

Abstract
More than 15 years ago the first generation of genetically-engineered mouse (GEM) models of prostate cancer was introduced. These transgenic models utilized prostate-specific promoters to express SV40 oncogenes specifically in prostate epithelium. Since the description of these initial models, there have been a plethora of GEM models of prostate cancer, representing various perturbations of oncogenes or tumor suppressors, either alone or in combination. This review describes these GEM models, focusing on their relevance for human prostate cancer and highlighting their strengths and limitations, as well as opportunities for the future.
Introduction
Prostate cancer is now the most prevalent cancer in aged men (reviewed in [1, 2]). Although the majority of newly diagnosed prostate cancers are relatively indolent and have good prognosis, a subset will progress to highly aggressive disease, which is usually lethal. Distinguishing prostate tumors that will progress to lethality from those that will remain indolent represents a major clinical challenge. Another major challenge is to understand the role of androgen receptor signaling in prostate tumorigenesis, which has proven to be much more complex than initially anticipated. Indeed, because prostate cancer is dependent on androgen receptor signaling, a prevalent treatment is androgen deprivation therapy. However, while androgen deprivation initially leads to tumor regression, ultimately the tumors recur and most continue to be dependent on androgen signaling, which is thereby is referred to as “castration resistant” disease [3]. A third major clinical challenge has been the propensity of prostate cancer to metastasize to bone, a primary contributor to its morbidity and mortality; however bone metastases have been exceedingly difficult to reliably model in mice.
These clinical challenges have provided the impetus for development of a plethora of genetically engineered mouse (GEM) models of prostate cancer. This review describes these genetically engineered mouse models, beginning with the “first generation” transgenic models and ultimately the “newest generation” inducible models (Table 1). These genetically engineered mouse models recapitulate the spectrum of prostate cancer phenotypes, ranging from those that display premalignant lesions to those that exhibit aggressive adenocarcinoma including, castration-resistance and metastases (Figure 1). Many of these GEM models are based on perturbations of genes or molecular pathways that are of known importance for human prostate cancer (reviewed in [1, 2]), which has enhanced their relevance for the disease. This review discusses the relationship of these models to human prostate cancer, highlighting their strengths and limitations, as well as opportunities for identifying biomarkers of disease progression and their use as preclinical models for therapeutic intervention. We provide a somewhat historical perspective, emphasizing models that have been most widely utilized and/or are most promising for biomarker discovery or preclinical applications. Primary references are provided mainly for the GEM models rather than for the molecular pathways that they are based on; for more extensive discussion of the molecular mechanisms of prostate cancer the reader is referred to our previous comprehensive reviews of this area [1, 2].
Table 1.
Representative genetically-engineered mouse models of prostate cancer
| Mouse Model | Strengths | Limitations | References |
|---|---|---|---|
| Group 1: SV40 transgenic models | |||
| TRAMP model Minimal probasin promoter driving SV40 large and small T antigens |
|
|
[10–12, 121] |
| LADY model Large probasin promoter driving SV40 large T antigen |
|
|
[31, 122] |
| T121 Model: Minimal probasin promoter driving SV40 small T antigen |
|
|
[35] |
| Group 2: Oncogene transgenic models | |||
| c-Myc models Probasin promoter driving c-Myc Lo-Myc Hi-Myc |
|
|
[43] |
| TMPRSS-ERG models: Probasin promoter driving expression of ERG or ETV1 |
|
|
[48–52] |
| Akt model Probasin promoter driving activated form of Akt |
|
|
[53] |
| Group 3: Developmental pathways and androgen receptor signaling | |||
| Nkx3.1 Germline loss of function; conditional loss of function |
|
|
[59, 61, 123] |
| Wnt/β-Catenin Probasin Cre to drive conditional inactivation of APC or β-Catenin |
|
|
[75, 76] |
| FGFR1 Chimeric protein having dimerization motif attached to FGFR1 |
|
|
[71] |
| TGFβ Stromal-Cre to conditionally inactivate TGFβ |
|
|
[78] |
| Androgen receptor Probasin promoter to express wild-type or mutated androgen receptor |
|
|
[81, 83] |
| Group 4: Pten tumor suppressor models | |||
| Pten germline |
|
|
[88, 89] |
Pten germline combined with:
|
|
|
[102–104, 107] |
| Pten conditional Probasin Cre to conditionally inactivate Pten in prostate |
|
|
[92, 95] |
| Pten inducible Nkx3.1CreERT2 or PSA CreERT2 to inducibly inactivate Pten in prostate |
|
|
[97, 98] |
| Pten; p53 conditional Probasin Cre to conditionally inactivate Pten and p53 in prostate |
|
|
[96] |
| Pten; Smad4 Probasin Cre to conditionally inactivate Pten and SMAD4 in prostate |
|
|
[111] |
| Pten; Braf Nkx3.1CreERT2 to inducibly inactivate Pten and activate Braf in prostate |
|
|
[115] |
| Pten; Kras Nkx3.1CreERT2 to inducibly inactivate Pten and activate Kras in prostate |
|
|
[117] |
Figure 1.
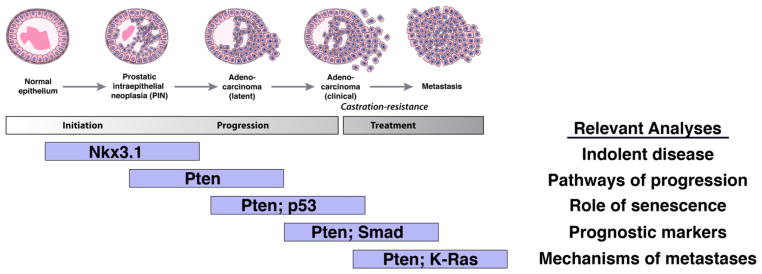
Representative models for specific stages of prostate cancer, indicating the types of analyses that can be addressed with each. Note that since these models are intended to be representative, rather than inclusive, they are biased towards Pten models.
Prostate biology in mice and man
A major consideration in “credentialing” GEM models is how closely they resemble important features of the human prostate gland and human prostate cancer. In humans, the prostate is a walnut-sized tissue surrounding the urethra at the base of the bladder, which produces important components of seminal fluid. The human prostate lacks discernible lobular structure, although it has a zonal architecture, corresponding to central, periurethral transition, and peripheral zones [4–7]. The outermost peripheral zone harbors the majority of prostate carcinomas, while benign prostatic hyperplasia (BPH), a common non-malignant condition, arises from the transition zone.
Unlike the human prostate, the mouse prostate consists of multiple lobes, corresponding to the ventral, lateral, dorsal, and anterior lobes, which have distinct patterns of ductal branching, histological appearance, gene expression, and secretory protein expression [8]. It is often asserted that the mouse dorsolateral lobe is most analogous to the human peripheral zone with respect to prostate cancer; although there is limited evidence to support this conclusion overall, gene expression profiling data supports this relationship [9].
At the histological level, both the mouse and human prostate contains three differentiated epithelial cell types: luminal, basal, and neuroendocrine. The luminal epithelial cells form a continuous layer of columnar cells that produce protein secretions; basal cells are located beneath the luminal epithelium, while neuroendocrine cells are rare cells of unknown function that express endocrine markers. Notably, most human prostate cancers are adenocarcinoma in origin and have primarily luminal features. However, neuroendocrine tumors may become increasingly more relevant in the most advanced tumors following clinical interventions.
“First generation” transgenic models expressing SV40 oncogenes
In the early days of modeling prostate cancer in mice, a major focus was based on expressing the human Simian Virus 40 (SV40) early genes, large T and small t-antigens specifically in the mouse prostatic epithelium using prostate-specific (or other) promoters. SV40 has been expressed in at least 10 prostate cancer transgenic mouse models. Here we will focus on those that have made the most impact in the field.
The TRAMP Model
Since its generation in 1996, the Transgenic Adenocarcinoma of the Mouse Prostate (TRAMP) mouse model has been one of the most widely used mouse in prostate cancer research [10–12]. This model represents a transgene comprising the minimal probasin promoter (−426 /+28) (Table 2) driving viral SV40 large-T and small t antigen antigens, which leads to prostate-specific inactivation of pRb and p53, specifically in the prostatic epithelium [11, 13]. TRAMP mice develop prostatic intraepithelial neoplasia (PIN) by 6 weeks, which progresses to high grade PIN by 12 weeks, and to poorly differentiated and invasive adenocarcinoma by 24 weeks of age with nearly 100% penetrance. Additionally, this model displays prevalent metastases to distant organs [10], although rarely to the skeleton. Notably, the lack of reliable metastases to bone represents a major limitation of most GEM models.
Table 2.
Promoter elements used to develop mouse models of prostate cancer
| Promoter/Cre Allele | Relevant mouse models |
|---|---|
|
| |
|
Rat Probasin (PB): Minimal PB Expression cassette carrying 426 basepairs of the rat probasin (PB) promoter and 28 base pairs of 5′-untranslated region (−426/+28) |
PB-T/tag(TRAMP) TRAMP crossed with BCL2 transgene TRAMP crossed with EGR1 null mice PB-mAR PB-Akt |
|
Large PB (LPB) A large (L) fragment of the PB promoter (from 11 500 to +28) that retains some enhancer elements to facilitate high gene expression |
LPB-Tag (LADY) LARGE T ANTIGEN |
|
ARR2PB A composite probasin promoter with androgen and glucocorticoid, prostate-specific, and achieves high levels of transgene expression |
ARR2PB-myc ARR2PB-FGFR1 (JOCK1) T121 ARR2PB CreERt2 |
|
PB-Cre/PB-Cre4 PB-Cre4 Cre gene under the control of the ARR2PB |
PB-Cre4; Ptenflox/flox PB-Cre4; Ptenflox/flox; p53flox/flox PB-Cre4; Ptenflox/flox; SMAD4flox/flox PB-Cre4; Apc flox/flox |
|
| |
|
Prostate Specific Antigen (PSA) PSA-CreERT2 6-kb fragment of the human PSA promoter driving expression of the nuclear-targeted modified Cre (tamoxifen-inducible) |
PSACreERt2; Ptenflox/flox |
|
| |
|
Nkx3.1 Prostate-specific homeobox gene regulated by androgen Nkx3.1CreERt2–modified Cre knocked into the Nkx3.1 gene to express in prostate; nuclear-targeted modified Cre (tamoxifen-inducible) |
Nkx3.1CreERt2; Ptenflox/flox Nkx3.1CreERt2; Ptenflox/flox; BrafLSLflox/+ Nkx3.1CreERt2; Ptenflox/flox; KrasLSLflox/+ |
|
| |
|
HoxB13 Prostate-specific homeobox gene not regulated by androgen Hoxb13-rtTA–tetracycline regulator driven by HoxB13 promoter |
Hoxb13-rtTA/TetO-H2BGFP |
A major limitation of the TRAMP mice is the inherent use of the probasin promoter (Table 2), which itself is regulated by androgens; in fact, this issue affects the many models driven by the probasin promoter. Therefore, interpreting the consequences of castration is confounded by the possibility that observed androgen sensitivity is due to the down-regulation of transgene expression.
Another concern regarding the TRAMP mice is that a majority of tumors are neuroendocrine in origin [14] and therefore this model is most likely to be relevant to the sub-population of patients with neuroendocrine disease [15]. Interestingly, the neuroendocrine phenotype of the TRAMP mice is suppressed by in mice lacking the ubiquitin ligase Siah2 via regulation of HIF-1alpha availability [16], which underscores the emerging significance of neuroendocrine phenotypes in prostate cancer. Furthermore, while a criticism of the TRAMP (and similar) models is the use of the non-physiological SV40 T antigen; prostate-specific inactivation of pRb and p53 (which are the major targets of SV40 T antigen) also leads to aggressive neuroendocrine tumors with metastases to distant organs [17]. Thus, the phenotype of the TRAMP mice may reflect the consequences of RB and p53 pathway inactivation.
Models combined with TRAMP
The TRAMP model has made a significant impact in our understanding of the molecular mechanisms of prostate cancer, in part because it has been extensively used to evaluate other molecules/pathways of interest. Below we discuss some of the molecular factors that have been investigated in the context of the TRAMP model.
BCL-2
Among the various mouse alleles that TRAMP mice have been crossed with is the anti-apoptotic gene, Bcl-2. The corresponding PB-BCl-2/TRAMP mice have an accelerated prostate cancer phenotype, but do not have increased incidence of metastases [18]. Although up-regulation of Bcl-2 has been associated with androgen-independent prostate cancer in humans, castration of the PB-BCl-2/TRAMP mice resulted in loss of Bcl-2 expression presumably because of the androgen dependence of the probasin promoter, which precluded the ability to assess whether Bcl-2 accelerated hormone-independent tumor growth.
Caveolin-1
Caveolin-1 is a major structural protein of the caveolae critical for mediating molecular transport and signal transduction and associated with advanced metastatic prostate cancer [19]. Although Caveolin-1 null mice do not display an evident prostate phenotype, when crossed with the TRAMP mice, cancer progression was accelerated in terms of the extent of tumor burden as well as metastases [20], supporting a tumor suppressor role for Caveolin-1 in prostate.
EGR-1
The early growth response protein 1 (EGR1) transcription factor is over-expressed in a majority of human prostate cancers and has been implicated in the regulation of several genes important for prostate tumor progression. The consequences of its deficiency were studied by combining the Egr1 null mice with the TRAMP or with an alternative SV20 model (CR2-T-Ag, discussed herein) [21]. Deletion of Egr1 in these contexts resulted in significantly delayed progression from PIN to invasive carcinoma, suggesting a role for Egr1 in the transition from localized to invasive carcinoma.
IGF-1
The role of insulin-like growth factor I (IGF-I) in prostate cancer has been controversial. Some studies have suggested that elevated serum IGF-I levels are associated with increased risk of developing prostate cancer [22], while other studies were not confirmatory [23]. Crossing TRAMP mice with mice that lack growth hormone-releasing hormone (GHRH) receptor function, which thereby have low levels of both serum growth hormone (GH) and IGF-I, results in significantly slower progression to prostate cancer [24]. Subsequent studies to evaluate the consequences of loss of IGF receptor or its forced expression in the prostate in combination with the TRAMP mice [25, 26] further support the role of IGF levels in prostate tumorigenesis.
SRC-3
The steroid receptor coactivator-3 (SRC-3) is a coactivator of the androgen receptor that is expressed primarily in basal and stromal cells, although TRAMP mice have increased expression of SRC-3 in luminal cells during cancer progression [27]. Combining the TRAMP mice with SRC-3 null mice resulted in a marked delay in tumorigenesis, with these mice exhibiting only PIN and early stage carcinomas [28], suggesting that SRC-3 contributes to tumor progression by influencing proliferation and survival of prostate cells.
The LADY Model
To circumvent the problem of variable expression levels of the initial version of the probasin promoter, the LADY series utilized a larger fragment of the probasin promoter (LPB; −11,500/+28) (Table 2) driving large-T antigen but lacking small-t antigen [29, 30]. Seven transgenic lines were derived, which form 3 groups based on the stage of the neoplasia and the timing of the initial occurrence of tumor phenotypes. Each of these lines develops hyperplasia, progressing to dysplasia, high-grade PIN and ultimately adenocarcinoma, and thereby models the various stages of prostate cancer. 12T-7f LADY mice are initially responsive to androgen, since tumors regress upon castration, while administration of androgens restores the epithelial/stromal cell ratio and tumor growth.
Involvement of distant organ metastases is a rare event in the LADY series. The exception is line 12T-10, which exhibits a neuroendocrine phenotype and develops prevalent metastasis by 9 months, although not to bone [31]. However, a double transgenic model with a probasin-driven Hepsin crossed with the LADY 12T-2f model (such that both the large T antigen and the hepsin were expressed specifically in the prostate) was reported to exhibit increased metastatic potential including to the skeleton [32]. However, since this initial report, there has been no further information on this model.
Another interesting application of the LADY model was to assess its collaboration with TGF-β signaling, which was achieved by crossing transgenic mice expressing a dominant negative TGFβ Receptor II with the LADY mice [33]. Although the prostate tumor phenotype was not affected by attenuation of TGF-β signaling, the mice displayed increased incidence of metastases. These findings support a role for TGF-β signaling particularly in prostate cancer metastases.
Other transgenic models expressing SV40 oncogenes
The T121 Model
A series of transgenic mouse lines were developed using a second-generation probasin promoter, the ARR2PB promoter, which has higher-level expression in prostatic epithelium [34] (Table 2). The ARR2PB promoter was used to express a truncated SV40 large-T antigen (T121), which inactivates pRb, without affecting the functionality of p53 or other T antigen targets [35]. These TgAPT121 mice develop PIN by 2 months of age, which by 4 months progresses to microinvasive and well-differentiated prostate adenocarcinoma [35]. Notably, as is the case for various other mouse alleles (discussed herein), this phenotype is accelerated by the Pten loss of function [35]. Given that dysfunctional pRB signaling is observed with high frequency in human prostate cancer [1, 2, 36], the TgAPT121 model may be useful to investigate the function of the RB pathway in prostate tumorigenesis.
The PSP94-TGMAP Model
Prostate secretory protein of 94 amino acids (PSP94) is one of the three abundant secretory proteins in the prostate. A 3.8 kb fragment of the PSP94 promoter region was used to drive SV40 large and small T-antigen expression in prostate epithelium, and the resulting mice developed hyperplasia by 10 weeks and well-differentiated adenocarcinomas by 24 weeks [37]. To overcome inherent variability of the transgenic approach, a knock-in model was generated by inserting the SV40 T-antigen coding sequence to the PSP94 locus (PSP-KIMAP) giving rise prostate cancer with higher tumor penetrance and rare occurrence of neuroendocrine differentiation [38]. These models have not been widely utilized, although this promoter may be beneficial for knock-in of genes other than SV40.
SV40 models that do not use prostate-specific promoters
Several models have been generated using promoters that are not prostate specific, although the resulting mice have prostate cancer phenotypes. Most of these express SV40 antigens and are discussed in this section. An example of one that does not express SV40 is the transgenic model expressing mutated B-RafV600E [39], which is discussed in a subsequent section.
CR2-Tag
The CR2-Tag model expresses SV40 T antigen under the control of the cryptidine-2 (CR2) promoter, which was generated to study intestinal epithelium, but unexpectedly the male mice developed prostate cancer [40]. Prostate cancer in the CR2-Tag mice exhibits PIN by 12 weeks of age, and invasive cancer with neuroendocrine features by 24 weeks with metastases to lymph node, liver, and lung.
C3(1)SV40
The C3(1)SV40 T mice were generated using a 4.5 Kb of rat [C3(1)] promoter to drive expression of SV40 T antigen [41]. These mice develop low-grade PIN by 2 months of age and high-grade PIN by 5 months, which eventually progresses to locally invasive adenocarcinoma.
Globin-SV40
Transgenic mice having the fetal globin promoter driving SV40 T-antigen expression develop prostate tumors that metastasize to lymph nodes and distant sites [42]. These tumors are resistant to castration and exhibit both neuroendocrine and epithelial phenotypes.
Proto-oncogene activation
An important category of genetically-engineered mouse models are those with activation of key oncogenic pathways of known relevance for human prostate cancer. In general, the phenotypes of these mice are less aggressive than the SV40 models, but have the important advantage that they tend not to develop neuroendocrine tumors. These transgenic models have been developed using variations of the probasin promoter (Table 2) to drive the transgene expression. Notably, in some cases, their phenotype is not as robust as more recent models using conditional activation of oncogene expression or deletion of tumor suppressors driving their activation (described herein).
c-Myc
A majority of human prostate cancers exhibit amplification and/or overexpression of c-Myc ([36] and reviewed in [1, 2]). To investigate its role in disease progression, transgenic mice were engineered to express human c-Myc under the control of a minimal (weaker) probasin promoter, called the Lo-Myc mice, or the stronger probasin promoter (ARR2PB; Table 2), called the Hi-Myc mice [43]. Both the Hi- and Lo-Myc transgenic models develop PIN that progresses to locally invasive adenocarcinoma by 3–6 or 10–12 months, respectively. The initial phenotypic observations of these Lo- and Hi-Myc models were further supported and extended by subsequent analyses [44]. Furthermore, castration of Hi-Myc mice at 2 months, but not at 8 months, causes tumor regression. This suggests that these early but not later tumors are more resistant to castration, although this interpretation is complicated by the dependence of the probasin promoter on androgen signaling. The castration-resistance of this Myc model was also shown by the collaboration of Myc with NF-kappaB signaling [45].
Notably, crossing the Hi-Myc mice with the ARR2PB-hepsin transgene (similar to the strategy used for the LADY model discussed above) resulted in tumors without metastases [46], raising concerns about the interpretation that hepsin is associated with bone metastases [32]. An important feature of the Myc models is that develop adenocarcinoma rather than neuroendocrine tumors. Additionally, expression-profiling analyses of the Myc mice was one of the first examples of cross-species analyses from GEM models to human prostate cancer to provide biomarkers of disease progression [43], a strategy that continues to be extremely valuable.
TMPRSS2-ERG
An important recent discovery was the observation that a majority of prostate cancers have translocations of members of the ETS gene family fused with the TMPRSS-2 promoter [47]. The functional role of these translocations has been evaluated in various transgenic mouse models. In an initial report, the ARR2PB promoter (Table 2) was used to express a fusion of the noncoding exon 1 of TMPRSS2 with two Ets genes, ERG or ETV1; these mice were reported to develop PIN by 12–14 weeks of age [48, 49]. The conclusion that overexpression of ETS genes lead to PIN was supported by another study of an independent study [50]. However, subsequent reports using analogous transgenic approaches concluded that mice expressing ETS genes do not develop PIN, although they do so when combined with loss of function of Pten [51, 52]. Given the presumed importance of ETS gene translocations for prostate tumorigenesis, this issue of their functional role in prostate cancer needs to be further addressed, potentially using alternative approaches to conditionally express ETS genes in the prostate.
Akt
Based on the considerable importance of activation of mTOR/Akt signaling for prostate tumorigenesis, a transgenic mouse was developed that expressed a constitutively activate form of Akt in the prostate using a probasin promoter [53]. These mice develop PIN but, interestingly, the severity of the phenotype is modest relative to that of Pten loss of function (described herein), despite the fact that Akt deficiency is sufficient to suppress tumor development in Pten null mice [54]. Subsequent analyses of these Akt transgenic mice revealed cooperativity with p27 via relief of senescence [55], which is notable since Pten loss also cooperates with loss of p27 (described herein).
Vav3
Based on its overexpression in prostate cancer, Vav3 function was investigated in GEM models, using the ARR2PB promoter to express the constitutively-active protein [56]. These mice display PIN by 3 months of age, which progresses to adenocarcinoma [56]. Interestingly, these Vav3 transgenic mice developed nonbacterial chronic prostatitis, which is of interest since prostatitis is thought to predispose to prostate cancer. Thus, these Vav3 transgenic mice may enable the study of prostatitis for prostate cancer development.
Her-2/neu
The role of the Her2/neu oncogene has been investigated in prostate cancer using the long probasin promoter (as used for the LADY series) to express a constitutively active Neu in prostate [57]. These mice develop hyperplasia or varying degrees of PIN, without invasion or metastases.
Ras/Raf
Ras and Raf signaling pathways are frequently deregulated in lethal tumors [36]. Transgenic mice expressing a mutated H-Ras (RasVal12) driven by a minimal probasin promoter (PB-Ras) develop PIN that does not progress to cancer [39]. However, these PB-Ras transgenic mice have interesting differentiation defects, as they develop multifocal intestinal metaplasia with vacuolated cells, resembling goblet cells that resemble cells found in the intestinal epithelium. The role of Ras pathway activation in prostate cancer has been investigated the context of Pten loss of function in more robust knock-in models (described herein).
An oncogenic mutation in the B-Raf protooncogene (B-RafV600E), which was engineered to express in the skin using a tyrosinase promoter, was found to develop prostate hyperplasia with progression to adenocarcinoma [39]. A limitation of this model is that the cell type in which the transgene is expressed in prostate is not known. The role of B-Raf activation in prostate cancer has been further investigated in collaboration with Pten (described herein).
Developmental and signaling pathways
In addition to evaluation of tumorigenic pathways, analyses of molecular pathways that are biologically relevant for the development and maturation of the prostate have provided insights into the role of essential pathways in prostate cancer, as well as important mouse models. Notably, these studies have also provided innovative and more sophisticated approaches for gene targeting in the prostate. These include models that perturb homeobox transcription factors that function in prostate development, and models that perturb key signaling pathways in the epithelium or stroma.
Homeobox genes and other transcription factors
Nkx3.1
The unique relationship of the Nkx3.1 homeobox gene as a regulator of prostate development whose loss of function is associated with prostate cancer initiation [58] has merited generation of both conventional and conditional knockout mouse models, as well as the development of new alleles to achieve targeted gene deletion in the prostate. Therefore, germline loss of function of Nkx3.1 leads to prostate epithelial defects, including aberrant branching morphogenesis and defective protein secretions [59, 60]. These mice develop dysplasia by 3 months, which becomes progressively more severe with increasing age, culminating in PIN by 1 year [59, 60]. A similar phenotype is observed in mice having conditional deletion of Nkx3.1 in the prostate [61]. However, neither the germline nor the conditional mice progress to invasive prostate cancer, suggesting that Nkx3.1 loss is not sufficient for cancer progression. Notably, Nkx3.1 heterozygotes display a similar, although less severe, phenotype as their null counterparts, indicating that Nkx3.1 is haploinsufficient and providing a model to evaluate the interplay between transcription factor dosage and tumor initiation.
Nkx3.1 has been shown to be expressed in a rare population of luminal epithelial stem cells called castration resistant Nkx3.1-expressing cells (CARNs) [62], and has therefore been important in studying prostate stem cells as well as cell or origin of prostate cancer. Furthermore, these analyses have been facilitated by the generation of an inducible Cre allele, Nkx3.1-CreERT2, which has a Cre-ERT2 cassette knocked in to the Nkx3.1 gene (Table 2) and thereby enables gene deletion in adult prostate epithelium following delivery of tamoxifen [62]. This Nkx3.1CreERT2 allele provides a valuable means to inactivate tumor suppressors or activate oncogene function in the prostate (see below). However, similar to probasin, the Nkx3.1 promoter is also dependent on androgens.
HoxB13
Another homeobox gene that is important in both prostate development and cancer is HoxB13. Unlike Nkx3.1, HoxB13 is not androgen regulated [63] while similar to Nkx3.1, the germline mutant mice display defects in prostate differentiation [64]. Recent studies showing the association of human HOXB13 with genetic risk of developing prostate cancer [65] have led to a resurgence of interest in its function in prostate cancer. Notably, the promoter region of HoxB13 required for expression in the prostate has been defined [66] (Table 2), which may be a meaningful alternative to other prostate specific promoters that are androgen regulated. Furthermore, analyses of the HoxB13 promoter has led to the development of a tetracycline-regulated mouse model for regulated gene expression in the prostate [67], which is of considerable importance since previous attempts to develop such Tet-regulated models for prostate have largely failed.
Sox9
The transcription factor Sox9 is expressed in the epithelia of all mouse prostatic lobes from the initial stages of their development. Using a conditional approach with mice expressing Cre recombinase under the control of Nkx3.1 regulatory sequences revealed a lack of ventral prostate development and abnormal anterior prostate differentiation and an early loss of expression of genes specific to the prostate epithelia such as Nkx3.1 [68]. Conversely overexpression of Sox9 accelerated the consequences of Pten loss of function in cancer progression [69].
Growth Factor Signaling
FGF
Based on studies that have implicated FGF signaling in prostate cancer (reviewed in [2, 70]), transgenic mice were generated expressing FGFR1 in the prostatic epithelium, using a novel dimerization approach to inducibly and reversibly activate receptor activity specifically in prostate [71]. The resulting mice engineered to express the modified FGFR1, referred to as JOCK1, exhibit hyper-proliferation and PIN, while aged JOCK1 mice progress to invasion and metastatic disease [72]. In addition to providing models to study FGF function in prostate cancer, this approach may provide a means to study “oncogene addiction” in prostate cancer, as there are currently few other suitable alleles that allow for inducible and reversible gene induction. Notably, in an alternative approach to evaluate FGF signaling in prostate, transgenic mice expressing FGF8b in the prostate together with Pten loss of function were shown to display poorly differentiated adenocarcinoma [73], which further underscores the significance of FGF pathway activation as well as its interaction with Pten loss of function.
Wnt/β-Catenin pathway
Canonical Wnt/β-catenin signaling plays a key role in prostate tumorigenesis, and particularly for advanced disease. Perturbation of Wnt/β--catenin signaling in the prostate has been investigated using a probasin-driven Cre allele (PB-Cre4; Table 2) to achieve conditional deletion in prostate epithelium [74]. This PB-Cre4 allele was crossed with a floxed allele of the adenomatous polyposis coli gene (APC), which is a negative regulator of the Wnt/β-catenin pathway. The corresponding PBCre4-APCf/f mice develop locally invasive adenocarcinoma without distant metastases by 7 months [75]. Castration of mice harboring advanced tumors result in partial regression, indicating that tumors in the PBCre4-APCf/f mice may be partially castration resistant. An alternative model to activate Wnt/B-catenin signaling used the PBCre4 combined with conditional deletion of exon 3 ofβ-catenin, in which the resulting PBCre4- β-Cateninf/f mice display progression from hyperplasia to high-grade PIN, with castration-resistance [76]. Given the importance of aberrant Wnt/β-catenin signaling in human prostate cancer, these models may help to investigate the role of this signaling pathway in cancer progression.
TGF-β
Analyses of the role of TGF-β signaling for prostate tumorigenesis using mouse models has provided insights into the role of stroma in cancer progression and the interaction of the epithelium and stroma for these processes [77, 78]. Notably, targeted disruption of TGF-β receptor II in mouse fibroblasts (although not specifically in to prostate) was found to result in PIN, which was accelerated when crossed with mice having cooperation with defects in the prostatic epithelium. These findings emphasize the importance of interactions of the epithelium and stroma for prostate tumorigenesis.
Androgen Receptor Signaling
Androgen receptor signaling is essential for all aspects of prostate development as well as prostate cancer (reviewed in [2, 70]). In fact, perturbation of androgen receptor signaling is arguably the most significant driving event in prostate tumorigenesis, and a critical target for new therapeutic approaches for treatment of prostate cancer. Therefore, a clear understanding of the role of androgen signaling in prostate cancer is essential; however, such analyses has been complicated by the fact that androgen receptor signaling occurs in both the epithelium and the stroma, where its functions in these compartments have been reported to be distinct [79, 80].
Androgen receptor function in the prostate epithelium has been evaluated using the minimal probasin promoter to express androgen receptor [81]. The resulting PB-mAR mice develop hyperplasia that progresses to microinvasive high-grade PIN, supporting the idea that AR is a positive regulator of prostate tumorigenesis. Moreover, an alternative approach in which AR was conditionally activated in the prostate also resulted in a PIN phenotype [82]. However, an alternative using the probasin promoter to express either wild-type androgen receptor or a prevalent mutated form (AR-E231G) in prostatic epithelium reported the rapid development of PIN with progression to invasion only in mice that expressed the mutated androgen receptor [83]. Conversely, inactivation of androgen receptor expression in the prostatic epithelium via conditional deletion using PB-Cre4 resulted in prostate differentiation defects as well as increased epithelial proliferation [84]. Furthermore, the phenotypic consequences of this androgen receptor knock-out were accelerated in the context of the TRAMP mice [79].
Furthermore, various lines of evidence suggest that androgen receptor, which is expressed in the prostatic stroma as well as epithelium, has different roles in these compartments, which is not unexpected from the classic work of Cunha and colleagues (reviewed in [1, 2]). In particular, deletion of both stromal and epithelial androgen receptor in the context of the TRAMP mice, result in reduced proliferation and abrogation of tumor defects [79]. Furthermore, conditional deletion of androgen receptor in fibroblasts or smooth muscle cells result in distinct defects in prostate differentiation or tumor growth [85, 86], suggesting a role for stromal AR not only in normal prostate development but also in prostate cancer. The complexity and significance of androgen receptor signaling warrants further study using systematic approaches to target androgen receptor in the epithelium or stroma in various normal and tumor contexts.
Pten and other tumor suppressor models
Of the many gain of function transgenic mouse models of prostate cancer, most transgenic models with the notable exception of the Myc transgenic model [43], are limited in terms of the robustness of their phenotype or their relevance to human prostate cancer (i.e., neuroendocrine features). Considerably more robust and versatile are GEM models based on loss of function of tumor suppressor function, and most notably, those based on Pten loss of function. Indeed, PTEN is of considerable importance for prostate cancer because of its relevance for regulation of androgen receptor signaling [1, 2, 87]. Reflecting its broad significance, the consequences of loss of function of Pten have been investigated in a variety of different types of GEM, including germline, conditional and inducible ones, in which the consequences of Pten loss have been investigated alone or in conjunction with loss of function of various tumor suppressors or with conditional activation of various oncogenes. These have enabled a progressive series of models to study prostate tumorigenesis (Figure 1).
Interestingly, as already introduced for the combination of Pten loss with TMPRSS-ERG (described above), many of these other alleles, including both gain and loss of function, have limited or minimal phenotypes on their own, while their consequences for tumorigenesis are unleashed in combination with Pten loss. It is likely that models based on Pten loss of function, and particularly the more sophisticated conditional or inducible models, will be a primary focus in future studies, particularly those focused on using GEMs for preclinical applications.
Germline deletion
Since Pten null homozygosity results in early embryonic lethality, the consequences of its germline loss of function have been studied in the heterozygotes, which are viable and display a broad range of cancer phenotypes [88, 89]. In particular, Pten heterozygotes display prostate hyperplasia with PIN, although they do not progress to cancer [88, 89]. Notably, an important observation from analyses of its germline deletions is that Pten loss is sufficient for hormone independence at least [90].
Conditional deletion
Although these initial models established a critical role for Pten in prostate cancer, and particularly for castration resistance, analyses of germline Pten loss is complicated by the other cancer phenotypes, most of which emerge much earlier than their prostate cancer phenotypes [88, 89]. Thus, various groups have investigated the consequences of Pten loss of function in prostate cancer via its conditional deletion of a floxed allele using the probasin PB-Cre allele [91, 92] or alternative approaches [93]. Although one initial study claimed to observe metastases following conditional Pten loss [92], this was not observed in the other initial reports [94, 95], nor have there been any subsequent reports showing that Pten loss alone leads to metastases. Further analyses have shown that conditional loss of Pten in the prostate leads to PIN that progress to high grade PIN with areas of microinvasion. Importantly, these conditional Pten mice develop senescence, which precludes their advancement to overt adenocarcinoma [96].
Inducible deletion
A primary limitation of the Pten conditional models described above is that the PB-Cre4 allele induces Pten deletion prior to complete differentiation of the prostate and is not restricted to the prostatic epithelium (M. Shen and C. Abate-Shen, unpublished observations), both of which confound the interpretation of the prostate phenotypes. To circumvent this problem, alternative Cre alleles have been developed which enable inducible deletion of Pten specifically in the epithelium of adult (fully mature) prostate [97] [98]. These models use, alternatively, a PSA-CreERT2 allele or the Nkx3.1-CreERT2 allele to achieve inducible expression following tamoxifen induction. Another inducible Cre allele has been described using the ARR2PB promoter, although not crossed with Pten [99].
The phenotypes of mice having inducible Pten deletion are similar to the conditional alleles in that they develop PIN that progresses to microinvasion but not metastases. However, the phenotype of the inducible deletion models is generally more severe than the constitutive deletion model (M. Shen and C. Abate-Shen, unpublished observations). Interestingly, the Nkx3.1CreERT2; Ptenflox/flox mice develop castration-resistant prostate tumors, which in contract to the non-castrated (intact) counterparts having robust senescence, virtually lack a senescence phenotype [98]; this suggests that castration-resistance promotes cancer progression by bypassing senescence.
Other PI3 Kinase pathway perturbations
The significance of Pten loss and concomitant activation of PI3 kinase pathway activation for prostate tumorigenesis is further underscored from analyses of other GEM models having perturbations of alternative components of this signaling pathway. In addition to the Akt transgenic mice described in the preceding section, alternative GEM models of the PI3 kinase signaling pathways include constitutive activation in the prostate of p110β, a downstream effector of Pten [100] and prostate-specific loss of function of the AKT-inactivating phosphatase PHLPP1 [101].
Pten combined with other genes
Combined with germline Pten loss
Several models have examined the consequences of germline loss of function of Pten together with loss of other tumor suppressor genes, which typically result in more aggressive prostate cancer phenotypes. In particular, analyses of germline loss of function of p27, a tumor suppressor involved in the in G1/S transition, has a modest phenotype on its own [102, 103], but together with Pten loss displays cooperativity resulting in PIN and progression to microinvasion [103]. Similarly, compound germline loss of Pten and Nkx3.1 results in high grade PIN that ultimately progresses to adenocarcinoma in aged mice, and castration-resistance following surgical depletion of androgens [104, 105]. Notably, the aged (>12 month) Nkx3.1; Pten compound mutant mice develop rare metastases to lymph node and lungs [105], which is a considerable progression from the germline Pten single mutants. Furthermore, analyses of PIN in the Nkx3.1; Pten compound mutant mice also led to a classification of PIN in genetically engineered mice that is still widely utilized [106]. Finally, combining the Pten, Nkx3.1 and p27 germline alleles led to the identification of haploinsufficiency of p27, which was not evident in the individual combinations [102].
In addition, combining Myc gain of function with germline loss of Pten revealed a complementary function of these key pathways, that together with loss of p53, drives prostate tumorigenesis [107, 108]. Another interesting combination is that of loss of function of Par, a negative regulator of NFκb, combined with loss of Pten, whose progressive defects accentuate the significance of NFκb activity for prostate tumorigenesis [109]. These examples underscore the value of analyzing molecular pathways of cancer in the context of Pten loss of function.
Combined with conditional or inducible Pten loss
Particularly informative have been a series of models having conditional or inducible deletion of Pten combined various alleles that result in deletion of tumor suppressors or activation of oncogenes. Analyses of these models have provided important insights regarding the molecular pathways of prostate tumorigenesis, as well as essential models of lethal, metastatic prostate cancer.
The role of loss of function of p53 in prostate cancer has been investigated in collaboration with Pten following their combined conditional deletion using the probasin Cre allele [96]. This important study showed that although senescence precluded the development of more aggressive phenotype in the Pten model, loss of function of p53 overcame this halt in tumor progression. The resulting compound mutant mice developed lethal prostate tumors that did not display senescence, although they were also not metastatic. This is an excellent example in which analyses of the prostate phenotype of mutant mice led to the identification of a key molecular pathway that influences prostate tumorigenesis. Furthermore, accelerating the phenotype of the Pten and p53 mice by perturbing telomerase function resulted in lethal prostate tumors that are locally invasive to bone [110]
SMAD4 was identified as a gene of interest in prostate cancer based on its expression in the Pten conditional mice as well as in human prostate cancer [111]. Its functional consequences for prostate tumorigenesis were investigated in mice having conditional loss of SMAD4 together with Pten driven by the probasin Cre allele [111]. The resulting mice develop lethal prostate tumors that are adenocarcinoma in origin; however, unlike the lethal tumors in the Pten; p53 conditional mice, the Pten; SMAD4 conditional mice are reported to also have metastases to distant organs in ~12% of the cases, although not to the bone. Additionally, analyses of the gene signature of these lethal tumors revealed a 4-gene signature of aggressive prostate tumors that is being evaluated for its prognostic significance for human prostate cancer [111].
As noted above, the RAS and RAF signaling pathways are activated in a high percentage of lethal prostate tumors in humans [36], and translocations of both RAS and RAF have been reported to occur in aggressive prostate tumors [112, 113]. Although transgenic models having activation of RAS or RAF have phenotypes that were either modest or difficult to analyze (discussed above), mice having conditional activation of either Braf or Kras in the mouse prostate together with loss of function of Pten have promising phenotypes. In particular, a conditionally activatable Braf allele [114] combined with inducible deletion of Pten using the Nkx3.1-CreERT2 allele resulted in lethal adenocarcinomas, which display metastases to distant organs in ~30% of the cases [115]. Interestingly, these mice display activation of Myc as well as MAP kinase signaling.
Finally, combining a conditionally activatable Kras allele [116] with inducible deletion of Pten using the Nkx3.1-CreERT2 allele results in lethal prostate tumors that display metastases to distant organs in 100% of the cases [117]; a similar model has been developed using the constitutive probasin Cre allele [118]. In addition to Pten, activated Kras also cooperates in tumorigenesis in combination with the Scribbled (SCRIB), which is involved in establishing and maintaining epithelial polarity [119], as well as with activation of Wnt signaling [120]. Despite its robust phenotype and prevalence of distant metastases, activation of Kras together with loss of Pten does not result in bone metastases. Although it is an important advance to have a model that displays full penetrance of metastases, it is intriguing, and at the a same time disappointing, that this very aggressive mouse model fails to display metastases to bone, which is the major site of metastases in humans.
Conclusions and perspectives
The reader of this review with its intentionally historical perspective will undoubtedly be impressed by the remarkable evolution of GEM models of prostate cancer in the short span of ~15 years. These models originated from simple transgenic ones that established the feasibility of studying prostate cancer phenotypes in mice. Now, the current standard is conditional or inducible models having 2 or more molecular perturbations. These models have not only evolved in terms of their sophistication, but also in terms of the sophisticated information that has been learned from their analyses, which is immediately relevant for understanding the pathogenesis of human prostate cancer, and the potential for impacting cancer treatment and diagnosis by through the identification of prognostics signature and the preclinical evaluation of new therapeutic approaches.
Yet, we still have a ways to go to develop the “ideal” GEM model. First of all, while most current models feature molecular pathways that are relevant for human prostate cancer, many of the actual mouse alleles do not accurately represent the precise molecular events that occur in humans. For example, few human prostate cancer has loss of both Pten alleles, yet this is the most frequently used GEM model. Secondly, we are limited by the availability of relatively few reporter CRE drivers, none of which is optimal either because of their early expression pattern, their dependence of androgens or, in the case of the tamoxifen regulated promoters, the potential impact from even short doses of tamoxifen. Finally, while we have advanced considerably in developing models of metastatic prostate cancer, the remaining challenge is to develop models that reliably target bone. So, while we have come a long way so far, we still have some significant challenges ahead.
Acknowledgments
We thank our colleagues in the NCI mouse models of human cancer consortium and the community of researchers who have been developing prostate cancer mouse models whose research have inspired us over the years. We are grateful to Drs. Robert Matusik, Michael Shen, Chee Wai Chua, Alvaro Aytes and Takashi Kobayashi for comments on the manuscript. Research in the CAS laboratory is supported in part by NIH grants (CA084294, CA141535 and CA154293), the T.J. Martell Foundation for Leukemia, Cancer and AIDS Research and the V-Foundation for Cancer Research. CAS is an American Cancer Society Research Professor supported in part by a generous gift from the F.M. Kirby Foundation.
References
- 1.Abate-Shen C, Shen MM. Molecular genetics of prostate cancer. Genes & Development. 2000;14(19):2410–2434. doi: 10.1101/gad.819500. [DOI] [PubMed] [Google Scholar]
- 2.Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 2010;24(18):1967–2000. doi: 10.1101/gad.1965810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23(32):8253–61. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 4.McNeal JE. Origin and development of carcinoma in the prostate. Cancer. 1969;23:24–34. doi: 10.1002/1097-0142(196901)23:1<24::aid-cncr2820230103>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 5.McNeal JE. The zonal anatomy of the prostate. Prostate. 1981;2(1):35–49. doi: 10.1002/pros.2990020105. [DOI] [PubMed] [Google Scholar]
- 6.McNeal JE. Normal histology of the prostate. Am J Surg Pathol. 1988;12(8):619–633. doi: 10.1097/00000478-198808000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Timms BG. Prostate development: a historical perspective. Differentiation. 2008;76(6):565–77. doi: 10.1111/j.1432-0436.2008.00278.x. [DOI] [PubMed] [Google Scholar]
- 8.Cunha GR, et al. The endocrinology and developmental biology of the prostate. Endocrine Rev. 1987;8(3):338–362. doi: 10.1210/edrv-8-3-338. [DOI] [PubMed] [Google Scholar]
- 9.Berquin IM, et al. Expression signature of the mouse prostate. J Biol Chem. 2005;280(43):36442–51. doi: 10.1074/jbc.M504945200. [DOI] [PubMed] [Google Scholar]
- 10.Gingrich JR, et al. Androgen-independent prostate cancer progression in the TRAMP model. Cancer Research. 1997;57(21):4687–91. [PubMed] [Google Scholar]
- 11.Gingrich JR, et al. Metastatic prostate cancer in a transgenic mouse. Cancer Research. 1996;56(18):4096–4102. [PubMed] [Google Scholar]
- 12.Greenberg NM, et al. Prostate-Cancer in a Transgenic Mouse. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(8):3439–3443. doi: 10.1073/pnas.92.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenberg NM, et al. The Rat Probasin Gene Promoter Directs Hormonally and Developmentally-Regulated Expression of a Heterologous Gene Specifically to the Prostate in Transgenic Mice. Molecular Endocrinology. 1994;8(2):230–239. doi: 10.1210/mend.8.2.8170479. [DOI] [PubMed] [Google Scholar]
- 14.Chiaverotti T, et al. Dissociation of epithelial and neuroendocrine carcinoma lineages in the transgenic adenocarcinoma of mouse prostate model of prostate cancer. American Journal of Pathology. 2008;172(1):236–246. doi: 10.2353/ajpath.2008.070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abrahamsson PA. Neuroendocrine differentiation in prostatic carcinoma. Prostate. 1999;39(2):135–148. doi: 10.1002/(sici)1097-0045(19990501)39:2<135::aid-pros9>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 16.Qi J, et al. Siah2-dependent concerted activity of HIF and FoxA2 regulates formation of neuroendocrine phenotype and neuroendocrine prostate tumors. Cancer Cell. 2010;18(1):23–38. doi: 10.1016/j.ccr.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou ZX, et al. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Research. 2006;66(16):7889–7898. doi: 10.1158/0008-5472.CAN-06-0486. [DOI] [PubMed] [Google Scholar]
- 18.Bruckheimer EM, et al. Bcl-2 accelerates multistep prostate carcinogenesis in vivo. Oncogene. 2000;19(46):5251–8. doi: 10.1038/sj.onc.1203881. [DOI] [PubMed] [Google Scholar]
- 19.Thompson TC. Metastasis-related genes in prostate cancer: The role of caveolin-1. Cancer and Metastasis Reviews. 1998;17(4):439–442. doi: 10.1023/a:1006110326366. [DOI] [PubMed] [Google Scholar]
- 20.Williams TM, et al. Caveolin-1 promotes tumor progression in an autochthonous mouse model of prostate cancer - Genetic ablation of Cav-1 delays advanced prostate tumor development in tramp mice. Journal of Biological Chemistry. 2005;280(26):25134–25145. doi: 10.1074/jbc.M501186200. [DOI] [PubMed] [Google Scholar]
- 21.Abdulkadir SA, et al. Impaired prostate tumorigenesis in Egr1-deficient mice. Nature Medicine. 2001;7(1):101–107. doi: 10.1038/83231. [DOI] [PubMed] [Google Scholar]
- 22.Chan JM, et al. Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study. Science. 1998;279(5350):563–6. doi: 10.1126/science.279.5350.563. [DOI] [PubMed] [Google Scholar]
- 23.Severi G, et al. Circulating insulin-like growth factor-I and binding protein-3 and risk of prostate cancer. Cancer Epidemiology Biomarkers & Prevention. 2006;15(6):1137–1141. doi: 10.1158/1055-9965.EPI-05-0823. [DOI] [PubMed] [Google Scholar]
- 24.Majeed N, et al. A germ line mutation that delays prostate cancer progression and prolongs survival in a murine prostate cancer model. Oncogene. 2005;24(29):4736–4740. doi: 10.1038/sj.onc.1208572. [DOI] [PubMed] [Google Scholar]
- 25.Sutherland BW, et al. Conditional deletion of insulin-like growth factorI receptor in prostate epithelium. Cancer Res. 2008;68(9):3495–504. doi: 10.1158/0008-5472.CAN-07-6531. [DOI] [PubMed] [Google Scholar]
- 26.Kaplan-Lefko PJ, et al. Enforced epithelial expression of IGF-1 causes hyperplastic prostate growth while negative selection is requisite for spontaneous metastogenesis. Oncogene. 2008;27(20):2868–76. doi: 10.1038/sj.onc.1210943. [DOI] [PubMed] [Google Scholar]
- 27.Tien JCY, Zhou SL, Xu JM. The Role of SRC-1 in Murine Prostate Carcinogenesis Is Nonessential due to a Possible Compensation of SRC-3/AIB1 Overexpression. International Journal of Biological Sciences. 2009;5(3):256–264. doi: 10.7150/ijbs.5.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chung AC, et al. Genetic ablation of the amplified-in-breast cancer 1 inhibits spontaneous prostate cancer progression in mice. Cancer Research. 2007;67(12):5965–75. doi: 10.1158/0008-5472.CAN-06-3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kasper S, et al. Development, progression, and androgen-dependence of prostate tumors in probasin-large T antigen transgenic mice: A model for prostate cancer. 6. Vol. 78. Laboratory Investigation; 1998. pp. 319pp. I–Xv. [PubMed] [Google Scholar]
- 30.Yan Y, et al. Large fragment of the probasin promoter targets high levels of transgene expression to the prostate of transgenic mice. Prostate. 1997;32(2):129–39. doi: 10.1002/(sici)1097-0045(19970701)32:2<129::aid-pros8>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 31.Masumori N, et al. A probasin-large T antigen transgenic mouse line develops prostate adenocarcinoma and neuroendocrine carcinoma with metastatic potential. Cancer Research. 2001;61(5):2239–49. [PubMed] [Google Scholar]
- 32.Klezovitch O, et al. Hepsin promotes prostate cancer progression and metastasis. Cancer Cell. 2004;6(2):185–195. doi: 10.1016/j.ccr.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 33.Tu WH, et al. The loss of TGF-beta signaling promotes prostate cancer metastasis. Neoplasia. 2003;5(3):267–277. doi: 10.1016/S1476-5586(03)80058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang JF, et al. A small composite probasin promoter confers high levels of prostate-specific gene expression through regulation by androgens and glucocorticoids in vitro and in vivo. Endocrinology. 2000;141(12):4698–4710. doi: 10.1210/endo.141.12.7837. [DOI] [PubMed] [Google Scholar]
- 35.Hill R, et al. Heterogeneous tumor evolution initiated by loss of pRb function in a preclinical prostate cancer model. Cancer Research. 2005;65(22):10243–10254. doi: 10.1158/0008-5472.CAN-05-1579. [DOI] [PubMed] [Google Scholar]
- 36.Taylor BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gabril MY, et al. Prostate targeting: PSP94 gene promoter/enhancer region directed prostate tissue-specific expression in a transgenic mouse prostate cancer model. Gene Ther. 2002;9(23):1589–99. doi: 10.1038/sj.gt.3301895. [DOI] [PubMed] [Google Scholar]
- 38.Duan W, et al. Knockin of SV40 Tag oncogene in a mouse adenocarcinoma of the prostate model demonstrates advantageous features over the transgenic model. Oncogene. 2005;24(9):1510–24. doi: 10.1038/sj.onc.1208229. [DOI] [PubMed] [Google Scholar]
- 39.Scherl A, et al. Prostatic intraepithelial neoplasia and intestinal metaplasia in prostates of probasin-RAS transgenic mice. Prostate. 2004;59(4):448–459. doi: 10.1002/pros.20020. [DOI] [PubMed] [Google Scholar]
- 40.Garabedian EM, Humphrey PA, Gordon JI. A transgenic mouse model of metastatic prostate cancer originating from neuroendocrine cells. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(26):15382–15387. doi: 10.1073/pnas.95.26.15382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maroulakou IG, et al. Prostate and Mammary Adenocarcinoma in Transgenic Mice Carrying a Rat C3(1) Simian-Virus-40 Large Tumor-Antigen Fusion Gene. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(23):11236–11240. doi: 10.1073/pnas.91.23.11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.PerezStable C, et al. Prostate cancer progression, metastasis, and gene expression in transgenic mice. Cancer Research. 1997;57(5):900–906. [PubMed] [Google Scholar]
- 43.Ellwood-Yen K, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4(3):223–38. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 44.Iwata T, et al. MYC overexpression induces prostatic intraepithelial neoplasia and loss of Nkx3.1 in mouse luminal epithelial cells. PLoS One. 2010;5(2):e9427. doi: 10.1371/journal.pone.0009427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jin RJ, et al. The nuclear factor-kappaB pathway controls the progression of prostate cancer to androgen-independent growth. Cancer Res. 2008;68(16):6762–9. doi: 10.1158/0008-5472.CAN-08-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nandana S, et al. Hepsin cooperates with MYC in the progression of adenocarcinoma in a prostate cancer mouse model. Prostate. 2010;70(6):591–600. doi: 10.1002/pros.21093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tomlins SA, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310(5748):644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 48.Tomlins SA, et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia. 2008;10(2):177–88. doi: 10.1593/neo.07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tomlins SA, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448(7153):595–9. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 50.Klezovitch O, et al. A causal role for ERG in neoplastic transformation of prostate epithelium. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(6):2105–2110. doi: 10.1073/pnas.0711711105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carver BS, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nature Genetics. 2009;41(5):619–624. doi: 10.1038/ng.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.King JC, et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nature Genetics. 2009;41(5):524–526. doi: 10.1038/ng.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Majumder PK, et al. Prostate intraepithelial neoplasia induced by prostate restricted Akt activation: the MPAKT model. Proc Natl Acad Sci U S A. 2003;100(13):7841–6. doi: 10.1073/pnas.1232229100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen ML, et al. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/− mice. Genes Dev. 2006;20(12):1569–74. doi: 10.1101/gad.1395006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Majumder PK, et al. A prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer Cell. 2008;14(2):146–55. doi: 10.1016/j.ccr.2008.06.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Y, et al. Targeted overexpression of Vav3 oncogene in prostatic epithelium induces nonbacterial prostatitis and prostate cancer. Cancer Research. 2008;68(15):6396–6406. doi: 10.1158/0008-5472.CAN-08-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li Z, et al. Prostatic intraepithelial neoplasia and adenocarcinoma in mice expressing a probasin-Neu oncogenic transgene. Carcinogenesis. 2006;27(5):1054–1067. doi: 10.1093/carcin/bgi324. [DOI] [PubMed] [Google Scholar]
- 58.Abate-Shen C, Shen MM, Gelmann E. Integrating differentiation and cancer: The Nkx3.1 homeobox gene in prostate organogenesis and carcinogenesis. Differentiation. 2008;76(6):717–727. doi: 10.1111/j.1432-0436.2008.00292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bhatia-Gaur R, et al. Roles for Nkx31 in prostate development and cancer. Genes & Development. 1999;13(8):966–977. doi: 10.1101/gad.13.8.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schneider A, et al. Targeted disruption of the Nkx3.1 gene in mice results in morphogenetic defects of minor salivary glands: parallels to glandular duct morphogenesis in prostate. Mechanisms of Development. 2000;95(1–2):163–174. doi: 10.1016/s0925-4773(00)00355-5. [DOI] [PubMed] [Google Scholar]
- 61.Abdulkadir SA, et al. Conditional loss of Nkx3.1 in adult mice induces prostatic intraepithelial neoplasia. Molecular and Cellular Biology. 2002;22(5):1495–1503. doi: 10.1128/mcb.22.5.1495-1503.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang X, et al. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature. 2009;461(7263):495–U61. doi: 10.1038/nature08361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sreenath T, et al. Androgen-independent expression of hoxb-13 in the mouse prostate. Prostate. 1999;41(3):203–7. doi: 10.1002/(sici)1097-0045(19991101)41:3<203::aid-pros8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 64.Economides KD, Capecchi MR. Hoxb13 is required for normal differentiation and secretory function of the ventral prostate. Development. 2003;130(10):2061–9. doi: 10.1242/dev.00432. [DOI] [PubMed] [Google Scholar]
- 65.Ewing CM, et al. Germline mutations in HOXB13 and prostate-cancer risk. N Engl J Med. 2012;366(2):141–9. doi: 10.1056/NEJMoa1110000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McMullin RP, Mutton LN, Bieberich CJ. Hoxb13 regulatory elements mediate transgene expression during prostate organogenesis and carcinogenesis. Dev Dyn. 2009;238(3):664–72. doi: 10.1002/dvdy.21870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rao V, et al. Prostate. 2012. A Hoxb13-driven reverse tetracycline transactivator system for conditional gene expression in the prostate. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thomsen MK, et al. Sox9 is required for prostate development. Dev Biol. 2008;316(2):302–11. doi: 10.1016/j.ydbio.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 69.Thomsen MK, et al. SOX9 elevation in the prostate promotes proliferation and cooperates with PTEN loss to drive tumor formation. Cancer Res. 2010;70(3):979–87. doi: 10.1158/0008-5472.CAN-09-2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Abate-Shen C, Shen MM. Molecular genetics of prostate cancer. Genes Dev. 2000;14(19):2410–34. doi: 10.1101/gad.819500. [DOI] [PubMed] [Google Scholar]
- 71.Freeman KW, et al. Inducible prostate intraepithelial neoplasia with reversible hyperplasia in conditional FGFR1-expressing mice. Cancer Research. 2003;63(23):8256–63. [PubMed] [Google Scholar]
- 72.Acevedo VD, et al. Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell. 2007;12(6):559–71. doi: 10.1016/j.ccr.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 73.Zhong C, et al. Cooperation between FGF8b overexpression and PTEN deficiency in prostate tumorigenesis. Cancer Res. 2006;66(4):2188–94. doi: 10.1158/0008-5472.CAN-05-3440. [DOI] [PubMed] [Google Scholar]
- 74.Wu XT, et al. Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mechanisms of Development. 2001;101(1–2):61–69. doi: 10.1016/s0925-4773(00)00551-7. [DOI] [PubMed] [Google Scholar]
- 75.Bruxvoort KJ, et al. Inactivation of Apc in the mouse prostate causes prostate carcinoma. Cancer Research. 2007;67(6):2490–2496. doi: 10.1158/0008-5472.CAN-06-3028. [DOI] [PubMed] [Google Scholar]
- 76.Yu XP, et al. Activation of beta-Catenin in Mouse Prostate Causes HGPIN and Continuous Prostate Growth After Castration. Prostate. 2009;69(3):249–262. doi: 10.1002/pros.20877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Placencio VR, et al. Stromal transforming growth factor-beta signaling mediates prostatic response to androgen ablation by paracrine Wnt activity. Cancer Research. 2008;68(12):4709–4718. doi: 10.1158/0008-5472.CAN-07-6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bhowmick NA, et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303(5659):848–851. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 79.Niu Y, et al. Targeting the stromal androgen receptor in primary prostate tumors at earlier stages. Proc Natl Acad Sci U S A. 2008;105(34):12188–93. doi: 10.1073/pnas.0804701105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Niu Y, et al. Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc Natl Acad Sci U S A. 2008;105(34):12182–7. doi: 10.1073/pnas.0804700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stanbrough M, et al. Prostatic intraepithelial neoplasia in mice expressing an androgen receptor transgene in prostate epithelium. Proc Natl Acad Sci U S A. 2001;98(19):10823–8. doi: 10.1073/pnas.191235898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhu C, et al. Conditional expression of the androgen receptor induces oncogenic transformation of the mouse prostate. J Biol Chem. 2011;286(38):33478–88. doi: 10.1074/jbc.M111.269894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Han GZ, et al. Mutation of the androgen receptor causes oncogenic transformation of the prostate. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(4):1151–1156. doi: 10.1073/pnas.0408925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu CT, et al. Increased prostate cell proliferation and loss of cell differentiation in mice lacking prostate epithelial androgen receptor. Proc Natl Acad Sci U S A. 2007;104(31):12679–84. doi: 10.1073/pnas.0704940104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Welsh M, et al. Smooth muscle cell-specific knockout of androgen receptor: a new model for prostatic disease. Endocrinology. 2011;152(9):3541–51. doi: 10.1210/en.2011-0282. [DOI] [PubMed] [Google Scholar]
- 86.Yu S, et al. Altered prostate epithelial development in mice lacking the androgen receptor in stromal fibroblasts. Prostate. 2012;72(4):437–49. doi: 10.1002/pros.21445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shen MM, Abate-Shen C. Pten inactivation and the emergence of androgen-independent prostate cancer. Cancer Research. 2007;67(14):6535–6538. doi: 10.1158/0008-5472.CAN-07-1271. [DOI] [PubMed] [Google Scholar]
- 88.Di Cristofano A, et al. Pten is essential for embryonic development and tumour suppression. Nature Genetics. 1998;19(4):348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 89.Podsypanina K, et al. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(4):1563–1568. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Banach-Petrosky W, et al. Prolonged exposure to reduced levels of androgen accelerates prostate cancer progression in Nkx3.1; Pten mutant mice. Cancer Research. 2007;67(19):9089–9096. doi: 10.1158/0008-5472.CAN-07-2887. [DOI] [PubMed] [Google Scholar]
- 91.Trotman LC, et al. Pten dose dictates cancer progression in the prostate. Plos Biology. 2003;1(3):385–396. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang SY, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4(3):209–221. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 93.Backman SA, et al. Early onset of neoplasia in the prostate and skin of mice with tissue-specific deletion of Pten. Proc Natl Acad Sci U S A. 2004;101(6):1725–30. doi: 10.1073/pnas.0308217100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Backman SA, et al. Early onset of neoplasia in the prostate and skin of mice with tissue-specific deletion of Pten. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(6):1725–1730. doi: 10.1073/pnas.0308217100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Trotman LC, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1(3):E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen Z, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436(7051):725–30. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ratnacaram CK, et al. Temporally controlled ablation of PTEN in adult mouse prostate epithelium generates a model of invasive prostatic adenocarcinoma. Proc Natl Acad Sci U S A. 2008;105(7):2521–6. doi: 10.1073/pnas.0712021105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Floc’h N, et al. Dual therapeutic targeting of the Akt/mTOR signaling pathway inhibits castration-resistant prostate cancer in a genetically engineered mouse model. Cancer Research. 2012 doi: 10.1158/0008-5472.CAN-12-0283. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Luchman HA, et al. Temporally controlled prostate epithelium-specific gene alterations. Genesis. 2008;46(4):229–34. doi: 10.1002/dvg.20386. [DOI] [PubMed] [Google Scholar]
- 100.Lee SH, et al. A constitutively activated form of the p110beta isoform of PI3-kinase induces prostatic intraepithelial neoplasia in mice. Proc Natl Acad Sci U S A. 2010;107(24):11002–7. doi: 10.1073/pnas.1005642107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen M, et al. Identification of PHLPP1 as a tumor suppressor reveals the role of feedback activation in PTEN-mutant prostate cancer progression. Cancer Cell. 2011;20(2):173–86. doi: 10.1016/j.ccr.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gao H, et al. A critical role for p27kip1 gene dosage in a mouse model of prostate carcinogenesis. Proc Natl Acad Sci U S A. 2004;101(49):17204–9. doi: 10.1073/pnas.0407693101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Di Cristofano A, et al. Pten and p27(KIP1) cooperate in prostate cancer tumor suppression in the mouse. Nature Genetics. 2001;27(2):222–224. doi: 10.1038/84879. [DOI] [PubMed] [Google Scholar]
- 104.Kim MJ, et al. Cooperativity of Nkx3.1 and Pten loss of function in a mouse model of prostate carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(5):2884–2889. doi: 10.1073/pnas.042688999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Abate-Shen C, et al. Nkx3.1; Pten mutant mice develop invasive prostate adenocarcinoma and lymph node metastases. Cancer Res. 2003;63(14):3886–90. [PubMed] [Google Scholar]
- 106.Park JH, et al. Prostatic intraepithelial neoplasia in genetically engineered mice. Am J Pathol. 2002;161(2):727–35. doi: 10.1016/S0002-9440(10)64228-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kim J, et al. Interactions between cells with distinct mutations in c-MYC and Pten in prostate cancer. PLoS Genet. 2009;5(7):e1000542. doi: 10.1371/journal.pgen.1000542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kim J, et al. A mouse model of heterogeneous, c-MYC-initiated prostate cancer with loss of Pten and p53. Oncogene. 2012;31(3):322–32. doi: 10.1038/onc.2011.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fernandez-Marcos PJ, et al. Simultaneous inactivation of Par-4 and PTEN in vivo leads to synergistic NF-kappaB activation and invasive prostate carcinoma. Proc Natl Acad Sci U S A. 2009;106(31):12962–7. doi: 10.1073/pnas.0813055106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ding Z, et al. Telomerase reactivation following telomere dysfunction yields murine prostate tumors with bone metastases. Cell. 2012;148(5):896–907. doi: 10.1016/j.cell.2012.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ding Z, et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature. 2011;470(7333):269–73. doi: 10.1038/nature09677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang XS, et al. Characterization of KRAS Rearrangements in Metastatic Prostate Cancer. Cancer Discov. 2011;1(1):35–43. doi: 10.1158/2159-8274.CD-10-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Palanisamy N, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nature Medicine. 2010;16(7):793–8. doi: 10.1038/nm.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dankort D, et al. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007;21(4):379–84. doi: 10.1101/gad.1516407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang J, et al. Braf activation cooperates with Pten loss to regulate c-Myc activation in advanced prostate cancer. Cancer Research. 2012 doi: 10.1158/0008-5472.CAN-12-0820. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tuveson DA, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5(4):375–87. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 117.Aytes A, et al. A mouse model of metastatic prostate cancer leads to activation of Ets signaling. 2012. submitted. [Google Scholar]
- 118.Mulholland DJ, et al. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Research. 2012;72(7):1878–89. doi: 10.1158/0008-5472.CAN-11-3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pearson HB, et al. SCRIB expression is deregulated in human prostate cancer, and its deficiency in mice promotes prostate neoplasia. J Clin Invest. 2011;121(11):4257–67. doi: 10.1172/JCI58509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pearson HB, Phesse TJ, Clarke AR. K-ras and Wnt Signaling Synergize to Accelerate Prostate Tumorigenesis in the Mouse. Cancer Research. 2009;69(1):94–101. doi: 10.1158/0008-5472.CAN-08-2895. [DOI] [PubMed] [Google Scholar]
- 121.Gingrich JR, et al. Pathologic progression of autochthonous prostate cancer in the TRAMP model. Prostate Cancer and Prostatic Diseases. 1999;2(2):70–75. doi: 10.1038/sj.pcan.4500296. [DOI] [PubMed] [Google Scholar]
- 122.Kasper S, et al. Development, progression, and androgen-dependence of prostate tumors in probasin-large T antigen transgenic mice: a model for prostate cancer. Laboratory Investigation. 1998;78(3):319–33. [PubMed] [Google Scholar]
- 123.Kim MJ, et al. Nkx3.1 mutant mice recapitulate early stages of prostate carcinogenesis. Cancer Research. 2002;62(11):2999–3004. [PubMed] [Google Scholar]