

Abstract
Aims: Accumulating evidence indicates that oxidative stress is associated with inflammation, and the cellular redox status can determine the sensitivity and the final outcome in response to inflammatory stimuli. To control the redox balance, mammalian cells contain a variety of oxidoreductases belonging to the thioredoxin superfamily. The large number of these enzymes suggests a complex mechanism of redox regulation in mammals, but the precise function of each family member awaits further investigations. Results: We generated mice deficient in transmembrane thioredoxin-related protein (TMX), a transmembrane oxidoreductase in the endoplasmic reticulum (ER). When exposed to lipopolysaccharide (LPS) and d-(+)-galactosamine (GalN) to induce inflammatory liver injury, mutant mice were highly susceptible to the toxicants and developed severe liver damage. LPS-induced production of inflammatory mediators was equivalent in both wild-type and TMX−/− mice, whereas neutralization of the proinflammatory cytokine tumor necrosis factor-α suppressed the toxic effects of LPS/GalN in the mutant mice. Liver transcriptional profiles revealed enhanced activation of the p53-signaling pathway in the TMX−/− mice after LPS/GalN treatment. Furthermore, TMX deficiency also caused increased sensitivity to thioacetamide, which exerts its hepatotoxicity through the generation of reactive oxygen species. Innovation: The present study is the first to address the role of the oxidoreductase TMX in inflammatory liver injury. The phenotype of mice deficient in TMX suggests a functional link between redox regulation in the ER and susceptibility to oxidative tissue damage. Conclusion: We conclude that TMX plays a major role in host defense under the type of inflammatory conditions associated with oxidative stress. Antioxid. Redox Signal. 18, 1263–1272.
Introduction
The inflammatory response is a part of a normal host defense for eliminating pathogens, harmful environmental agents, or tumor cells (13). Excessive reactions or prolonged activation of the pathway, however, can cause adverse clinical effects. Accumulating evidence shows that oxidative stress is associated with inflammation, and the modulation of the cellular redox status has emerged as a potential therapeutic strategy to block uncontrolled inflammatory processes (31, 37).
To control the redox balance, cells are equipped with the protective mechanisms consisting of a variety of antioxidants and redox enzymes. Thioredoxin is one of the most important regulators of cellular redox homeostasis (30). With the huge availability of sequence data in public databases, a number of oxidoreductases of the thioredoxin superfamily have been identified. Such oxidoreductases are characterized by thioredoxin-like domains with CXXC active-site motifs that are responsible for catalysis of the thiol–disulfide exchange (5). The large number of these redox modulators suggests a complex mechanism of redox regulation, but their physiological functions in the pathogenesis of inflammatory diseases await further clarification.
Innovation.
In the present study, we examined the physiological significance of redox regulations in an animal model of drug-induced inflammation. We created the mutant mice deficient in the oxidoreductase transmembrane thioredoxin-related protein (TMX), a transmembrane protein in the endoplasmic reticulum. The phenotype of TMX-null mutant mice suggests a potential role for TMX in the host defense against inflammatory conditions associated with oxidative stress. Understanding the physiological function of TMX should have medical implications in the pathogenesis of many acute and chronic inflammatory disorders, and the modulation of the cellular redox status has emerged as a potential therapeutic strategy to block uncontrolled inflammatory processes.
The mammalian endoplasmic reticulum (ER) contains a number of oxidoreductases that are proposed to catalyze the formation and rearrangement of protein disulfides (3). Among them, protein disulfide isomerase (PDI) and ERp57 have been extensively investigated as general folding catalysts in the ER. Although most ER oxidoreductases, including PDI and ERp57, are soluble luminal proteins, some are integral membrane proteins classified as members of the TMX subfamily. To date, four family members (TMX, TMX2, TMX3, and TMX4) have been reported (14, 24, 27, 36, 40). All contain one thioredoxin-like domain, but are otherwise not closely related in regions outside this domain. TMX is the first functionally characterized member of the membrane-bound ER oxidoreductases. It has an N-terminal signal peptide that is cleaved to produce mature proteins of ∼30 kDa (26). Topological studies suggest that TMX is a type I transmembrane protein, and the thioredoxin-like domain containing a CPAC active-site motif is oriented to the luminal side of the ER. Recently, we have shown that TMX interacts with the major histocompatibility complex (MHC) class I heavy chains that are produced under stress conditions, and protects misfolded proteins from proteasomal degradation (25).
The presence of multiple oxidoreductases in the mammalian genome implies that a complex mechanism of redox signaling exists in the ER. Although a considerable body of research has been devoted to this protein family, the in vivo functions of its individual members are still poorly understood. In this study, we have created TMX-null mutant mice by targeted gene disruption. We found that TMX-deficient mice have increased sensitivity to toxic agents, such as lipopolysaccharide (LPS) or thioacetamide (TAA), which cause acute liver failure. Our results suggest a potential role for TMX in controlling the progression of hepatitis. We discuss the significance of our findings in the context of the pathophysiology associated with oxidative stress in mammals.
Results
Generation of TMX gene-deficient mice
The murine TMX gene was inactivated by targeted disruption of the coding sequence. The first two exons containing the translation initiator ATG codon and the region encoding the thioredoxin-like active site were replaced by a neomycin resistance gene (Fig. 1A). Correct gene targeting in embryonic stem cells was verified by Southern blot analysis (Fig. 1B). Chimeric male mice were mated with wild-type females, and the resultant heterozygous TMX mutant mice were intercrossed to generate homozygotes. Polymerase chain reaction (PCR) analysis of genomic DNA was used to identify the mouse genotype (Fig. 1C). In northern blot (Fig. 1D) and reverse transcription–polymerase chain reaction analyses (Fig. 1E) of the TMX−/− mice, the TMX gene transcripts were not detected. The lack of TMX protein in mutant mice was confirmed by immunoblotting using an anti-TMX polyclonal antibody (Fig. 1F). TMX−/− mice were born at the expected frequency and developed normally. Both sexes were fertile and showed no major histological abnormalities.
FIG. 1.

Generation of TMX-deficient mice. (A) Structure of the targeting vector (top), the wild-type allele (middle), and the mutant allele (bottom). Exons are represented by filled boxes. The locations of the PCR primers (arrowheads) used for screening of ES cells and genotyping, and the probes for Southern blotting are indicated. (B) Southern blot analysis of BamHI-digested genomic DNA from wild-type (+/+) and targeted (+/−) ES cells using the 5′ probe. wt, wild-type allele; mt, mutant allele. (C) PCR analysis of genomic DNA from wild-type (+/+), heterozygous (+/−), and homozygous mice (−/−). (D) Northern blot analysis of TMX. Total RNA from liver tissues with the indicated genotypes was electrophoresed and transferred to a membrane. The blot was hybridized with TMX cDNA as the probe. The ethidium bromide-stained RNA blot is shown in the lower panel. (E) RT-PCR analysis. Total RNA extracted from liver tissues was used to examine the expression of TMX, using glyceraldehyde-3-phosphate dehydrogenase as an internal standard. (F) Immunoblot analysis of extracts obtained from whole embryos. Protein samples from wild-type and TMX−/− mice were analyzed by immunoblotting with antibodies against TMX. α-tubulin (TUB) was used as a loading control. ES, embryonic stem; RT-PCR, reverse transcription–polymerase chain reaction; TMX, transmembrane thioredoxin-related protein.
Absence of TMX does not cause severe ER stress
We investigated the expression profiles of several ER-resident proteins functionally related to TMX. Tissue lysates prepared from the liver, kidney, and spleen from wild-type and TMX−/− mice were analyzed by immunoblotting (Fig. 2A and data not shown). The lack of TMX in the TMX−/− mice did not affect the expression of the ER molecular chaperone-binding protein (BiP) and calnexin. Neither did we detect a compensatory upregulation of other oxidoreductases such as PDI and ERp57. These results suggest that TMX deficiency did not alter the expression of genes activated by ER stress, which were further confirmed by microarray analysis as described below. In Figure 2B, we examined the effect of TMX depletion on the expression levels of several disulfide-bonded proteins on the cell surface. The TMX−/− cells showed no defects in cell surface expression of immunoglobulin family members, including MHC class I, T cell receptors, cluster-of-differentiation antigen 4 (CD4), CD8, and CD19 or the integrin family member CD11b, suggesting that TMX is not essential for oxidative protein folding and transport of these molecules. The unchanged expression levels of these cell surface markers confirmed that there were no apparent differences in the immune cell populations between the two genotypes.
FIG. 2.

TMX deficiency does not cause severe ER dysfunction. (A) Liver extracts from wild-type (+/+) and TMX−/− mice were analyzed by immunoblotting with antibodies specific for calnexin (CNX), BiP, TUB, PDI, and ERp57. (B) Surface expression patterns of disulfide-bonded membrane proteins. Spleen cells were stained with the indicated antibodies and analyzed by flow cytometry. Wild-type (WT), shaded; TMX−/−, thick lines; unstained cells (NC), dotted lines. BiP, binding protein; ER, endoplasmic reticulum; PDI, protein disulfide isomerase.
TMX−/− mice exhibit increased susceptibility to liver injury induced by LPS/d-(+)-galactosamine
We investigated the expression pattern and tissue distribution of TMX in wild-type mice. We found that the expression level of the murine TMX gene was especially high in the liver (data not shown), which is consistent with the expression pattern of human TMX (24). The strikingly high expression of TMX observed in liver tissue led us to examine a potential role for this redox enzyme in drug-induced liver damage. We employed an LPS/d-(+)-galactosamine (GalN)-induced liver injury, a well-characterized model of endotoxin shock in mice, for this purpose (1, 11, 18, 32). Administration of LPS in conjunction with GalN is known to sensitize mice to LPS-induced inflammatory cytokines and causes acute liver failure. Mice were injected intraperitoneally with the combination of LPS and GalN, and their serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities were determined as an index for hepatic injury. Even at a low dose of LPS (which does not induce severe liver damage in wild-type mice), the serum levels of these hepatocellular injury markers were substantially elevated in the TMX-deficient mice (Fig. 3A). GalN was required for sensitization to the toxic effects of LPS, and no evidence of liver damage was observed in the mutant mice treated with LPS alone (data not shown). TMX−/− mice had normal liver histology indistinguishable from the wild-type mice (Fig. 3B, C). Upon treatment with LPS/GalN, however, histological evidence of significant liver injury was found in the TMX−/− mice group (compare Fig. 3D, E).
FIG. 3.

Increased sensitivity to LPS/GalN-induced liver damage in TMX−/− mice. (A) Mice were injected intraperitoneally with LPS/GalN and their serum levels of AST and ALT measured as indicated. Data represent mean values±SD of four to seven animals. *p<0.05; **p<0.001 (Student's t-test). (B–E) Liver sections from untreated (B, C) or LPS/GalN-treated mice [24 h post-injection (D, E)] of the wild-type (+/+) and TMX−/− genotypes were stained with hematoxylin and eosin. Bars represent 100 μm. ALT, alanine aminotransferase; AST, aspartate aminotransferase; GalN, d-(+)-galactosamine; LPS, lipopolysaccharide; SD, standard deviation.
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay of the LPS/GalN-treated liver sections revealed increased numbers of apoptotic cells in the mutant mice compared with the wild-type mice (Fig. 4A, B). Hepatocyte apoptosis was evaluated by measuring caspase activation, which is a common feature of apoptotic cell death. Administration of LPS/GalN induced activation of initiator caspase 8 and effector caspase 3 in the TMX−/− mice alone (Fig. 4C). Cleavage of poly(ADP–ribose) polymerase, a target of effector caspases, and upregulation of Bcl2-associated X protein (Bax), a proapoptotic Bcl-2 family member, occurred only in the mutant mice (Fig. 4C, D). The level of FLICE inhibitory protein (c-FLIP) did not change upon LPS/GalN treatment in both groups. From these results, we conclude that the TMX−/− mice are more susceptible to liver injury and apoptosis induced by LPS/GalN.
FIG. 4.

LPS/GalN-induced liver apoptosis in TMX−/− mice. (A, B) Apoptotic cells were detected by TUNEL assays performed on liver sections prepared from LPS/GalN-treated wild-type (+/+) and TMX−/− mice. Bars 100 μm. (C, D) Liver extracts were prepared from mice 24 h post-LPS/GalN administration and analyzed by immunoblotting with the indicated antibodies. The full-length (asterisk) and the cleaved fragments of PARP (arrowhead) are indicated (C). The relative expression levels of Bax and FLIP were quantified and normalized to TUB (D). Bax, Bcl2-associated X protein; FLIP, FLICE inhibitory protein; PARP, poly(ADP–ribose) polymerase; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling.
Tumor necrosis factor-α is a critical effector of LPS/GalN-induced hepatotoxicity observed in TMX−/− mice
LPS induces production of inflammatory mediators that are believed to be the major effectors of hepatotoxicity. One hour after administering the injection, serum levels of interleukin-6 (IL-6), IL-10, monocyte chemotactic protein-1 (MCP-1), and tumor necrosis factor-α (TNF-α) increased in response to LPS. Unexpectedly, no statistically significant differences were observed between wild-type and TMX−/− mice (Fig. 5A). IL-6, IL-10, and TNF-α levels rapidly decreased, returning to normal within 6 h postchallenge with LPS/GalN in both the wild-type and TMX−/− mice (Fig. 5B). MCP-1 levels peaked at 3 h and were still elevated 6 h post-treatment. No detectable IL-12p70 and interferon-γ (IFN-γ) were observed throughout the study period. Similar results were obtained from the in vitro studies using primary macrophages, and LPS-induced TNF-α production was equivalent in both wild-type and TMX-deficient cells (Fig. 5C). In addition, there were no significant differences observed between the two genotypes in their surface expression levels of Toll-like receptor 4, which mediates LPS-induced immune responses (2) (Fig. 5D). Taken together, these results indicate that loss of TMX did not substantially affect the magnitude and duration of the early inflammatory response induced by LPS/GalN.
FIG. 5.

Cytokine production in LPS/GalN-treated mice. (A) Serum concentrations of IL-6, IL-10, MCP-1, and TNF in wild-type (+/+) and TMX−/− mice were determined 1 h after LPS/GalN injections. Data represent the mean values±SD of five animals for each group. None of the cytokines were detectable in untreated mice. (B) After LPS/GalN treatment, blood samples were collected periodically and serum cytokine levels measured. Data are expressed as the mean values±SD of three animals. (C) TNF concentrations in the culture supernatants of peritoneal macrophages stimulated with LPS (10 ng/ml). Data represent the mean values±SD of two independent experiments. (D) Surface expression of TLR4 in the phagocyte fraction of liver cells. Nonparenchymal cells prepared from the livers of wild-type and TMX−/− mice were stained with allophycocyanin-anti-CD11b and phycoerythrin-anti-TLR4. The expression of TLR4 was quantified by flow cytometry on the CD11b-positive phagocyte fraction. IL, interleukin; MCP-1, monocyte chemotactic protein-1; TLR4, Toll-like receptor 4; TNF, tumor necrosis factor.
TNF-α has been implicated as a critical early mediator of acute liver injury induced by endotoxin (6, 29). To test whether the increased liver damage in TMX−/− mice was mediated by TNF-α-induced toxicity, we examined the protective effect of neutralizing antibodies against TNF-α in the LPS/GalN model of liver injury. As shown in Figure 6A, LPS/GalN-induced liver damage in TMX−/− mice was almost completely suppressed by pretreatment with anti-TNF-α antibody. The high degree of protection afforded by the anti-TNF-α antibody clearly indicates that the liver damage observed was mainly mediated by the toxic effects of secreted TNF-α in response to LPS. Since the expression levels of TNF receptor 1 (TNF-R1), which has been shown to mediate the majority of TNF-α activities (7, 22, 42), were equivalent both in wild-type and TMX−/− mice (Fig. 6B, C), the increased liver damage observed in the mutants may be due to defects in the hepatocellular response against TNF-α toxicity, most likely in the signaling pathway downstream of the TNF receptor.
FIG. 6.

TNF-α is a critical effector of hepatotoxicity induced by LPS/GalN in TMX−/− mice. (A) Inhibition of TNF-α protects the liver from LPS/GalN-induced damage. Mice were intraperitoneally injected with saline (−) or anti-TNF-α-neutralizing antibodies (+) 6 h before LPS/GalN administration. Serum levels of AST and ALT were measured 24 h after the LPS/GalN challenge. Data represent the mean values±SD of two to three animals. (B) Immunoblots of liver extracts for TNF-R1 and TUB. The protein expression of TNF-R1 was quantified and normalized to TUB. (C) Serum levels of soluble TNF-R1 (sTNF-R1) were determined by enzyme-linked immunosorbent assay before and 1 h after injection of LPS/GalN. sTNF-R1 generated by the proteolytic cleavage of the molecule on the cell surface has been shown to bind TNF-α and regulate circulating TNF-α activity. Concentrations of sTNF-R1 were measured in duplicate, and data represent the mean values±SD of six animals.
Upregulation of p53 target genes in LPS/GalN-treated TMX−/− mice
TNF-α treatment results in the activation of multiple responses involving an apoptotic pathway and a survival pathway mediated by nuclear factor-κB (NF-κB). We examined the TNF-α-mediated activation of NF-κB and c-Jun N-terminal kinase (JNK), both of which play crucial roles in TNF-α-induced survival and the apoptotic pathway, respectively (4, 43, 44). TNF-α induced the phosphorylation and subsequent degradation of inhibitor of κB-α in a similar way in the wild-type and TMX−/− fibroblasts; transient activation of JNK was also not affected by TMX deficiency (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/ars). These results suggest that the TMX−/− cells were not severely impaired in activating the key components of the TNF signaling pathway.
To identify the signaling pathway that may be associated with LPS/GalN-induced liver failure, transcriptional profiles were compared using a microarray dataset from untreated or LPS/GalN-treated wild-type and TMX−/− mice. Proinflammatory mediators, including S100 proteins (12, 41), annexins (28, 34), and C–C motif chemokines such as MCP-1/Ccl2, were upregulated in the TMX−/− group, but not in the wild type (Fig. 7A and data not shown). Upregulation of these previously identified markers for inflammation-associated disorders validated the experimental results. Endotoxin and TNF-α have been shown to induce ER stress in a reactive oxygen species (ROS)-dependent manner (19, 45). In our study, LPS/GalN treatment induced a slight upregulation of at least two known ER stress markers, C/EBP homologous protein (CHOP) and BiP, to a similar extent in both genotypes (data not shown).
FIG. 7.
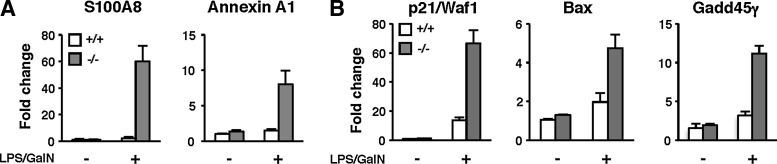
Upregulation of p53-target genes in LPS/GalN-treated TMX−/− mice. (A, B) Induction of S100A8, annexin A1, p21/Waf1, Bax, and Gadd45γ was verified by real-time quantitative PCR performed in triplicate. mRNA expression levels were normalized to that of 18S ribosomal RNA. The graphs show the fold changes for each gene compared with the untreated wild-type mice. Data are the mean values±SD of two animals.
Next, we focused on genes whose expression level increased more than twofold after LPS/GalN treatment. We compared the fold changes of the expression of such genes in both genotypes, and extracted 28 genes exhibiting a higher expression level (at least twofold) in TMX−/− mice than in wild type (Supplementary Table S1). To exclude false positives, some of the differentially expressed genes were verified by quantitative real-time PCR. Intriguingly, LPS/GalN treatment induced the expression of several p53 target genes, including Bax, p21, and members of growth arrest and the DNA damage-inducible gene family (Gadd45β and Gadd45γ); induction of these p53 targets by LPS/GalN was considerably increased in the TMX−/− mice (Fig. 7B and data not shown). Given that the activation of p53 and its downstream pathway is implicated in cell cycle arrest and cell death, it is suggested that enhanced expression of such target genes contribute to the type of inflammatory tissue injury observed in TMX−/− mice.
TMX protects against oxidative stress-mediated cellular damage
Taking these findings together, we hypothesized that the change in the redox environment potentially caused by TMX deficiency might render hepatocytes more sensitive to the inflammatory conditions associated with oxidative stress, which could contribute to an increased severity of liver failure. Oxidative damage in the liver was assessed by measuring the formation of protein adducts with the lipid peroxidation product 4-hydroxy-2-nonenal (HNE). The immunohistochemical staining of liver sections shows that administration of LPS/GalN resulted in a marked accumulation of HNE-modified proteins in TMX−/− mice compared with wild-type animals (Fig. 8C, D). The extensive staining observed in LPS/GalN-treated TMX−/− mice clearly suggests increased oxidative stress leading to a severe damage in the liver tissues.
FIG. 8.

Loss of TMX enhances liver damage associated with oxidative stress. (A–D) Increased lipid peroxidation in LPS/GalN-treated TMX−/− mice. Representative images of HNE staining in liver sections from wild-type (+/+) or TMX−/− mice treated with vehicle (A, B) or LPS/GalN (24 h postinjection (C, D). Bars represent 100 μm. (E) Loss of TMX enhances liver damage induced by TAA. Serum levels of AST and ALT were measured 24 h after TAA injection. Data represent the mean values±SD of four animals. *p<0.05 (Student's t-test). HNE, 4-hydroxy-2-nonenal; TAA, thioacetamide.
To further explore the protective role for TMX in oxidative stress-mediated cellular damage, we examined the effect of TMX deficiency in a second animal model of liver injury using TAA (17). TAA is a thionosulfur-containing compound, and intraperitoneal injection with it causes acute liver failure in mice; ROS involvement in this process has been postulated (8, 20, 35). Twenty-four hours after TAA injection, AST and ALT levels were significantly higher in the TMX−/− mice than in the wild-type animals (Fig. 8E). Thus, as shown in the LPS/GalN model of liver injury, TMX−/− mice exhibited a significantly higher sensitivity to TAA.
We have searched for proteins that potentially interact with TMX to gain more information of its protective roles in the inflammatory response. Using TMX-conjugated affinity microparticles, proteins were purified from mouse liver membrane fractions and identified by mass spectrometry (Supplementary Fig. S2; Supplementary Table S2). The ER-associated proteins identified include a member of the long-chain acyl-CoA synthetase family, a fatty acid transport protein, and Derlin-1, a component of the ER-associated degradation machinery (21). Additional studies should evaluate the possibility that TMX contributes to the regulation of liver homeostasis and stress response through cooperative interactions with such membrane proteins.
Discussion
In this article, we found a hypersensitivity of TMX-deficient mice to LPS/GalN, resulting in marked aggravation of oxidative stress-induced hepatitis. LPS-induced liver injury is a classical model of endotoxin shock. The tissue damage is mediated mainly through the cytotoxic effect of TNF-α (10), which can induce a variety of mechanisms to initiate cell death, and the signaling events activated seem to be closely interlinked. Although the downstream signaling pathways have been examined rather intensively, the mechanism of aggressive and fatal hepatitis has not been clarified completely neither in an animal model nor clinically.
Accumulating evidence shows that oxidative stress is associated with inflammation. During liver injury, hepatocytes are exposed to ROS generated from inflammatory cells. ROS are also produced intracellularly upon activation of inflammatory responses and play a critical role in a TNF-α-induced cell death pathway (33, 39). Among a variety of mechanisms and cellular responses against inflammation, we have attributed the increased liver damage in TMX−/− mice to elevated oxidative stress, which was clearly demonstrated by a marked accumulation of HNE–protein adducts in the mutant mice. The production of ROS and the resulting oxidative protein modifications can impose diverse effects of protein function and promote cell death (9). In addition, several antioxidant genes were downregulated in TMX−/− mice treated with LPS/GalN (Supplementary Table S3). Microarray data also revealed enhanced activation of the p53 signaling in TMX−/− mice after an LPS/GalN challenge. Excess ROS can cause oxidative damage to cellular macromolecules such as DNA, and induce genotoxic effects leading to activation of the p53-dependent cell death pathway. Although TMX localized on the ER membrane would not possibly regulate the p53 signaling via direct interaction, we consider it one of the important pathways contributing to the oxidative liver damage. In agreement with this study, a recent microarray analysis indicates that transcriptional regulation by p53 is involved in hepatotoxicant-induced liver injury (16). Furthermore, Schafer et al. showed that inhibition of p53 attenuated hepatocellular death induced by LPS (38). Although the precise mechanisms by which ROS exert their toxic effects in inflammatory liver injury are not fully understood, our work, together with the previous studies, shows the crucial involvement of oxidative stress-induced cell death in inflammatory liver damage.
Expression profiles in the liver showed that TMX deficiency in mice did not significantly induce the unfolded protein response, suggesting that the mutant mice are not exposed to persistent ER stress at a steady state. Furthermore, after an LPS/GalN challenge, only a slight upregulation of ER stress markers, such as CHOP and BiP, was observed to a similar extent in wild-type and TMX−/− mice. These observations indicate that the lack of TMX does not cause obvious phenotypes of ER dysfunction, and ER stress would not be a major contributing factor to the liver injury in our LPS/GalN model. Elevated oxidative stress in TMX−/− mice receiving LPS/GalN suggests that the mutant mice may fail to control the intracellular redox balance under inflammatory conditions. Mitochondria are the primary source of ROS, and their dysfunction has been implicated in oxidative stress-mediated liver injury (10). It has been reported very recently that a group of ER proteins, including TMX, are enriched on the mitochondrion-associated membrane (MAM), a subdomain of the ER forming close contacts with mitochondria (15, 23). Although the physiological significance of ER–mitochondria interactions in inflammatory liver injury has not been elucidated thus far, TMX could potentially participate in the cellular protection from oxidative damage through the targeting to MAM.
Our results also show that the absence of TMX augments the cellular response against oxidative stress evoked not only by LPS/GalN but also by TAA. Collectively, these results may suggest that TMX might have broader roles in the host defense against inflammatory conditions associated with oxidative stress such as viral infection, ischemia reperfusion damage, and even cancer. Considering that the cellular redox status might determine the sensitivity and the final outcome in response to inflammatory stimuli, understanding the protective role of TMX should have medical implications in the pathogenesis of such acute and chronic inflammatory diseases.
The present study is the first to address the role of the oxidoreductase TMX in inflammatory liver injury. The phenotype of mice deficient in TMX suggests a functional link between redox regulation in the ER and susceptibility to oxidative stress-mediated tissue damage. This animal model might be useful for targeting therapeutic agents for the treatment of hepatitis progression, and also for dissecting the role of TMX in the redox-signaling network connecting the ER and the extra-ER environment.
Materials and Methods
Reagents
LPS from Escherichia coli 0111:B4 (L4391) and GalN hydrochloride (G1639) were purchased from Sigma and TAA from Wako.
Generation of TMX-deficient mice
The TMX-mutant mice (Acc. No. CDB0445K: http://cdb.riken.jp/arg/mutant%20mice%20list.html) were generated as described (http://cdb.riken.jp/arg/Methods.html; for a detailed description, please see Supplementary Materials and Methods). TMX-mutant mice were backcrossed for more than 10 generations to the C57BL/6J background, and maintained in a specific pathogen-free facility. All experiments were conducted according to the Institutional guidelines and regulations.
Protein extraction and immunoblotting
Tissue samples were homogenized in a lysis buffer containing 50 mM Tris–HCl, pH 7, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.5% sodium dodecyl sulfate. Lysates were clarified by centrifugation at 20,000 g for 10 min, and immunodetection was carried out as described previously (25). The antibodies used are listed in the Supplementary Materials and Methods.
Flow cytometry
Spleen cell suspensions were obtained by passing the tissues through a 40-μm nylon cell strainer (BD Falcon). After removal of erythrocytes using a red blood cell lyzing buffer (Sigma), cells were stained with the fluorescence-labeled antibodies for 20 min at 4°C, and analyzed using FACSCalibur (Becton Dickinson) and FlowJo software (Tree Star).
Liver injury model
Female mice at 11–15-week old (20–23 g) were used. Mice were injected intraperitoneally with a combination of 3 ng LPS and 10 mg GalN in a total volume of 0.2 ml per animal. Blood samples were obtained before and after LPS/GalN challenge for serum analysis. Liver samples were collected 24 h postinjection. In the TAA-induced liver injury model, mice were given a 75 mg/kg body weight intraperitoneal injection of 10 mg/ml TAA solution.
Histological analysis
Liver specimens fixed in 10% neutral formalin were embedded in paraffin, sectioned, and stained with hematoxylin and eosin. Apoptotic cells were detected via TUNEL assays performed on paraffin sections using the MEBSTAIN apoptosis kit II (MBL). Lipid peroxidation was assessed using mouse monoclonal antibodies specific for HNE-modified protein (NOF Corporation) and Histofine Mousestain kit (Nichirei Biosciences).
Determination of cytokine concentrations
The concentrations of selected cytokines (i.e., IL-6, IL-10, MCP-1, IFN-γ, TNF-α, and IL-12p70) were determined using a cytometric bead array mouse inflammation kit (BD Biosciences) following the manufacturer's protocol.
Enzyme-linked immunosorbent assay
Release of soluble TNF-R1 (sTNF-R1) was measured by enzyme-linked immunosorbent assay using the Quantikine mouse sTNF-R1 immunoassay kit (R&D Systems).
Quantitative real-time PCR
For cDNA synthesis, 1 μg of total RNA was reverse transcribed using High-Capacity RNA-to-cDNA Master Mix (Applied Biosystems). Gene expression was quantitated using the Power SYBR Green PCR Master Mix and ABI PRISM 7000 Sequence Detection System (Applied Biosystems). The sequences of the primers used are provided in the Supplementary Materials and Methods (Supplementary Table S4).
Supplementary Material
Abbreviations Used
- ALT
alanine aminotransferase
- APC
allophycocyanin
- AST
aspartate aminotransferase
- Bax
Bcl2-associated X protein
- BiP
binding protein
- CHOP
C/EBP homologous protein
- ER
endoplasmic reticulum
- ES
embryonic stem
- FITC
fluorescein isothiocyanate
- FLIP
FLICE inhibitory protein
- GalN
d-(+)-galactosamine
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HNE
4-hydroxy-2-nonenal
- IFN-γ
interferon-γ
- IL
interleukin
- JNK
c-Jun N-terminal kinase
- LPS
lipopolysaccharide
- MAM
mitochondrion-associated membrane
- MCP-1
monocyte chemotactic protein-1
- MHC
major histocompatibility complex
- NF-κB
nuclear factor-κB
- PARP
poly(ADP–ribose) polymerase
- PDI
protein disulfide isomerase
- PE
phycoerythrin
- ROS
reactive oxygen species
- RT-PCR
reverse transcription–polymerase chain reaction
- SD
standard deviation
- sTNF-R1
soluble TNF-R1
- TAA
thioacetamide
- TLR4
Toll-like receptor 4
- TMX
transmembrane thioredoxin-related protein
- TNF-α
tumor necrosis factor-α
- TNF-R1
TNF receptor 1
- TTF
tail-tip fibroblasts
- TUB
α-tubulin
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
Acknowledgments
We thank Akie Teratani, Suzuyo Furukawa, Masahiro Takenaka, Yoshimi Yamaguchi, and Ryoko Otsuki for technical assistance, and Takamasa Nomura, Osamu Hori, Satoshi Ogawa, and Shinya Toyokuni for invaluable technical advice and discussion. This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and in part by the Program for Promotion of Fundamental Studies in Health Sciences of National Institute of Biomedical Innovation, a Grant-in-Aid for Scientific Research on Innovative Areas (Research in a proposed research area) Molecular Basis and Disorders of Control of Appetite and Fat Accumulation, and the World Class University Grant R31-10010 through the Ewha Womans University. YM was supported by a Research Fellowship of the Japan Society for the Promotion of Science for Young Scientists.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Akashi-Takamura S. Furuta T. Takahashi K. Tanimura N. Kusumoto Y. Kobayashi T. Saitoh S. Adachi Y. Doi T. Miyake K. Agonistic antibody to TLR4/MD-2 protects mice from acute lethal hepatitis induced by TNF-alpha. J Immunol. 2006;176:4244–4251. doi: 10.4049/jimmunol.176.7.4244. [DOI] [PubMed] [Google Scholar]
- 2.Akira S. Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 3.Appenzeller-Herzog C. Ellgaard L. The human PDI family: versatility packed into a single fold. Biochim Biophys Acta. 2008;1783:535–548. doi: 10.1016/j.bbamcr.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 4.Beg AA. Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 5.Berndt C. Lillig CH. Holmgren A. Thioredoxins and glutaredoxins as facilitators of protein folding. Biochim Biophys Acta. 2008;1783:641–650. doi: 10.1016/j.bbamcr.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 6.Beutler B. Rietschel ET. Innate immune sensing and its roots: the story of endotoxin. Nat Rev Immunol. 2003;3:169–176. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- 7.Chen G. Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296:1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- 8.Chieli E. Malvaldi G. Role of the microsomal FAD-containing monooxygenase in the liver toxicity of thioacetamide S-oxide. Toxicology. 1984;31:41–52. doi: 10.1016/0300-483x(84)90154-9. [DOI] [PubMed] [Google Scholar]
- 9.Circu ML. Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48:749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding WX. Yin XM. Dissection of the multiple mechanisms of TNF-alpha-induced apoptosis in liver injury. J Cell Mol Med. 2004;8:445–454. doi: 10.1111/j.1582-4934.2004.tb00469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galanos C. Freudenberg MA. Reutter W. Galactosamine-induced sensitization to the lethal effects of endotoxin. Proc Natl Acad Sci U S A. 1979;76:5939–5943. doi: 10.1073/pnas.76.11.5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gebhardt C. Nemeth J. Angel P. Hess J. S100A8 and S100A9 in inflammation and cancer. Biochem Pharmacol. 2006;72:1622–1631. doi: 10.1016/j.bcp.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 13.Grivennikov SI. Greten FR. Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haugstetter J. Blicher T. Ellgaard L. Identification and characterization of a novel thioredoxin-related transmembrane protein of the endoplasmic reticulum. J Biol Chem. 2005;280:8371–8380. doi: 10.1074/jbc.M413924200. [DOI] [PubMed] [Google Scholar]
- 15.Hayashi T. Rizzuto R. Hajnoczky G. Su TP. MAM: more than just a housekeeper. Trends Cell Biol. 2009;19:81–88. doi: 10.1016/j.tcb.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang L. Heinloth AN. Zeng ZB. Paules RS. Bushel PR. Genes related to apoptosis predict necrosis of the liver as a phenotype observed in rats exposed to a compendium of hepatotoxicants. BMC Genomics. 2008;9:288. doi: 10.1186/1471-2164-9-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hunter AL. Holscher MA. Neal RA. Thioacetamide-induced hepatic necrosis. I. Involvement of the mixed-function oxidase enzyme system. J Pharmacol Exp Ther. 1977;200:439–448. [PubMed] [Google Scholar]
- 18.Kaufmann T. Jost PJ. Pellegrini M. Puthalakath H. Gugasyan R. Gerondakis S. Cretney E. Smyth MJ. Silke J. Hakem R. Bouillet P. Mak TW. Dixit VM. Strasser A. Fatal hepatitis mediated by tumor necrosis factor TNFalpha requires caspase-8 and involves the BH3-only proteins Bid and Bim. Immunity. 2009;30:56–66. doi: 10.1016/j.immuni.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozlov AV. Duvigneau JC. Miller I. Nurnberger S. Gesslbauer B. Kungl A. Ohlinger W. Hartl RT. Gille L. Staniek K. Gregor W. Haindl S. Redl H. Endotoxin causes functional endoplasmic reticulum failure, possibly mediated by mitochondria. Biochim Biophys Acta. 2009;1792:521–530. doi: 10.1016/j.bbadis.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 20.Lee JW. Shin KD. Lee M. Kim EJ. Han SS. Han MY. Ha H. Jeong TC. Koh WS. Role of metabolism by flavin-containing monooxygenase in thioacetamide-induced immunosuppression. Toxicol Lett. 2003;136:163–172. doi: 10.1016/s0378-4274(02)00333-8. [DOI] [PubMed] [Google Scholar]
- 21.Lilley BN. Ploegh HL. A membrane protein required for dislocation of misfolded proteins from the ER. Nature. 2004;429:834–840. doi: 10.1038/nature02592. [DOI] [PubMed] [Google Scholar]
- 22.Lotz M. Setareh M. von Kempis J. Schwarz H. The nerve growth factor/tumor necrosis factor receptor family. J Leukoc Biol. 1996;60:1–7. doi: 10.1002/jlb.60.1.1. [DOI] [PubMed] [Google Scholar]
- 23.Lynes EM. Bui M. Yap MC. Benson MD. Schneider B. Ellgaard L. Berthiaume LG. Simmen T. Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. EMBO J. 2012;31:457–470. doi: 10.1038/emboj.2011.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsuo Y. Akiyama N. Nakamura H. Yodoi J. Noda M. Kizaka-Kondoh S. Identification of a novel thioredoxin-related transmembrane protein. J Biol Chem. 2001;276:10032–10038. doi: 10.1074/jbc.M011037200. [DOI] [PubMed] [Google Scholar]
- 25.Matsuo Y. Masutani H. Son A. Kizaka-Kondoh S. Yodoi J. Physical and functional interaction of transmembrane thioredoxin-related protein with major histocompatibility complex class I heavy chain: redox-based protein quality control and its potential relevance to immune responses. Mol Biol Cell. 2009;20:4552–4562. doi: 10.1091/mbc.E09-05-0439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsuo Y. Nishinaka Y. Suzuki S. Kojima M. Kizaka-Kondoh S. Kondo N. Son A. Sakakura-Nishiyama J. Yamaguchi Y. Masutani H. Ishii Y. Yodoi J. TMX, a human transmembrane oxidoreductase of the thioredoxin family: the possible role in disulfide-linked protein folding in the endoplasmic reticulum. Arch Biochem Biophys. 2004;423:81–87. doi: 10.1016/j.abb.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 27.Meng X. Zhang C. Chen J. Peng S. Cao Y. Ying K. Xie Y. Mao Y. Cloning and identification of a novel cDNA coding thioredoxin-related transmembrane protein 2. Biochem Genet. 2003;41:99–106. doi: 10.1023/a:1022073917044. [DOI] [PubMed] [Google Scholar]
- 28.Monastyrskaya K. Babiychuk EB. Draeger A. The annexins: spatial and temporal coordination of signaling events during cellular stress. Cell Mol Life Sci. 2009;66:2623–2642. doi: 10.1007/s00018-009-0027-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morikawa A. Sugiyama T. Kato Y. Koide N. Jiang GZ. Takahashi K. Tamada Y. Yokochi T. Apoptotic cell death in the response of D-galactosamine-sensitized mice to lipopolysaccharide as an experimental endotoxic shock model. Infect Immun. 1996;64:734–738. doi: 10.1128/iai.64.3.734-738.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakamura H. Thioredoxin and its related molecules: update 2005. Antioxid Redox Signal. 2005;7:823–828. doi: 10.1089/ars.2005.7.823. [DOI] [PubMed] [Google Scholar]
- 31.Nakamura H. Hoshino Y. Okuyama H. Matsuo Y. Yodoi J. Thioredoxin 1 delivery as new therapeutics. Adv Drug Deliv Rev. 2009;61:303–309. doi: 10.1016/j.addr.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Okuyama H. Nakamura H. Shimahara Y. Araya S. Kawada N. Yamaoka Y. Yodoi J. Overexpression of thioredoxin prevents acute hepatitis caused by thioacetamide or lipopolysaccharide in mice. Hepatology. 2003;37:1015–1025. doi: 10.1053/jhep.2003.50203. [DOI] [PubMed] [Google Scholar]
- 33.Osawa Y. Nagaki M. Banno Y. Yamada Y. Imose M. Nozawa Y. Moriwaki H. Nakashima S. Possible involvement of reactive oxygen species in D-galactosamine-induced sensitization against tumor necrosis factor-alpha-induced hepatocyte apoptosis. J Cell Physiol. 2001;187:374–385. doi: 10.1002/jcp.1088. [DOI] [PubMed] [Google Scholar]
- 34.Parente L. Solito E. Annexin 1: more than an anti-phospholipase protein. Inflamm Res. 2004;53:125–132. doi: 10.1007/s00011-003-1235-z. [DOI] [PubMed] [Google Scholar]
- 35.Porter WR. Neal RA. Metabolism of thioacetamide and thioacetamide S-oxide by rat liver microsomes. Drug Metab Dispos. 1978;6:379–388. [PubMed] [Google Scholar]
- 36.Roth D. Lynes E. Riemer J. Hansen HG. Althaus N. Simmen T. Ellgaard L. A di-arginine motif contributes to the ER localization of the type I transmembrane ER oxidoreductase TMX4. Biochem J. 2009;425:195–205. doi: 10.1042/BJ20091064. [DOI] [PubMed] [Google Scholar]
- 37.Rubartelli A. Sitia R. Stress as an intercellular signal: the emergence of stress-associated molecular patterns (SAMP) Antioxid Redox Signal. 2009;11:2621–2629. doi: 10.1089/ars.2009.2377. [DOI] [PubMed] [Google Scholar]
- 38.Schafer T. Scheuer C. Roemer K. Menger MD. Vollmar B. Inhibition of p53 protects liver tissue against endotoxin-induced apoptotic and necrotic cell death. FASEB J. 2003;17:660–667. doi: 10.1096/fj.02-0774com. [DOI] [PubMed] [Google Scholar]
- 39.Shen HM. Pervaiz S. TNF receptor superfamily-induced cell death: redox-dependent execution. FASEB J. 2006;20:1589–1598. doi: 10.1096/fj.05-5603rev. [DOI] [PubMed] [Google Scholar]
- 40.Sugiura Y. Araki K. Iemura S. Natsume T. Hoseki J. Nagata K. Novel thioredoxin-related transmembrane protein TMX4 has reductase activity. J Biol Chem. 2010;285:7135–7142. doi: 10.1074/jbc.M109.082545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vogl T. Tenbrock K. Ludwig S. Leukert N. Ehrhardt C. van Zoelen MA. Nacken W. Foell D. van der Poll T. Sorg C. Roth J. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13:1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 42.Wajant H. Pfizenmaier K. Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y. Singh R. Lefkowitch JH. Rigoli RM. Czaja MJ. Tumor necrosis factor-induced toxic liver injury results from JNK2-dependent activation of caspase-8 and the mitochondrial death pathway. J Biol Chem. 2006;281:15258–15267. doi: 10.1074/jbc.M512953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wullaert A. Heyninck K. Beyaert R. Mechanisms of crosstalk between TNF-induced NF-kappaB and JNK activation in hepatocytes. Biochem Pharmacol. 2006;72:1090–1101. doi: 10.1016/j.bcp.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 45.Xue X. Piao JH. Nakajima A. Sakon-Komazawa S. Kojima Y. Mori K. Yagita H. Okumura K. Harding H. Nakano H. Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J Biol Chem. 2005;280:33917–33925. doi: 10.1074/jbc.M505818200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.