

Abstract
Purpose
Osteoprotegerin (OPG) is a decoy receptor for the Receptor of NF-κB (RANK) ligand that can inhibit osteoclastogenesis. Previous studies have suggested that Mammalian Target of Rapamycin (mTOR) inhibition upregulates OPG production. We tested the hypothesis that the mTOR inhibitor rapamycin could inhibit neuroblastoma bone metastases through its action on OPG.
Experimental Design
An orthotopic model of bone metastasis was established. Mice with established disease were subsequently treated with rapamycin (5 mg/kg IP daily) or vehicle control (DMSO 1:1000). X-rays were obtained twice a week to detect pathologic fractures. Serum OPG levels were measured by ELISA after two weeks of treatment.
Results
Mice with bone disease receiving rapamycin had increased serum levels of OPG in the CHLA-20 mice compared to controls (36.89 pg/mL±3.90 vs 18.4 pg/mL±1.67, p=0.004) and NB1691 tumor-bearing groups (46.03±2.67 pg/mL vs 17.96±1.84 pg/mL, p=0.001), and a significantly longer median time to pathologic fractures with CHLA-20 (103 days vs 74.5 days, p=0.014) and NB1691 xenografts.
Conclusion
In a xenograft model, increased OPG expression correlated with a delay to pathologic fracture suggesting a potential role for mTOR inhibitors in the treatment of neuroblastoma bone metastases.
Keywords: Rapamycin, Osteoprotegerin, Bone metastasis, Neuroblastoma
Neuroblastoma is the most common extracranial solid tumor of childhood. Stage 4 disease in these patients involves the presence of distant metastasis and portends a very poor prognosis in children older than 18 months of age. Approximately 65% of all neuroblastoma patients will present with stage 4 disease. Despite some successes in therapy, the 5-year survival rate of these patients remains a dismal 30%–40%. Bone and bone marrow are common sites of distant metastasis. In a large multi-institutional review, patients with skeletal disease at diagnosis had a 5 year event free survival of only 25% [1]. In addition to a very low survival rate, bone metastases have added morbidities including pathologic fractures, nerve compression, intractable pain, and metabolic derangements. Present therapy which includes aggressive chemotherapy and bone marrow transplantation does little to treat bone lesions or prevent their progression [2].
Neuroblastoma bone lesions are osteolytic and form through the recruitment and activation of osteoclasts which break down native bone. Following invasion, growth factors released from bone breakdown promote further tumor growth and further stimulation of osteoclasts. This “vicious cycle” of positive feedback ultimately leads to rapid growth of the tumor and rapid breakdown of native bone [3]. There are multiple factors which contribute to the activation of osteoclasts in bone metastasis. One of these factors, receptor of NF-κ-B ligand (RANKL) has been shown to play a key role in neuroblastoma bone metastasis. Neuroblastoma lesions appear to follow a progression in bone which is primarily driven by RANKL secreted by invasive neuroblastoma cells. RANKL is also secreted by mesenchymal cells and osteoblasts in the presence of tumor cells. RANKL acts on osteoclast progenitors to promote differentiation and activation [4]. Denosumab, a monoclonal antibody that blocks RANKL has shown some promise in early clinical trials against other tumors metastatic to bone, confirming this pathway as a major therapeutic target in treating bone metastasis [5].
Osteoprotegerin (OPG) is soluble protein in the TNF family that acts as a decoy receptor to RANKL and serves to inhibit osteoclast differentiation. Although high levels of both OPG and RANKL transcripts are found in many established neuroblastoma cell lines, RANKL appears to be preferentially secreted in all lines, promoting osteoclastogenesis [6]. The addition of recombinant OPG in clinical trials involving multiple myeloma and breast cancer metastatic to bone decreased the effect of RANKL and slowed the progression of skeletal lesions [7]. In experiments looking specifically at bone metastases of neuroblastoma, the upregulation of OPG using neuroprogenitor cells decreased the incidence of osteolytic lesions and the overall growth of metastases [8].
Rapamycin is a macrocyclic lactone antibiotic which has multiple effects in vivo. At high doses, it has been demonstrated to have direct inhibition of the growth of some neuroblastoma xenografts through the down-regulation of the mammalian target of rapamycin (mTOR) [9]. Inhibition of mTOR inhibits the proliferation and survival of cancer cells as well as having a profound anti-angiogenic effect. In addition, rapamycin has been shown to slow the early healing of fractures in experimental mice, presumably due to anti-angiogenic and anti-proliferative effects [10]. Lastly, a mechanism has been proposed whereby inhibition of mTOR directly upregulates OPG secretion in mouse bone marrow stromal cells thereby rendering protection from osteoclastogenesis [11].
Thus, rapamycin may act through several different mechanisms which appear to have a beneficial effect on the treatment of neuroblastoma bone metastases. While its anti-proliferative and anti-angiogenic effects are well established, an anti-osteoclastogenic effect through an increased secretion of OPG has not yet been described in neuroblastoma xenografts. Similarly, the mechanism of increased secretion of OPG through inhibition of mTOR remains unknown. We sought to determine whether rapamycin would have an anti-tumor effect on a model of established neuroblastoma bone metastasis both as a single agent and in combination with a modeled chemotherapy regimen.
1. Methods
1.1. Cell lines
The human neuroblastoma cell line NB1691 was provided by P. Houghton (Columbus, OH). The CHLA-20 cell line was provided by C.P. Reynolds (Los Angeles, CA). These cells were engineered to constitutively express firefly luciferase as previously described [12].
1.2. In Vitro proliferation
Both cell lines were plated in a 96-well plate at a concentration of 1×105 cells per well. Rapamycin (LKT Labs, St Paul MN) was diluted in concentrations of 10–1000 nM in DMSO. The final concentration of DMSO in all wells was 5%. The cells were treated for 48 h. Following this, cell proliferation was measured using a MTT assay (Cell Titer 96 Aqueous One Cell Proliferation Assay, Promega, Madison, WI). All experiments were repeated in triplicate.
1.3. OPG RT-PCR
Neuroblastoma cell lines (NB1691 and CHLA-20) were co-cultured with human mesenchymal stem cells (hMSC) (Lonza, Walkersville, MD). Twenty four hours after plating hMSC at a concentration of 106 cells per well in a 6-well plate, neuroblastoma cells were added at a concentration of 106 cells per well. Treatment began 24 h after the addition of the neuroblastoma cells. In one group, the cells were treated with rapamycin 100 nM; a control group was treated with DMSO only. After 3 days, the cells were harvested and RNA extracted. RNA was reverse transcribed using Superscript II First Strand Synthesis (Life Technologies, Grand Island, New York). RT-PCR was performed using primers for human OPG; Hs00171068_m1 TNFRSF11B (Applied Biosystems, Carlsbad, CA).
1.4. In Vitro anti-osteoclastic effect of rapamycin
A co-culture of neuroblastoma cells with bone marrow harvested from CB-17 SCID mice (Taconic Farms, Hudson, NY) was used to determine the anti-osteoclastic effects of rapamycin. Neuroblastoma cells were plated at a density of 1×105 cells per well in a 48 well plate. 24 h later, bone marrow was harvested from the femoral cavity of CB-17 SCID mice. The harvested marrow was filtered through a 40μm cell strainer and centrifuged. Collected cells were resuspended in α-MEM media (Mediatech Cellgro, Herndon, VA) and incubated at 37°C for 2 h in a 10 cm culture plate. Following this, non-adherent cells were plated along with neuroblastoma cells in 48 well plates at a density of 1×106 cells per well. 24 h after the addition of bone marrow cells, increasing concentrations of rapamycin (50–1000 nM) were added to wells. After maintaining the culture for 5 days, the cells were fixed and stained for tartrate resistant acid phosphatase (TRAP) activity (Sigma, St. Louis, MO). Osteoclasts were identified as multi-nucleated TRAP-positive cells. All experiments were performed in triplicate.
1.5. In Vivo experiments
All experiments were approved by the institutional IUCAC committee (protocol # 273) prior to initiation. Tumors were established in the femoral canal of CB17-SCID mice as previously described [8]. The presence of active tumor growth was confirmed at two weeks using bioluminescence imaging. Following this the mice were size-matched into groups based on bioluminescent signal (n=5 for all groups). The treatment group received rapamycin at 5 mg/kg prepared in solution using 5% DMSO and 5% TWEEN-80 (Sigma) in PBS, via intraperitoneal injection. The control group received 5% DMSO and 5% TWEEN-80 in PBS only. All mice received daily rapamycin injections for 3 weeks. One group was sacrificed immediately after 3 weeks of treatment and the mouse femurs were collected in formalin. Serum was also collected at this time for measurement of mouse or human OPG by ELISA (RayBiotech Inc, Norcross, GA). These femurs were analyzed using μCT ex vivo CT scanning. 4μm cuts were obtained through the distal femur. A region of interest (ROI) was created with the same volume for all samples. Within this ROI, cortical bone thickness was calculated using MicroView CT Analysis Software (Parallax Innovations, Allentown, PA). In a second group of mice, x-rays of the affected femur were taken twice a week using a Faxitron MX20 system. The endpoint for this experiment was determined to be grade 4 or complete pathologic fracture. Once the mice reached this endpoint they were sacrificed and the date noted. A third group of mice received AAV2/8-OPG (Vector Production Core, St Jude Children’s Hospital, Memphis, TN). Human OPG cDNA (Invivogen, San Diego, CA) was excised from pORF9-hOPG using NheI and AgeI. The resulting 1.3 kb fragment was ligated onto pAV CAGG/MCS. The packaging plasmid AAV8-2 was used to make the AAV2/8-OPG pseudotyped vector [13]. Once tumors were established, a cohort of mice was injected with 2×1011 vg of AAV2/8-OPG via the tail vein. Following this, the mice were followed via x-ray as described above with the endpoint being grade 4 fracture. A grade 4 or pathologic fracture was defined as a fracture that clearly crossed both sides of the bony cortex or a fracture that was obviously displaced.
2. Results
2.1. Rapamycin treatment has little effect on neuroblastoma proliferation in Vitro
As previously noted, rapamycin can have a direct growth inhibitory effect in vivo [4]. However previous and current experience in our lab demonstrates a relative resistance to rapamycin with the two tested cell lines at low concentrations of rapamycin. The neuroblastoma cell lines CHLA-20 and NB1691 were plated in a 96-well plate at concentrations of 1×105 cells per well and were subjected to increasing concentrations of rapamycin. Following 48 h of exposure, the viable cells were measured using an MTT assay. Rapamycin treatments up to 100 nM demonstrated minimal inhibition of proliferation in both NB1691 and CHLA-20 cell lines (Fig. 1A).
Fig 1.

Rapamycin Increases OPG Production and Decreases Osteoclastogenesis at Doses not Directly Toxic to Neuroblastoma Cells. (A) Rapamycin at doses tested (50–100 nM) has minimal effect on the proliferation of CHLA-20 and NB1691 cells after 48 h of exposure. (B) Treatment with rapamycin results in a 1.3-fold (*p=0.025) and 2.5-fold (**p<0.001) increase in OPG expression in CHLA-20 and NB1691 respectively as measured by RT-PCR after 48 h exposure. (C) Treatment with rapamycin decreases osteoclastogenesis 2-fold when tested in a co-culture of CHLA-20 (*p=0.004) or NB1691 (**p=0.001) cells and harvested mouse bone marrow cells.
2.2. RT-PCR of rapamycin treated neuroblastoma cells grown in co-culture with human mesenchymal stem cells demonstrates an upregulation of OPG
Co-cultures of NB1691 and CHLA-20 neuroblastoma cell lines and hMSC were harvested and RNA extracted for RT-PCR after 3 days of treatment with rapamycin or DMSO vehicle. hMSCs were chosen based on the hypothesis that rapamycin would upregulate OPG production in both human neuroblastoma and differentiated stem cells which would be detectable by RT-PCR against human OPG. The CHLA-20 co-culture treated with rapamycin demonstrated a mean 1.3-fold increase (p= 0.025) in OPG expression compared to control. Similarly the NB1691 co-culture treated with rapamycin demonstrated a mean 2.5-fold increase (p<0.001) in OPG expression compared to control (Fig. 1B). This demonstrates that rapamycin acts to upregulate OPG expression in an in vitro model of the cell macro-environment present in neuroblastoma bone metastases.
2.3. Rapamycin treatment of neuroblastoma–bone marrow co-culture demonstrates a dose-dependent decrease in osteoclasts as measured by TRAP staining
Co-cultures of NB1691 and CHLA-20 neuroblastoma cell lines and harvested mouse bone marrow cells were treated with 100 nM rapamycin. Following 5 days incubation, the plates were fixed and stained for Tartrate-Resistant Acid Phosphatase (TRAP) positive cells. Positively stained osteoclasts were counted and demonstrated a dose-dependent decrease for given rapamycin concentrations. In CHLA-20 co-culture, the number of osteoclasts in wells treated with 100 nM rapamycin was approximately 50% less than control (mean 14.3±1.39 to 30.07±3.49, p=0.004). In NB1691 co-culture, osteoclasts in wells treated with 100 nM rapamycin were approximately 55% less than control (mean 17.75± 2.19 to 32.38±1.73, p=0.001) (Fig. 1C). This demonstrates a distinct effect of decreased differentiation of osteoclasts due to increased secretion of OPG by neuroblastoma cells despite the lack of a direct toxic effect to the tumor cells.
2.4. Rapamycin increased serum OPG levels in treated mice
Serum was collected immediately following a three week treatment course from the control and treated groups of tumor-bearing mice. Levels of human OPG increased in the rapamycin treated groups compared to control for CHLA-20 (36.89±3.9 pg/ml vs. 18.41±1.67 pg/mL, p=0.0004) and NB1691 (46.03±2.67 pg/mL vs 17.96±1.84 pg/mL, p= 0.001). This represents OPG contribution by the neuroblastoma xenograft as this ELISA is specific for human OPG. Similarly mouse OPG was increased compared to control in the CHLA-20 (30.29±2.34 ng/mL vs 8.27±0.765 ng/mL, p=0.0001) and NB1691 (21.2±4.69 ng/mL vs. 8.59± 0.986 ng/mL, p=0.011) groups (Fig. 2A). This represents the contribution of host bone marrow stroma, specifically osteoblasts, to increased mouse OPG secretion in response to rapamycin treatment. It is important to note that while the individual ELISAs are specific to species, there is evidence that both human OPG and mouse OPG exert similar effects on mouse osteoclasts.
Fig 2.
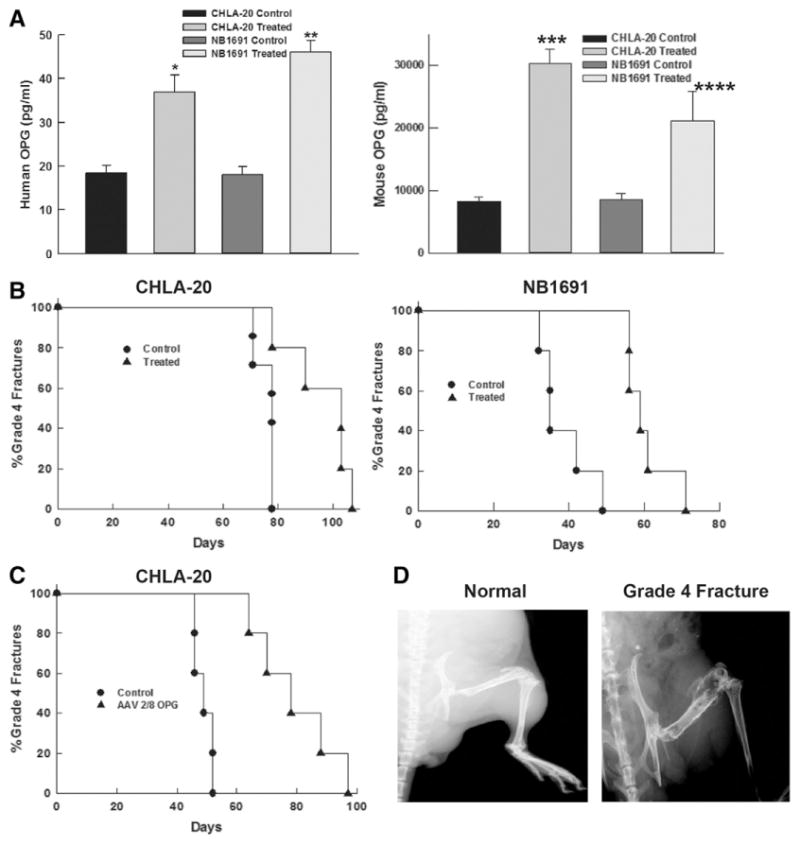
Rapamycin Increases Serum OPG and Delays Time to Grade 4 Fracture in Mouse Neuroblastoma Xenografts. (A) Treatment with rapamycin increased serum levels of human OPG 2-fold (*p=0.0004) and 2.5-fold (**p=0.001) in mice implanted with CHLA-20 and NB1691 xenografts respectively. Similarly, mouse OPG (from host bone marrow stroma) was increased 3.65-fold (***p=0.0001) and 2.5-fold (****p=0.0105) in the CHLA-20 and NB1691 implanted mice respectively. (B) Treatment with rapamycin delayed time to grade 4 fracture in CHLA-20 and NB1691 xenografts. (C) AAV2/8-OPG delayed time to grade 4 fracture in a CHLA-20 xenograft. (D) Comparison of normal and grade 4 fracture in bone metastasis xenograft.
2.5. Rapamycin Increases the time to grade 4 fracture in mice with neuroblastoma xenografts
Xenograft tumor models were established by injecting 2×105 cells into the femoral cavity of SCID mice. The injected cells were engineered to express luciferase and established tumors were confirmed using bioluminescence. Once established tumors had been confirmed (approximately two weeks after injection), the mice were treated with rapamycin (5 mg/kg via intraperitoneal injection) for two weeks. Immediately after injections began, the mouse femurs were imaged bi-weekly using a Faxitron MX20 machine. The end result of the experiment was defined as a grade 4 fracture, or a fracture that was obviously displaced on x-ray. Two different neuroblastoma cell lines were injected into mice femurs. In the CHLA-20 group, the median increase in time to fracture was 30 days (103 days vs. 74.5 days, p=0.001). In mice injected with NB1691 tumor cells, the median increase in time to fracture was 24 days (59 days vs. 35 days, p=0.002) (Fig. 2B). This suggests that the treatment effect from rapamycin carries over to in vivo experiments with an overall effect of decreased tumor-driven bone breakdown.
2.6. Treatment with AAV2/8-OPG results in delay to grade 4 fracture
As noted previously, rapamycin has been proven to be both anti-proliferative and anti-angiogenic. Both of these effects may have contributed to the increased time to fracture. In order to determine if the upregulation of OPG was a primary driver of protection against pathologic fracture, we injected SCID mice that had established bone metastasis from CHLA-20 xenografts with adeno-associated virus vector encoding human OPG (AAV2/8-OPG). Tail vein injection with AAV2/8-OPG resulted in high level transduction of the host hepatocytes with subsequent expression and systemic release of OPG. Mice treated with AAV2/8-OPG had a median increase of 29 days to grade 4 fracture compared to control (78 vs 49 days, p=0.003) (Fig. 2C). AAV2/8-OPG treated mice demonstrated a similar delay to fracture as those treated with rapamycin (Fig. 2D). While rapamycin has been noted to have some anti-angiogenic properties when used in vivo [8], this suggests that the primary effect in bone metastases is due to an upregulation of OPG, as delivery of OPG alone would not be expected to have anti-proliferative or anti-angiogenic effects.
2.7. Treatment with rapamycin decreases the number of osteoclasts in cortical bone
Neuroblastoma xenografts were established in SCID mice from CHLA-20 and NB1691 cell lines as described above. At the end of a 3 week treatment course of rapamycin, the mice were sacrificed and the femurs were collected in formalin. The bones were decalcified and paraffin embedded. Following this, the slides were stained for tartrate resistant acid phosphatase (TRAP Staining kit, Sigma St Louis, MO). The resulting slides were viewed under a HPF microscope and the numbers of positive staining cells within the respective cortical bone of each sample were counted. Samples from mice treated with rapamycin had approximately half as many osteoclasts as controls. (CHLA-20 mean 14.8±2.42 osteoclasts/hpf vs. 31±2.74 osteoclasts/hpf, p= 0.002, NB1691 mean 17.2±1.24 osteoclasts/hpf vs. 27.4± 2.09 osteoclasts/hpf, p=0.003) (Fig. 3A). A typical example of TRAP-stained femurs is shown in Fig 3B. A separate set of mouse femurs was collected from animals which received neither tumor xenografts nor treatment with any drugs. These samples were prepared as above and stained for TRAP. These femurs had a mean osteoclast number/hpf of 20.2 which is closer in number to the mice treated with rapamycin. These results suggest that treatment with rapamycin blocks tumor directed osteoclastogenesis.
Fig 3.
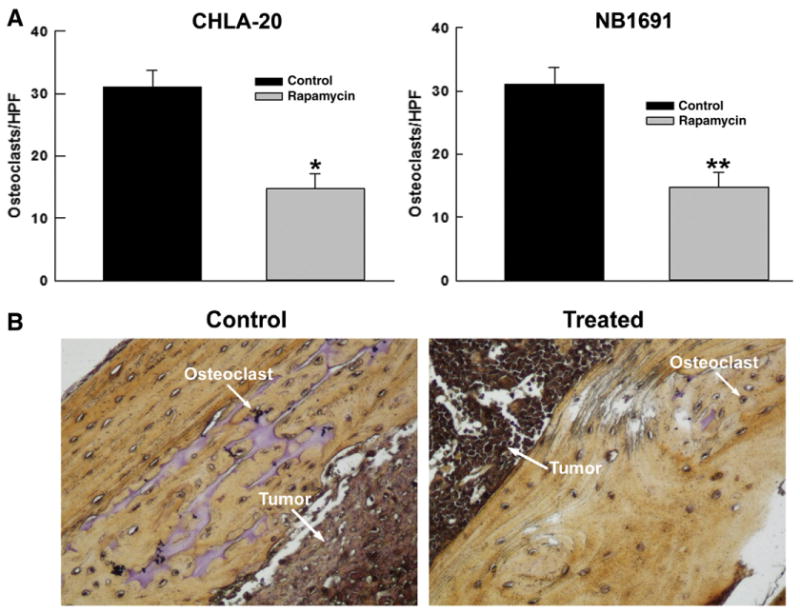
Rapamycin Decreases Osteoclastogenesis in a Mouse Neuroblastoma Xenograft. (A) Treatment with rapamycin (5 mg/kg) daily for 3 weeks decreased the number of osteoclasts in mouse femurs by 2-fold (*p=0.022) and 1.6-fold in CHLA-20 and NB1691 (**p=0.003) xenografts respectively as detected by TRAP staining, indicating a decrease in RANKL signaling in treated mice through an upregulation of OPG. (B) Example of TRAP stained control and treated mouse femurs. Treated femurs demonstrate a significant decrease in osteoclasts by TRAP staining compared to controls.
2.8. Treatment with rapamycin increases bone volume fraction and cortical bone thickness in a xenograft model
Neuroblastoma xenografts were established in SCID mice as noted above using CHLA-20 and NB1691 cell lines. Once confirmed by bioluminescence, groups (n=5) were subjected to a 3 week treatment course of 5 mg/kg rapamycin daily. Following this all mouse femurs were collected and analyzed by ex vivo CT. Analysis of the results revealed an increase in bone volume fraction (BVF) and cortical thickness in mice treated with rapamycin. In the CHLA-20 group, mean BVF was increased in the treated group (0.572±0.027 vs. 0.477± 0.029, p=0.042) (Fig. 4A), and mean cortical thickness was also higher in the rapamycin treated group (0.045±0.012 mm vs. 0.0163±0.003 mm, p=0.0306) (Fig. 4B). Similarly, in the NB1691 group, mean BVF increased in the mice with rapamycin treatment (0.62±0.012 vs. 0.545±0.009, p= 0.0001) and mean cortical thickness was higher for the treated group (0.0432±0.011 vs. 0.0149±0.002 mm, p= 0.017) (Fig. 4C). These results suggest that the increase to fracture times demonstrated in the rapamycin treated groups is due to an overall decrease in osteoclast activity and a decrease in bone breakdown due to tumor cells.
Fig 4.
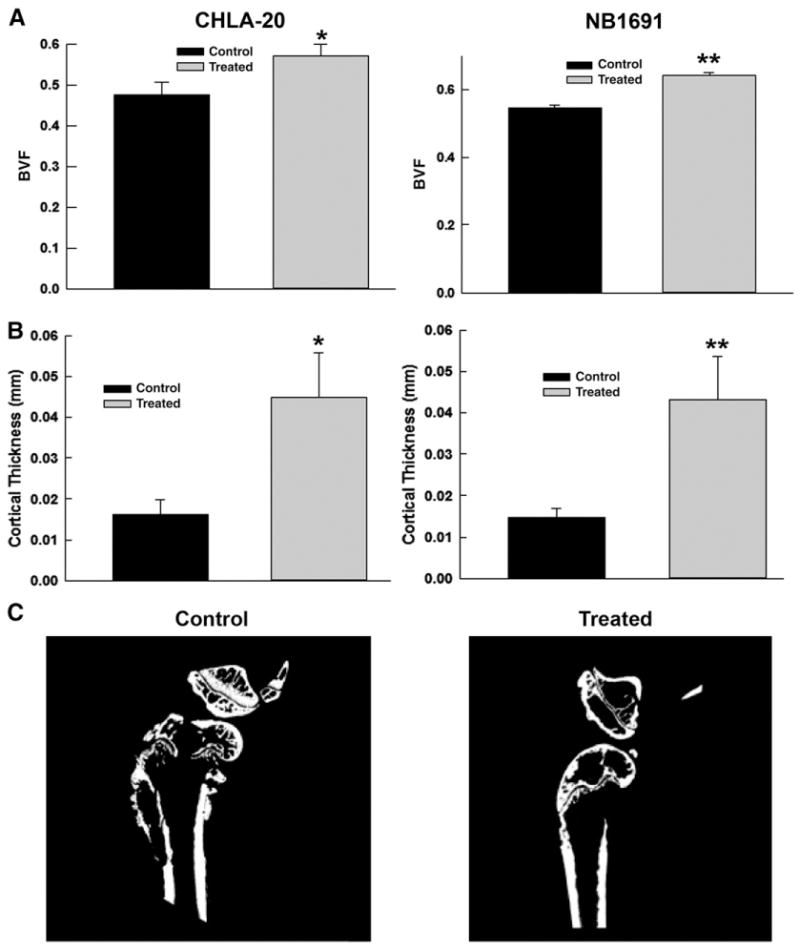
Rapamycin Increased Bone Volume Fraction and Cortical Thickness in Treated Mice. (A) Bone volume fractions (BVF) in CHLA-20 (*p=0.042) and NB1691 (**p=0.001) xenografts increased as a result of rapamycin treatment indicating an inhibition of osteoclastogenesis by the neuroblastoma xenografts. (B) Cortical thickness in CHLA-20 (*p=0.031) and NB1691 (**p=0.017) xenografts was also increased relative to control, similarly indicating decreased osteoclastogenesis by neuroblastoma xenografts. (C) Representative μCT Images of control and treated mouse femurs. Control femurs demonstrate obvious cortical breakdown by tumor, while treated femurs demonstrate a preservation of cortical architecture after two weeks.
3. Discussion
Bone metastases add significant morbidity and disability to patients suffering from neuroblastoma. The mechanism driving cancerous invasion of the bone is multifaceted but appears to involve RANKL signaling by the tumor cells to induce osteoclastogenesis thereby breaking down native bone. Osteoprotegerin is a competitive inhibitor of RANKL and has been proposed as a target to block tumor driven osteoclastogenesis. Here we propose that mTOR inhibition, through treatment with rapamycin promotes the production of OPG, interfering with RANKL driven bone breakdown, thereby slowing the progression of bony metastases.
We have demonstrated that in both in vitro and in vivo models, rapamycin effectively upregulates the production of OPG in neuroblastoma cells. This serves to inhibit RANKL signaling and decrease osteoclast differentiation thereby restricting the breakdown of native bone by metastatic neuroblastoma. This paper and many others have described the important effects of RANKL signaling in bone metastasis.
Using the established neuroblastoma cell lines CHLA-20 and NB1691, rapamycin treatment at doses that were not directly toxic to the cells stimulated the expression of OPG, as detected by RT-PCR. Similarly, co-cultures with harvested mouse bone marrow treated with rapamycin demonstrated decreased osteoclastogenesis compared to untreated controls. In a mouse model, treatment with rapamycin resulted in increased levels of serum human OPG, increased cortical bone thickness, and delayed time to pathologic fracture. These results are indicative of a significant protective effect against progression of bony metastatic disease in neuroblastoma. The cell lines chosen (CHLA-20 and NB-1691) were known to be resistant to rapamycin at the doses tested. This was confirmed in an in vitro MTT assay. Previous researchers have demonstrated an antiangiogenic effect of rapamycin on tumor xenografts. Treatment of neuroblastoma bone metastasis xenografts with the anti-angiogenic agent Bevacizumab (Genentech, San Francisco CA) had no effect on the progression to grade IV fracture (data not shown). However treatment with an AAV-OPG construct had similar results when compared to the mice treated with rapamycin. These results imply but do not prove that treatment with rapamycin affects the growth of neuroblastoma exclusively through upregulation of OPG.
We also sought to determine if this trend of OPG upregulation was preserved in human patients in human neuroblastoma (IRB approval # XPD10-043). The NB2005 and NB2008 neuroblastoma trials were designed and administered at St Jude Children’s Research Hospital. The NB2005 trial included oral gefitinib and irinotecan as upfront therapy prior to induction chemotherapy and ultimately stem cell transplantation for stage 4 neuroblastoma patients. NB2008 had a similar design but substituted temsirolimus for gefitinib. Temsirolimus is an mTOR inhibitor with similar biologic effects to rapamycin. In our pre-clinical studies, temsirolimus had a similar effect to rapamycin in upregulating OPG in cultured cells. After obtaining appropriate IRB approval, we were able to obtain plasma samples from five patients on the NB2005 protocol, who did not receive temsirolimus, as well as four patients on the NB2008 trial who did receive temsirolimus. We chose a time point approximately fifty days into therapy where all patients had received two cycles of their assigned treatment. We then analyzed plasma samples for OPG using ELISA. Patients treated with temsirolimus had a three-fold higher level of OPG in their plasma compared to those patients not treated with temsirolimus (1299±29.98 pg/mL vs. 702.7±37.1 pg/mL, p=0.025) (Fig. 5). Unfortunately, given the small patient size and the retrospective nature of the study, it was not feasible to screen these patients for bone metastases or to further compare the two treatments.
Fig 5.
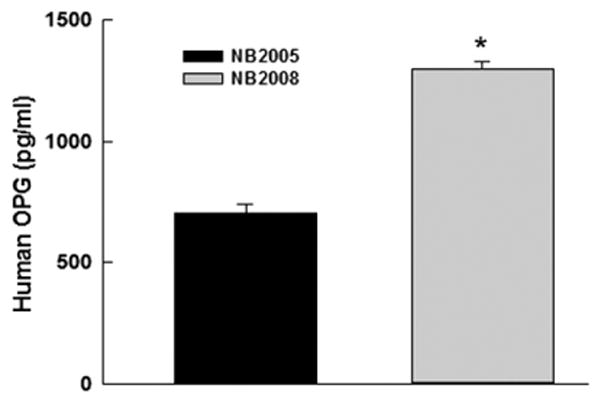
Temsirolimus Increased Serum OPG Levels in Treated Patients In a cohort of stage 4 neuroblastoma patients, the addition of temsirolimus (NB2008) increased OPG secretion 3-fold (*p= 0.025) compared to controls (NB2005) after two cycles of therapy as detected by ELISA.
Bisphosphonates have also demonstrated a delay to fracture in preclinical neuroblastoma models [14] and have been used in clinical trials with limited success. It is unclear whether these two therapies act via similar or different pathways. Current clinical trials using RANKL decoy antibodies in multiple cancers metastatic to bone have shown great promise in preventing pathologic fractures as well as the spread of the disease [6]. Based on our results and the results of others, these clinical studies should be extended into pediatric solid tumors that are metastatic to bone; in particular neuroblastoma.
In conclusion treatment with rapamycin as a single agent restricted the growth of bone metastasis in a mouse neuroblastoma xenograft model. We propose that the mechanism for this effect is the upregulation of OPG and the disruption of normal tumor RANKL signaling which decreased osteoclast directed bone resorption. Both OPG signaling and RANKL signaling provide excellent targets for the prevention of new bony metastasis and to slow the progression of existing bone metastases. The role of mTOR inhibition in this pathway is as yet not fully delineated. However using the mTOR inhibitor rapamycin we have been able to demonstrate an upregulation of OPG. Further study into this intricate signaling mechanism and the direct role that mTOR inhibition may play is certainly warranted. Our results suggest further investigation of both RANKL inhibitors and mTOR inhibitors in neuroblastoma bone metastasis may be of value.
References
- 1.Ladenstein R, Phillip T, Lasset C, et al. Multivariate analysis of risk factors in stage 4 neuroblastoma patients over the age of one year treated with megatherapy and stem-cell transplantation: a report from the European Bone Marrow Transplantation Solid Tumor Registry. J Clin Oncology. 1998;16(3):953–65. doi: 10.1200/JCO.1998.16.3.953. [DOI] [PubMed] [Google Scholar]
- 2.Park JR, Villablanca JG, London WB, et al. Outcome of high-risk stage 3 neuroblastoma with myeloablative therapy and 13-cis retinoic acid: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2009;52:44–50. doi: 10.1002/pbc.21784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wittrant Y, Theoleyre S, Chipoy C, et al. RANKL/RANK/OPG: new therapeutic targets in bone tumors and associated osteolysis. Biochem Biophys Acta. 2004;1704:49–57. doi: 10.1016/j.bbcan.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 4.Michigami T, Ihara-Watanabe M, Yamazaki M, et al. Receptor activator of nuclear factor κB ligand (RANKL) is a key molecule of osteoclast formation for bone metastasis in a newly developed model of human neuroblastoma. Cancer Res. 2001;61:1637–44. [PubMed] [Google Scholar]
- 5.Santini D, Fratto ME, Vincenzi B, et al. Denosumab: the era of targeted therapies in bone metastatic diseases. Curr Cancer Drug Targets. 2009;9:834–42. doi: 10.2174/156800909789760375. [DOI] [PubMed] [Google Scholar]
- 6.Granchi D, Amato I, Battistelli L, et al. In Vitro blockade of receptor activator of nuclear factor-κB ligand prevents osteoclastogenesis induced by neuroblastoma cells. Int J Cancer. 2004;111:829–38. doi: 10.1002/ijc.20308. [DOI] [PubMed] [Google Scholar]
- 7.Body JJ, Greipp P, Coleman RE, et al. A phase I study of AMGN-0007 a recombinant osteoprotegerin construct in patients with multiple myeloma or breast carcinoma related bone metastases. Cancer. 2003;97(3 Suppl):887–92. doi: 10.1002/cncr.11138. [DOI] [PubMed] [Google Scholar]
- 8.Sims TL, Hamner JB, Bush RA, et al. Neural progenitor-cell mediated delivery of osteoprotegerin limits disease progression in a preclinical model of neuroblastoma bone metastasis. J Pediatr Surg. 2009;44:204–11. doi: 10.1016/j.jpedsurg.2008.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Houghton PJ, Morton CL, Kolb EA, et al. Initial testing (stage I) of the mTOR inhibitor rapamycin by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008;50:799–805. doi: 10.1002/pbc.21296. [DOI] [PubMed] [Google Scholar]
- 10.Holstein JH, Klein M, Garcia P, et al. Rapamycin affects early fracture healing in mice. Brit J Pharm. 2008;154:1055–62. doi: 10.1038/bjp.2008.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mogi M, Kondo A. Down-regulation of mTOR leads to up-regulation of osteoprotegerin in bone marrow cells. Biochem Biophys Rsch Comm. 2009;384:82–6. doi: 10.1016/j.bbrc.2009.04.084. [DOI] [PubMed] [Google Scholar]
- 12.Dickson PV, Hamner B, Ng CY, et al. In vivo bioluminescence imaging for early detection and monitoring of disease in a murine model of neuroblastoma. J Pediatr Surg. 2007;42:1172–9. doi: 10.1016/j.jpedsurg.2007.02.027. [DOI] [PubMed] [Google Scholar]
- 13.Gao GP, Alvira MR, Wang L, et al. Novel adenoassociated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci USA. 2002;99:11854–9. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dickson PV, Hamner JB, Cauthen LA, et al. Efficacy of zoledronate against neuroblastoma. Surgery. 2006;140(2):227–35. doi: 10.1016/j.surg.2006.02.004. [DOI] [PubMed] [Google Scholar]