKFB3/iPFK2 is disrupted. In other words, treatment with rosiglitazone has a limited role in altering intestine proliferation of Lactobacillus and Bifidobacterium in HFD-fed mice.
Fig. 5.

Alterations of intestine microbiota composition. Male PFKFB3+/− mice and wild-type littermates, at 5–6 weeks of age, were fed an HFD for 12 weeks and treated with rosiglitazone (Rosi, 10 mg/kg/day in PBS) or vehicle (PBS) orally for the last 4 weeks of the feeding regimen. Before, during and after treatment with rosiglitazone or PBS, fecal samples of HFD-fed mice were collected, pooled and homogenized. Total genomic DNA was isolated and subjected to quantitative real-time PCR using primers specific to Lactobacillus (A) and Bifidobacterium (B). Ct, cycle threshold.
3.6. PFKFB3/iPFK2 disruption blunts the insulin-sensitizing effect of PPARγ activation
Feeding an HFD to mice induces intestine inflammatory response, which contributes to the development of systemic insulin resistance [34]. Upon treatment with rosiglitazone, HFD-fed wild-type mice displayed a marked decrease in HOMA-IR (Fig. 6), an indicator of insulin resistance. Significantly, the decrease in HOMA-IR in HFD-fed and rosiglitazone-treated wild-type mice was accompanied by decreased intestine inflammatory response as described above (Fig. 4). In contrast, treatment with rosiglitazone only caused an insignificant decrease in HOMA-IR in HFD-fed PFKFB3+/− mice (Fig. 6), which was accompanied by increased intestine inflammatory response. The latter was at a much greater degree than that in HFD-fed and rosiglitazone-treated wild-type mice. These results, in combination, indicate a positive correlation between systemic insulin resistance and intestine inflammatory response, which is regulated by PFKFB3/iPFK2.
Fig. 6.
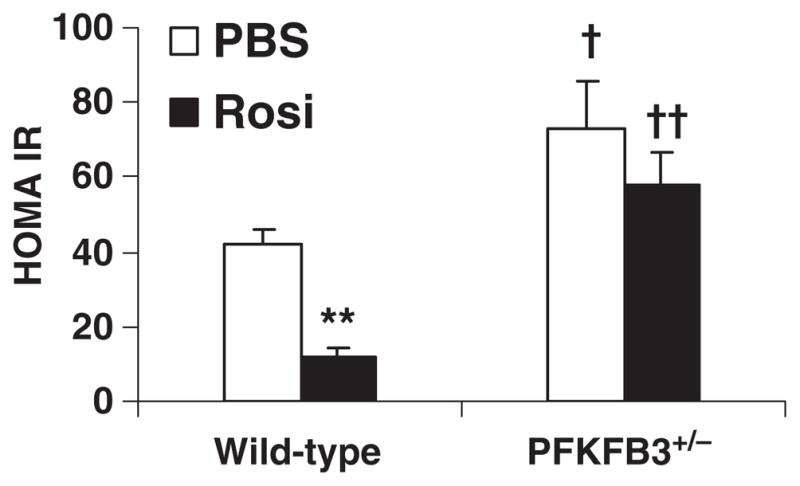
PFKFB3/iPFK2 regulation of systemic insulin sensitivity. Male PFKFB3+/− mice and wild-type littermates, at 5–6 weeks of age, were fed an HFD for 12 weeks and treated with rosiglitazone (Rosi, 10 mg/kg/day in PBS) or vehicle (PBS) orally for the last 4 weeks of the feeding regimen. At the end of feeding/treatment regimen, blood samples were collected and used to measure plasma levels of glucose and insulin. HOMA-IR was calculated using the following equation: HOMA-IR=basal glucose (mmol/L)×basal insulin (mU/L)/22.5. Data are means±S.E.; n=4–6. **P<.01, rosiglitazone vs. vehicle for the same genotype; †P<.05 and ††P<.01, PFKFB3+/− vs. wild type for the same treatment (rosiglitazone or vehicle).
4. Discussion
The mechanisms by which overnutrition induces intestine inflammation in relation to systemic insulin resistance and the actions of PPARγ activation remain to be elucidated. The present study provides evidence to support a critical role for PFKFB3/iPFK2, a regulator that links glucose and fatty acid metabolism and inflammatory responses, in protecting against diet-induced intestine inflammatory response. Notably, major intestine inflammatory biomarkers, including the mRNA levels of TLR4, TNFα and IL-6, as well as the phosphorylation of JNK1 and NF-κB p65, in PFKFB3/iPFK2-disrupted mice were higher than their respective levels in wild-type controls under the condition of overnutrition, i.e., HFD feeding. In addition, PFKFB3/iPFK2 appears to be needed for PPARγ activation to fully suppress diet-induced intestine inflammatory response.
It has been previously shown that PFKFB3/iPFK2 disruption exacerbates diet-induced adipose tissue inflammatory response [26,28]. At the cellular level, PFKFB3/iPFK2 disruption-associated increase in fatty acid oxidation leads to increased production of ROS and oxidative stress, thereby triggering adipocyte inflammatory response. This mechanism may also exist in intestine, given that PFKFB3/iPFK2 is abundantly expressed in intestine. In the present study, intestine iPFK2 amount in PFKFB3/iPFK2-disrupted mice was lower than that in wild-type mice and did not respond to HFD feeding as did intestine iPFK2 in wild-type mice in a defensive way. Furthermore, intestine iPFK2 amount negatively correlated with the degree of intestine inflammatory response. Because of this, it appears to be clear that intact PFKFB3/iPFK2 is required for full protection of overnutrition-induced intestine inflammatory responses. At this point, however, the extent to which the PFKFB3/iPFK2 in intestine cells, in particular the PFKFB3/iPFK2 in intestinal epithelial cells, protects against diet-induced intestine inflammatory response is unknown. What should also be noted is that adipose tissue inflammatory response is elevated in PFKFB3/iPFK2-disrupted mice [26]. Of importance, the status of adipose tissue inflammatory response determines the outcome of inflammatory responses in distal tissues including the liver [35]. As such, a possible contribution of elevated adipose tissue inflammation to an increase in diet-induced intestine inflammatory response in PFKFB3/iPFK2-disrupted mice cannot be ruled out. Considering this, there may exist a vicious cycle for inflammatory responses between adipose tissue and intestine, regardless of how an initiator triggers inflammatory responses.
The essential role for PFKFB3/iPFK2 in protecting against diet-induced intestine inflammatory response is further supported by the involvement of PFKFB3/iPFK2 in the anti-inflammatory effect of PPARγ activation. As a target gene of PPARγ, PFKFB3/iPFK2 is stimulated by TZDs [22,23,28]. Of significance, intact PFKFB3/iPFK2 is needed for actions of active PPARγ on channeling fatty acids to triglyceride synthesis to reduce excessive fatty acid oxidation-associated production of ROS, thereby suppressing inflammatory signaling through the JNK1 and NF-κB pathways and decreasing proinflammatory cytokine expression in adipocytes/adipose tissue. In the present study, in addition to causing an increase in major intestine proinflammatory indicators, PFKFB3/iPFK2 disruption also partially blunted the effect of rosiglitazone on suppressing diet-induced intestine inflammatory response. These changes in intestine inflammatory response were nearly identical to those observed in adipose tissue in PFKFB3/iPFK2-disrupted mice upon treatment with rosiglitazone [28]. Because of this, it is conceivable that intact PFKFB3/iPFK2 is needed for PPARγ activation to suppress overnutrition-induced intestine inflammatory response. On the other hand, although PPARγ activation brought about an anti-inflammatory effect in intestine in a manner consistent with that reported previously [20,21], suppressing diet-induced adipose tissue inflammatory response may be a prerequisite for PPARγ activation to suppress diet-induced intestine inflammatory response, given the primary role played by adipose tissue in the actions of PPARγ activation. Indeed, failure of rosiglitazone in fully suppressing diet-induced intestine inflammatory response reported herein was accompanied by defects in actions of rosiglitazone on reserving adipose tissue inflammatory response in PFKFB3/iPFK2-disrupted mice [28].
Suppressing inflammatory responses by active PPARγ underlies the insulin-sensitizing and antidiabetic effects of TZDs [10,13,28]. In the present study, the degree of intestine inflammatory response in rosiglitazone- and/or control-treated PFKFB3/iPFK2-disrupted mice and wild-type mice positively correlated with insulin resistance, indicated by HOMA-IR results. This observation argues in favor of the notion that PFKFB3/iPFK2 protection of diet-induced intestine inflammatory response is of importance to systemic insulin sensitivity and glucose homeostasis, as well as insulin sensitization brought about by PPARγ activation. As discussed before, there may exist a vicious cycle for inflammatory responses between adipose tissue and intestine during overnutrition. Considering this, intact PFKFB3/iPFK2 likely enables PPARγ activation to suppress inflammatory responses in both adipose tissue and intestine to achieve a systemic insulin-sensitizing effect. In other words, intact PFKFB3/iPFK2 enables PPARγ activation to suppress intestine inflammatory response to contribute to systemic insulin sensitization by working with or without PPARγ suppression of adipose tissue inflammatory response in a manner involving PFKFB3/iPFK2 [28].
In intestine, microbiota not only control energy absorption but also critically regulate inflammatory responses of intestine cells [17,18]. New evidence further demonstrates that interactions between HFD and microbiota promote inflammation in small intestine, which precedes and correlates with obesity and insulin resistance [16]. In the present study, HFD-fed PFKFB3/iPFK2-disrupted mice showed a decrease in the proliferation of intestine Lactobacillus in response to treatment of either rosiglitazone or PBS compared with HFD-fed wild-type mice. These results, on the one hand, suggest that PFKFB3/iPFK2 disruption creates an intestinal environment that allows oral dosing to alter intestine microbiota composition. On the other hand, treatment with rosiglitazone has a limited role in altering the proliferation of intestine Lactobacillus. Although it is unknown whether or not decreased proliferation of Lactobacillus contributes to an increase in intestine inflammatory response in PFKFB3/iPFK2-disrupted mice, a potential role for PFKFB3/iPFK2 in modulating the interactions between microbiotas and intestine cells could serve as additional mechanism(s) by which PFKFB3/iPFK2 regulates intestine inflammatory response. This point is worthy of further investigation.
In summary, the present study demonstrates a novel role for PFKFB3/iPFK2 in regulating intestine inflammatory response and provides data to support the involvement of PFKFB3/iPFK2 in the effect of PPARγ activation on suppressing diet-induced intestine inflammatory response. Furthermore, PFKB3/iPFK2 protection of intestine inflammatory response correlates well with systemic insulin sensitivity. Because of this, activation of intestinal PFKFB3/iPFK2 may be an approach to reversing overnutrition-associated intestine inflammatory response and to improving systemic insulin sensitivity.
Acknowledgments
This work was supported, in whole or in part, by NIH HL105114 (Y.E.C.) and by ADA grant 1-10-JF-54 and AHA 12BGIA9050003 (to C.W.).
Footnotes
This work was supported in part by William W. Allen Foundation (PI: C.W.; Endowment Recipient: Joanne Lupton), American Diabetes Association grant (1-10-JF-54) and American Heart Association grant (12BGIA9050003) (to C.W.) and National Institutes of Health (HL105114 to Y.E.C.).
References
- 1.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–30. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest. 2008;118:2992–3002. doi: 10.1172/JCI34260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ota T, Takamura T, Kurita S, Matsuzawa N, Kita Y, Uno M, et al. Insulin resistance accelerates a dietary rat model of nonalcoholic steatohepatitis. Gastroenterology. 2007;132:282–93. doi: 10.1053/j.gastro.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 4.Ohman MK, Shen Y, Obimba CI, Wright AP, Warnock M, Lawrence DA, et al. Visceral adipose tissue inflammation accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2008;117:798–805. doi: 10.1161/CIRCULATIONAHA.107.717595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor γ (PPAR γ) J Biol Chem. 1995;270:12953–6. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 6.Berger J, Bailey P, Biswas C, Cullinan C, Doebber T, Hayes N, et al. Thiazolidine-diones produce a conformational change in peroxisomal proliferator-activated receptor-gamma: binding and activation correlate with antidiabetic actions in db/db mice. Endocrinology. 1996;137:4189–95. doi: 10.1210/endo.137.10.8828476. [DOI] [PubMed] [Google Scholar]
- 7.Lebovitz HE, Dole JF, Patwardhan R, Rappaport EB, Freed MI. Rosiglitazone monotherapy is effective in patients with type 2 diabetes. J Clin Endocrinol Metab. 2001;86:280–8. doi: 10.1210/jcem.86.1.7157. [DOI] [PubMed] [Google Scholar]
- 8.Phillips LS, Grunberger G, Miller E, Patwardhan R, Rappaport EB, Salzman A. Once-and twice-daily dosing with rosiglitazone improves glycemic control in patients with type 2 diabetes. Diabetes Care. 2001;24:308–15. doi: 10.2337/diacare.24.2.308. [DOI] [PubMed] [Google Scholar]
- 9.Krentz AJ, Bailey CJ, Melander A. Thiazolidinediones for type 2 diabetes. BMJ. 2000;321:252–3. doi: 10.1136/bmj.321.7256.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evans RM, Barish GD, Wang Y-X. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–61. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 11.Grundy SM. Drug therapy of the metabolic syndrome: minimizing the emerging crisis in polypharmacy. Nat Rev Drug Discov. 2006;5:295–309. doi: 10.1038/nrd2005. [DOI] [PubMed] [Google Scholar]
- 12.Sugii S, Olson P, Sears DD, Saberi M, Atkins AR, Barish GD, et al. PPARγ activation in adipocytes is sufficient for systemic insulin sensitization. Proc Natl Acad Sci U S A. 2009;106:22504–9. doi: 10.1073/pnas.0912487106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qatanani M, Lazar MA. Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes Dev. 2007;21:1443–55. doi: 10.1101/gad.1550907. [DOI] [PubMed] [Google Scholar]
- 14.Drucker DJ. The biology of incretin hormones. Cell Metab. 2006;3:153–65. doi: 10.1016/j.cmet.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 15.Drucker DJ. The role of gut hormones in glucose homeostasis. J Clin Invest. 2007;117:24–32. doi: 10.1172/JCI30076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding S, Chi MM, Scull BP, Rigby R, Schwerbrock NMJ, Magness S, et al. High-fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS One. 2010;5:e12191. doi: 10.1371/journal.pone.0012191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102:11070–5. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Membrez M, Blancher F, Jaquet M, Bibiloni R, Cani PD, Burcelin RG, et al. Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. FASEB J. 2008;22:2416–26. doi: 10.1096/fj.07-102723. [DOI] [PubMed] [Google Scholar]
- 19.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–131. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 20.Adachi M, Kurotani R, Morimura K, Shah Y, Sanford M, Madison BB, et al. Peroxisome proliferator activated receptor γ in colonic epithelial cells protects against experimental inflammatory bowel disease. Gut. 2006;55:1104–13. doi: 10.1136/gut.2005.081745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohapatra SK, Guri AJ, Climent M, Vives C, Carbo A, Horne WT, et al. Immunoregulatory actions of epithelial cell PPARγ at the colonic mucosa of mice with experimental inflammatory bowel disease. PLoS One. 2010;5:e10215. doi: 10.1371/journal.pone.0010215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nielsen R, Pedersen TA, Hagenbeek D, Moulos P, Siersbaek R, Megens E, et al. Genome-wide profiling of PPARγ:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 2008;22:2953–67. doi: 10.1101/gad.501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Atsumi T, Nishio T, Niwa H, Takeuchi J, Bando H, Shimizu C, et al. Expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase/PFKFB3 isoforms in adipocytes and their potential role in glycolytic regulation. Diabetes. 2005;54:3349–57. doi: 10.2337/diabetes.54.12.3349. [DOI] [PubMed] [Google Scholar]
- 24.Okar DA, Wu C, Lange AJ. Adv Enzyme Regul. Elsevier; 2004. Regulation of the regulatory enzyme, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphate; pp. 123–54. [DOI] [PubMed] [Google Scholar]
- 25.Rider MH, Bertrand L, Vertommen D, Michels PA, Rousseau GG, Hue L. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochem J. 2004;381:561–79. doi: 10.1042/BJ20040752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huo Y, Guo X, Li H, Wang H, Zhang W, Wang Y, et al. Disruption of inducible 6-phosphofructo-2-kinase ameliorates diet-induced adiposity but exacerbates systemic insulin resistance and adipose tissue inflammatory response. J Biol Chem. 2010;285:3713–21. doi: 10.1074/jbc.M109.058446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chesney J, Telang S, Yalcin A, Clem A, Wallis N, Bucala R. Targeted disruption of inducible 6-phosphofructo-2-kinase results in embryonic lethality. Biochem Biophys Res Commun. 2005;331:139–46. doi: 10.1016/j.bbrc.2005.02.193. [DOI] [PubMed] [Google Scholar]
- 28.Guo X, Xu K, Zhang J, Li H, Zhang W, Wang H, et al. Involvement of inducible 6-phosphofructo-2-kinase in the anti-diabetic effect of PPARγ activation in mice. J Biol Chem. 2010;285:23711–20. doi: 10.1074/jbc.M110.123174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu C, Okar DA, Newgard CB, Lange AJ. Overexpression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase in mouse liver lowers blood glucose by suppression of hepatic glucose production. J Clin Invest. 2001;107:91–8. doi: 10.1172/JCI11103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu C, Kang JE, Peng L, Li H, Khan SA, Hillard CJ, et al. Enhancing hepatic glycolysis reduces obesity: differential effects on lipogenesis depend on site of glycolytic modulation. Cell Metab. 2005;2:131–40. doi: 10.1016/j.cmet.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 31.Wu C, Khan SA, Peng LJ, Li H, Camela S, Lange AJ. Perturbation of glucose flux in the liver by decreasing fructose-2,6-bisphosphate levels causes hepatic insulin resistance and hyperglycemia. Am J Physiol Endocrinol Metab. 2006;291:E536–43. doi: 10.1152/ajpendo.00126.2006. [DOI] [PubMed] [Google Scholar]
- 32.Matsuki T, Watanabe K, Fujimoto J, Kado Y, Takada T, Matsumoto K, et al. Quantitative PCR with 16S rRNA-gene-targeted species-specific primers for analysis of human intestinal bifidobacteria. Appl Environ Microbiol. 2004;70:167–73. doi: 10.1128/AEM.70.1.167-173.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walter J, Hertel C, Tannock GW, Lis CM, Munro K, Hammes WP. Detection of Lactobacillus, Pediococcus, Leuconostoc, and Weissella species in human feces by using group-specific PCR primers and denaturing gradient gel electrophoresis. Appl Environ Microbiol. 2001;67:2578–85. doi: 10.1128/AEM.67.6.2578-2585.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Wit N, Bosch-Vermeulen H, de Groot P, Hooiveld G, Bromhaar M, Jansen J, et al. The role of the small intestine in the development of dietary fat-induced obesity and insulin resistance in C57BL/6J mice. BMC Med Genomics. 2008;1:14. doi: 10.1186/1755-8794-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huo Y, Guo X, Li H, Xu H, Halim V, Zhang W, et al. Targeted overexpression of inducible 6-phosphofructo-2-kinase in adipose tissue increases fat deposition but protects against diet-induced insulin resistance and inflammatory responses. J Biol Chem. 2012 doi: 10.1074/jbc.M112.370379. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]