

Abstract
The evolution of a species involves changes in its genome and its transcriptome. Divergence in expression patterns may be more important than divergence in sequences for determining phenotypic changes, particularly among closely related species. We examined the relationships between organ evolution, sequence evolution, and expression evolution in Arabidopsis thaliana, rice (Oryza sativa), and maize (Zea mays). We found correlated divergence of gene sequences and expression patterns, with distinct divergence rates that depend on the organ types in which a gene is expressed. For instance, genes specifically expressed in reproductive organs (i.e., stamen) evolve more quickly than those specifically expressed in vegetative organs (e.g., root). The different rates in organ evolution may be due to different degrees of functional constraint associated with the different physiological functions of plant organs. Additionally, the evolutionary rate of a gene sequence is correlated with the breadth of its expression in terms of the number of tissues, the number of coregulation modules, and the number of species in which the gene is expressed, as well as the number of genes with which it may interact. This linkage supports the hypothesis that constitutively expressed genes may experience higher levels of functional constraint accumulated from multiple tissues than do tissue-specific genes.
INTRODUCTION
The idea that species evolution occurs at two levels, DNA sequence variation and changes in gene expression, was recognized as early as 1975 by King and Wilson (King and Wilson, 1975). Phenotypic divergence among closely related species with conserved coding sequences is primarily governed by changes in regulatory systems that generate variation in the magnitude, timing, and spacing of gene expression (Wittkopp and Kalay, 2012). Compared with the well-established theories of sequence evolution, the evolution of expression patterns and the methods needed to study this process have received little attention. Large-scale transcriptome profiling allows for interspecific comparison across homologous tissues to understand expression evolution in conjunction with sequence evolution. These two processes jointly shape the phenotypic diversity of the animal and plant kingdoms.
In molecular evolution, the neutral model serves as a null hypothesis for evaluating whether functional sequence variation is due to natural selection or genetic drift (Kimura, 1983). An equivalent null model is needed for the analysis of the evolution of expression patterns of orthologs across species. In their study of intraspecific and interspecific gene expression variation between humans and four nonhuman primates, Khaitovich et al. (2004) proposed that the neutral theory is also applicable to expression evolution (Khaitovich et al., 2004). They found that the levels of divergence in expression between species accumulated linearly with species divergence times, suggesting that the majority of variation in gene expression may be functionally neutral and may contribute little to phenotypic divergence. Thus, this neutral model of expression evolution provides the first case of a theoretical framework for identifying genes with accelerated expression divergence resulting from positive selection (Khaitovich et al., 2005a).
Whether sequence evolution and expression evolution occur in parallel or independently is also of interest in this context. In yeast species of the Saccharomyces sensu stricto complex and Caenorhabditis elegans, studies of orthologs and gene duplicates showed no correlation between sequence divergence and expression divergence, suggesting that sequence and expression evolution may occur independently (Wagner, 2000; Tirosh and Barkai, 2008). Conversely, interspecific studies among mammals and Drosophila melanogaster supported the coevolution model with the expression divergence rates of orthologs being positively correlated with sequence divergence rates (Khaitovich et al., 2005b; Good et al., 2006). To determine the evolutionary force responsible for correlated sequence and expression divergence, Good et al. (2006) compared the evolutionary rates (nonsynonymous substitution rate [Ka]/synonymous substitution rate [Ks]) of genes with differential expression patterns between D. melanogaster and Drosophila simulans with those of genes with similar expression. The proportions of positively selected genes were found to be similar between the two groups (Good et al., 2006). This result suggests that the correlated divergence of gene sequence and expression, if driven by the same evolutionary force, is unlikely due to positive selection; instead, the relaxation of functional constraint (i.e., decreased negative selection) may play a central role in the positive correlation between expression and sequence divergence.
The breadth of a gene’s expression across tissues can be quantitatively defined as “tissue specificity.” An ortholog’s tissue specificity may vary from species to species. This type of interspecific expression divergence may be more important than sequence variation in explaining phenotypic differences between species, since sequence changes at the protein level may lead to functional changes in all of the cells in an organism, whereas expression changes may only affect specific types of cells in which a gene is expressed (Wittkopp and Kalay, 2012). This principle is especially true among closely related species of the same family or genus. The developmental relationships of tissues can be illustrated as a tree structure (tissue tree), constructed based upon the similarity in expression patterns of genes. A tree constructed from the expression profiles of multiple species may reflect the evolutionary relationships of those species (Brawand et al., 2011). Interesting questions may be posed that center on the information delivered by an interspecific tissue tree. First, is the evolutionary relationship of species inferred from expression divergence of tissues consistent with that deduced from sequence divergence? Second, how can one infer the relative rates of tissue and/or organ evolution? Third, can one mathematically model the expression evolution of a gene and infer the evolutionary force driving the expression change, such as due to decreased negative selection or positive selection? These questions have been addressed in humans and chimpanzees using expression data from five homologous organs (i.e., brain, heart, liver, kidney, and testis). By comparing the evolutionary rates of housekeeping genes with those of tissue-specific genes, Khaitovich et al. (2004) found that the latter group evolves more quickly than the former group. Hence, they hypothesized that broadly expressed genes might be subject to higher levels of functional constraint, which accumulated from multiple tissues, than tissue-specific genes that face weaker functional constraint from single or fewer tissues. This proposition was formulated as a “tissue-driven hypothesis” to explain the correlated evolution of sequence and expression (Gu and Su, 2007). In particular, different degrees of functional constraint associated with tissue types affect the distinct rates of sequence and expression divergence. For instance, genes specific to the testis evolve at the highest rates in the body, whereas genes expressed in the brain evolve at the lowest rates (Khaitovich et al., 2006; Gu and Su, 2007; Brawand et al., 2011).
Most knowledge of expression evolution has been obtained from studies of animal species. Whether the evolution of plant transcriptomes resembles that of animal transcriptomes has not been explored. With the availability of expression data in plants, we are able to investigate the evolution of plant gene expression. In this study, we analyzed the relationships between sequence evolution and expression evolution in plants with expression data from Arabidopsis thaliana, rice (Oryza sativa), and maize (Zea mays). We developed a mathematical quantity, “tissue specificity,” that may be used to evaluate the relative contribution of a gene’s expression to tissue-specific phenotypes. Based on this metric, tissue-specific genes were selected to infer the relationships between organs, sequences, and expression patterns. Looking across organs, we found a significant positive correlation of gene sequence and expression pattern evolution. Based on the gene coregulation modules inferred from the interspecific expression data, we derived another expression-based quantity to characterize the functional constraint acting on a gene. This model incorporates several factors, including the breadth of tissue expression, the number of modules in which a gene participates, the number of other genes with which a gene is potentially coregulated, and the conservation of interspecific expression. Finally, we validated that this expression-based functional constraint (eFC), which may serve as an estimate of the selection pressure on a gene and the evolutionary potential of the gene to generate new functions and phenotypic characters, is negatively correlated with the gene’s rate of sequence evolution.
RESULTS
Compilation of Interspecific Expression Data
Our analysis focused on the transcriptomes of Arabidopsis thaliana, rice (Oryza sativa), and maize (Zea mays). whose genomes are well annotated and whose expression data sets cover most tissues and developmental stages with high quality. The data set was compiled from 63 samples of Arabidopsis, 75 samples of rice, and 60 samples of maize, profiled at different developmental stages in a total of 46 tissue types from the three species with microarrays (see Supplemental Data Set 1 online) (Schmid et al., 2005; Wang et al., 2010; Sekhon et al., 2011). We then compiled the gene sets with the alignments of amino acid sequences in the three species using relatively stringent criteria, resulting in a total of 5698 groups of putative one-to-one orthologs (hereafter, “orthologs”) that were the best reciprocal BLAST hits shared by the three species (see Methods; Figure 1A). After aligning the probes in the Arabidopsis, rice, and maize microarrays to the 5698 ortholog sequences from the most updated genome releases, 4117 orthologs present on the microarrays were retained to construct the interspecific expression data set. To maximally retain biological differences and to remove technical variation, we employed a two-step normalization procedure: First, the global expression levels of the 4117 orthologs were adjusted to a comparable baseline using median-based scaling normalization; next, the quantile normalization was performed on the 198 samples to generate a uniform distribution across all the samples. Finally, the median value of an ortholog of the samples profiled at different developmental stages of the same group of tissue was used to represent a gene’s expression level. Thus, the final interspecific ortholog expression matrix for this analysis was composed of 4117 orthologs in 15, 14, and 17 tissue groups in rice, maize, and Arabidopsis, respectively.
Figure 1.

Global Pattern of Tissue Expression in Arabidopsis, Maize, and Rice.
(A) The numbers of species-specific and orthologous genes in the three species.
(B) The PCA analysis of the interspecific tissue expression profiles.
Global Patterns of Tissue Expression
To obtain a primary pattern of tissue expression in the three species, we performed a principal component analysis (PCA) on the compiled data set (Figure 1B). The first two principle components cumulatively explained 63% of the total variance. The 15, 14, and 17 tissue groups in rice, maize, and Arabidopsis were distinctly grouped according to species rather than tissues (Figure 1B). To rule out the influence from the genes with strong species-specific expression, we further performed the PCA analysis on a group of 1000 genes that are expressed in all of the three species with the least coefficient of variation in terms of their expression levels. The tissues were still clustered by species, suggesting that tissue expression within species is more concordant than that between species (see Supplemental Figure 1 online). This pattern is inconsistent with a prior study of the six homologous organs in 10 animal species, in which the organs were clustered together (Brawand et al., 2011), suggesting that the transcriptional networks of animals are more highly conserved at the organ level than those of plants.
In order to group tissues into homologous organs in the three plant species, we constructed a dendrogram of the 46 tissue groups with hierarchical clustering (see Supplemental Figure 2A online). Consistent with the PCA result, the tissue tree depicted three distinct clades of Arabidopsis, rice, and maize tissues. Within each species, the tissue tree was consistent with the tree structures built based on genome-wide expression data in the original articles, indicating that the interspecific normalization on the 4117 orthologs did not significantly alter the original expression patterns (Schmid et al., 2005; Wang et al., 2010; Sekhon et al., 2011). Moreover, the fine tree architecture demonstrated concordant relationships of tissues, in which similar tissue groups were agglomerated to form homologous organs (see Supplemental Figure 2B online). For example, the vegetative tissues, such as leaf, rosette, cauline, and sheath, were clustered together, separated from reproductive tissues, such as seed, endosperm, embryo, and germinating seed. Interestingly, the sexual organs, such as the stamen in rice, the anther in maize, and pollen in Arabidopsis, were clearly separated from other tissues, suggesting that the transcriptomes in sexual tissues are substantially distinct from those of vegetative tissues (see Supplemental Figure 2B online). The tissue tree allowed us to categorize the tissues into the seven major homologous organs, which we named root, stem, leaf, seedling, flower, seed, and stamen (see Supplemental Table 1 online).
Relative Rates of Organ Evolution Deduced from Interspecific Expression Divergence
To estimate the degree of expression divergence of tissues in the three species, we used the neighbor-joining (NJ) algorithm (Saitou and Nei, 1987) to construct an unrooted interspecific tissue tree based on expression profiles in Arabidopsis, rice, and maize. The elements of the distance matrix used in the NJ method were derived as 1-r, where r is the Pearson’s correlation coefficient of the gene expression profiles between any pair of tissues. The expression divergence was defined as the divergence in terms of the expression profiles across the tissues in the three species. The NJ method follows the notion of minimal evolution, generating a tree whose total branch length (in the same units as the pairwise distances) should be the smallest possible to account for the observed pairwise distances. The branch length in an expression-based NJ tree summarizes the expression divergence of tissues among the three species, with longer branches indicating higher levels of species- and tissue-specific expression. While vegetative tissues usually have shorter terminal branches, reproductive tissues, such as stamen of rice, pollen of Arabidopsis, and anther of maize, generally have longer terminal branches (Figure 2A).
Figure 2.

Estimation of the Rates of Expression Divergence across the Seven Organs.
(A) Unrooted phylogeny tree constructed with the NJ algorithm to infer the evolutionary distances for tissue expression. The NJ branch length may represent the degree of expression divergence.
(B) Expression divergences of the seven organs computed based on the NJ tree lengths. The red dots and the corresponding boxes represent the observed values and the simulated distributions using the bootstrapping method, respectively. The left and right panels show the expression divergences deduced from all of the orthologs and the transcription factor genes, respectively.
Subsequently, we inferred the relative rates of organ evolution in Arabidopsis, rice, and maize, based on the degree of expression divergence at the organ level. As one homologous organ may contain various numbers of tissue samples in the three species, the NJ algorithm was applied to each possible combination of the three tissues belonging to a species to calculate the total branch length. An average branch length of all the combinations was then calculated to represent the global expression divergence of an organ. For instance, assuming one homologous organ consisted of A and B tissues in rice, C and D tissues in maize, and E and F tissue in Arabidopsis, the combinations for building the three-taxon NJ trees are ACE, ACF, BCE, BCF, ADE, ADF, BDE, and BDF, based on the distance matrix converted from the Pearson’s correlations. The interspecific expression divergence of this organ is the average of the total branch length of the eight three-taxon species trees. Finally, the bootstrap method was used to estimate a confidence interval for each organ by randomly sampling genes 1000 times with replacement to generate a distribution of expression divergence. This allowed us to place confidence intervals around our estimates of the interspecific expression divergences across the seven major organs. We found a range of divergence values, ranging from the lowest in root and the highest in stamen (Figure 2B). Examination of the expression of just transcription factors showed a concordant pattern (Figure 2B). The rapid evolution of the reproductive tissues in plants matches the pattern of organ evolution in animals, for which the testis is the fastest evolving organ in the body (Khaitovich et al., 2006).
Correlated Evolution of Gene Sequence and Gene Expression Drives Organ Evolution
With a quantitative measurement of expression divergence across the seven organs in plants, we were able to examine the relationships between expression evolution and sequence evolution. Instead of using an arbitrary cutoff to determine a fixed number of tissue-specific genes, our method considered all of the genes expressed in an organ. We assumed that a tissue phenotype is governed by all of the genes expressed in the tissue but that genes may have different expression specificity and that this might be how they contribute to specific phenotypic characteristics of a tissue. The contribution of each gene to the total expression in a tissue, relative to the gene’s expression in other tissues, was then defined as its tissue specificity. In this way, we may determine the degree of tissue specificity of the genes that are present in one specific organ and determine whether sequence divergences and expression divergences of these genes occur in parallel in organ evolution. Thus, we devised a scoring algorithm to measure the tissue specificity (TS) of a gene contributing to an organ by comparing the maximum expression value of a gene among the tissues within an organ against the maximum expression values in the other six organs. The detailed algorithm is described in Methods.
Our method assigned seven TS scores to each individual gene to indicate its expression specificity in the seven organs. For each organ, the genes were ranked based on their TS scores associated with the organ. A higher TS rank indicates a gene’s greater contribution to the phenotype of an organ. Our analysis showed that 400 to 500 genes with high tissue specificity were absolutely unique to each of the seven organs, indicating that ∼10% of the genes are specifically expressed in the organ (Figure 3A). Gene Ontology (GO) analysis of the genes with top tissue specificity in the seven organs showed significantly differential enrichments that are relevant to the basic physiological function of an organ (see Supplemental Data Set 2 online). Subsequently, we computed the average evolutionary rates of the genes sorted from high tissue specificity to low tissue specificity, in increments of 50 genes (i.e., average rates of the top 50, 100, 150, 200…4117 genes). The relative evolutionary rate of an ortholog was measured by the average of the Poisson-corrected distances of three pairs of proteins. Within each TS range, the seven organs had seven values indicating the rates of sequence divergence averaged from the genes expressed in the corresponding organs, as well as seven values indicating the rates of expression divergence of the seven organs deduced from NJ tree analysis. Then, we calculated the Pearson correlation between the two variables within each TS range. If the sequence divergence rates are significantly positively correlated with the expression divergence rate of the seven organs, we may infer that these two levels of evolution may occur in parallel; otherwise, sequence and expression may evolve independently. With this method, we may also estimate the fraction of genes (out of the 4117 orthologs) whose sequence divergences and tissue-specific expression may play a dominant role in organ evolution in plants.
Figure 3.

Correlated Evolution of Gene Sequence and Expression Drives Organ Evolution in Plants.
(A) Each organ contains ∼400 to 500 genes with top ranks of tissue specificity unique to this organ. With a decrease in tissue specificity, the proportion of genes shared among the seven organs gradually increases. The box plot represents the distribution of the fractions of the genes that were not shared by any pair of organs at different tissue specificity ranks.
(B) Correlation between the expression divergences of the seven organs and the sequence divergences of tissue-specific genes in the seven organs. Significant correlations (Pearson r > 0.7 with P values < 0.05) were found within the range represented by ∼800 to 1200 of the most highly tissue-specific genes.
(C) Percentages of the shared genes between any pair of organs among the top 800 tissue-specific genes in each organ.
(D) Inference of the relative evolutionary rates of the seven organs. The y axis represents the average evolutionary rates of protein sequences of the top 50, 100, 150 … genes ranked by tissue specificity in the seven organs. The dashed line represents the average evolutionary rates of all 4117 orthologs.
In Figure 3B, the highest correlations between sequence and expression divergence (0.7 to 0.8) with significant P values (P ≤ 0.05) correspond to the 800 to 1200 most highly tissue-specific genes. This range potentially suggests that the divergence of organs among the three species is primarily governed by ∼20 to 30% of all the genes considered, including those genes that are expressed specifically in one organ and those that are expressed in more than one organ. While the genes with top tissue specificity are absolutely unique to each organ, with the decrease of tissue specificity, more and more genes were shared among the seven organs, but the proportions of shared genes varied between different pairs of organs. For instance, among the top 800 tissue-specific genes in each organ, the genes in stamen and seed rarely overlapped with the genes in other organs (<10%), whereas leaf and seedling shared nearly 50% of their genes (Figure 3C). Among flower, root, and stem, 30 to 40% genes were shared (Figure 3C).
To illustrate the relative rates of organ evolution in plants, we examined the relationship between the sequence evolutionary rates of the 4117 orthologs and their tissue specificities associated with each of the seven organs (Figure 3D). Notably, the top 100 to 200 tissue-specific genes showed the highest evolutionary rates in all seven organs; with the addition of more genes that are less tissue specific, the average evolutionary rates gradually decreased (Figure 3D). Because more and more genes were shared among the seven organs as tissue specificity decreased, the average evolutionary rates in the seven organs converged to 0.405, the average of all 4117 orthologs (Figure 3D). Therefore, using 0.405 as a reference line, the relative evolutionary rates of the seven organs can be illustrated from the relative positions of their respective curves. Again, stamen appeared to be the fastest evolving organ because its curve was almost entirely above the reference line. Seed and leaf were the second and third most quickly evolving organs, respectively, whereas flower, seedling, and stem showed comparable slow evolutionary rates. Root was the most conserved organ and showed the lowest evolutionary rate. Collectively, our analysis supports the speculation that tissue and/or organ evolution in plants occurs via the parallel evolution of both gene expression and gene sequence. This hypothesis is consistent with the results of previous studies in animals.
Relaxed Functional Constraint Causes Rapid Evolution of Male Reproductive Genes in Plants
In animals, the rapid evolution of sexual organs (i.e., testis) as well as the genes involved in sexual reproduction is linked to sex-related positive selection (sexual selection), such as sperm competition (Dorus et al., 2004). In plants, although sexual selection, such as pollen competition, may also exist, whether it is the dominant force that drives the accelerated evolution of stamen-specific genes is as yet unresolved. A possible solution is to compare interspecific sequence divergence with intraspecific sequence variations (i.e., single nucleotide polymorphisms [SNPs]) using population genomic data. If the ratio of interspecific divergence versus intraspecific diversity is substantially higher among the stamen-specific genes than among the genes specific to other organs, positive selection may explain their rapid evolution; otherwise, the rapid evolution of sexual organs would be considered a result of relaxed functional constraint, or in another words, decreased negative selection.
We obtained a total of ∼114,000 biallelic SNPs associated with the 4117 orthologs identified from 80 Arabidopsis strains to perform the comparison (Cao et al., 2011). We profiled the relationships of SNP density per gene, the ratio of nonsynonymous versus synonymous SNPs, and the proportion of fast-evolving genes (nonsynonymous SNPs > synonymous SNPs) with the ranked tissue specificity of the 4117 orthologs in the seven organs (Figure 4). The seven organs showed no obvious gradient of changes in SNP density, indicating that the frequencies of natural mutations are not biased toward any specific organ type (Figure 4A). However, the ratios of nonsynonymous to synonymous SNPs and the proportions of rapidly evolving genes varied across the seven organs. The highest rates were found in stamen and seed, indicating that intraspecific variation in amino acid sequences occurs more frequently in sexual organs than in vegetative organs (Figures 4B and 4C). This pattern is consistent with the interspecific study revealing the rapid evolution of genes specific to sexual organs. The ratios of average protein divergence rates between Arabidopsis, rice, and maize and the protein variation rates (nonsynonymous to synonymous SNPs) in Arabidopsis populations did not vary significantly across the seven organs (Figure 4D). Thus, this analysis suggests that positive selection (including sexual selection) is unlikely the dominant force driving the rapid evolution of stamen; rather, it is more likely a result of the relaxed functional constraints acting on genes specific to sexual organs.
Figure 4.

Analysis of Population Genomic Data in Arabidopsis Indicates That Rapid Evolution of Stamen in Plants May Be Due to Relaxed Functional Constraint.
(A) SNP densities (number of SNPs per gene per 100 bp) identified from the 80 strains of Arabidopsis show no bias across the seven organs. However, genes with higher tissue specificity contain more SNPs than those with lower tissue specificity.
(B) Genes expressed in stamen and seed have higher ratios of nonsynonymous SNPs to synonymous SNPs than do those expressed in vegetative organs.
(C) The fractions of rapidly evolving genes in stamen and seed are higher than in vegetative organs. The rapidly evolving genes were defined as the genes with more nonsynonymous SNPs than synonymous SNPs.
(D) The ratios of interspecific protein divergence rates versus the intraspecific variation rates (rates of nonsynonymous SNPs versus synonymous SNPs) in the seven organs.
Identification of Interspecific Gene Coregulation Modules
Although the PCA analysis of tissue expression patterns revealed little conservation in the global transcriptional networks in Arabidopsis, rice, and maize at the organ level (Figure 1B), gene expression may be conserved at the pathway level. To understand the evolution of expression at the pathway level, we used the iterative signature algorithm (ISA) to decompose the interspecific expression matrix into individual modules in which each module contains a group of potentially coregulated genes that may plausibly function in the same pathways (Bergmann et al., 2003). An ISA module shows the tissues and species in which a gene is expressed and may indicate the genes with which it may interact. The function of the module can be further inferred from GO enrichment analysis. Moreover, unlike the traditional classification methods that only allow a gene to be assigned to one module, the ISA method can assign a gene to multiple modules, as a gene may participate in multiple functional pathways. This analysis provides another layer of information to illustrate the functional conservation of a gene. These representations of information about the modules in which a gene participates can be mathematically formulated to derive a quantity to describe the degree of expression divergence (or expression conservation) for a gene.
The ISA identified a total of 1181 modules, including 1917 genes whose expression patterns were subject to significant changes across the 46 tissue groups in the three species (Figure 5A). Arabidopsis, maize, and rice contained 179 (492 genes), 224 (859 genes), and 173 (735 genes) species-specific modules, respectively, and only 135 (670 genes) modules were shared by the three species (Figures 5B and 5C). Moreover, the two grasses shared more genes (959 genes) than they each shared with Arabidopsis (534 rice genes; 621 maize genes). An ISA module contains a group of genes with both up- and downregulation trends in a set of tissues. For instance, module 645 contains 31 genes upregulated in rice shoots and maize leaves but downregulated in stamen, radicle, and root tissues of rice (Figure 5D). Module 992 is specific to rice, containing genes upregulated in endosperm and germinating seeds with a function related to starch biosynthesis (Figure 5D). The 1181 ISA modules assigned clear functions based on GO analysis are publicly accessible at http://www.cmbb.arizona.edu/ISA.
Figure 5.

Identification of the ISA Modules with Coregulated Genes.
(A) The number of modules identified with various gene thresholds and condition thresholds.
(B) The numbers of modules unique to each species or shared between species.
(C) The numbers of genes in the modules unique to each species or shared between species.
(D) Two representative modules showing the coregulated orthologs across tissues and species. Each square represents a gene with upregulation (red) or downregulation in a module. “Os” is rice, and “Zm” is maize.
eFCs
Functional constraint refers to the pressure of purifying selection that restricts the variations of functional sequences in the genome so that the expression and biochemical activity of these molecules cannot be freely changed. By contrast, the relaxation of the functional constraint acting on a gene may increase the rate of sequence and expression divergence. It has been found in animals that genes expressed in multiple tissues usually evolve slower than those specifically expressed in a single tissue, plausibly due to the higher degree of functional constraint accumulated from multiple tissues than that from fewer tissues (Khaitovich et al., 2006). A tissue-driven hypothesis was also made to explain the differential rates of gene evolution (Gu and Su, 2007). To test these hypotheses in plants, we sought to formulate a metric of eFC with expression data, as the expression profile of a gene may directly indicate the biological functions it is involved in. Specifically, the eFC integrates the information from the ISA modules, including the number of functional modules a gene is assigned to, the breadth of tissues in which a gene is expressed, the interspecific conservation of expression, and the number of genes with which the gene is considered coregulated in a module. Presumably, the genes involved in multiple biological functions may exhibit higher eFC scores due to the accumulated functional constraints and conversely may exhibit lower rates of sequence evolution. This formula was implemented as an additive function that considered the abovementioned factors. Specifically, for gene i participating in n modules, the overall functional constraint is the sum of the subconstraint of the gene in each module. The subconstraint of gene i in module j is then dependent on the number of tissues and species in which it is expressed and the number of other genes in module j with which it is coregulated. The subconstraint is scaled by the average module size and condition size. The detailed model for calculating the eFC of a gene is described in Methods.
We tested the correlation of the eFC with the gene sequence divergence rate. The average sequence divergence rate of a gene was negatively correlated with the numbers of organs in which the gene is expressed (Pearson r = −0.0833, P value = 0.040; Spearman ρ = −0.0915, P value = 0.024) (Figure 6A). This result suggests that sequence evolution is cumulatively restrained by the functional constraints from the tissues in which it is expressed. Additionally, a weak but statistically significant negative correlation was observed between functional constraints and average gene sequence divergence (Pearson r = −0.0892, P value = 0.028; Spearman ρ = −0.105, P value = 0.009) (Figures 6B to 6D). This analysis showed that the eFC of a gene can be used as a metric to bridge tissue expression and sequence evolution.
Figure 6.

Expression-based functional constraint (eFC)
(A) Genes expressed in more organs are more conserved in protein sequence than those expressed in fewer organs.
(B) to (D) Protein evolutionary rates are negatively correlated with eFCs. The evolutionary rates are computed as the Poisson-corrected protein distance between the orthologs in any pair of Arabidopsis, rice, and maize.
[See online article for color version of this figure.]
DISCUSSION
Global Regulatory Networks Are Highly Divergent between Plant Species
To explore the role of expression evolution in conjunction with sequence evolution in organ evolution in plants, we analyzed the relationship of these two factors in Arabidopsis, rice, and maize. We identified several commonalities between animals and plants, such as the rapid evolution of sexual organs, the correlation between the rates of sequence and expression evolution, and evidence that tissue functions influence the different divergence rates of gene sequences and expression patterns. On the other hand, we found that the global regulatory networks in plants appear to be less conserved at the organ level than the analogous networks in mammals reported by Brawand et al. (2011) using high-throughput transcriptome sequencing data, but this difference is most likely true. Although the divergence times of monocot-dicot (∼200 million years ago [MYA]) and rice-maize (∼60 MYA) in our study are comparable with the divergence times of primate-platypus (∼200 MYA) and primate-macaque (∼60 MYA) in Brawand et al.’s study, the variations in genome size, gene content, and transposon composition in Arabidopsis (∼125 Mb), rice (∼420 Mb), and maize (∼2.8 Gb) are much larger than those among animals. Thus, the huge differences in the genomic sequences of Arabidopsis, rice, and maize, especially for those occurring in noncoding regions with regulatory functions, might potentially cause substantial changes in gene regulation systems across plant species. However, we cannot rule out the possibility of artifact because the expression data sets in the three evolutionarily distant plant species were produced by different microarray platforms and by different groups. These technical issues have limited our studies to a focus on the organ level. Thus, interspecific expression data profiled within the same family, such as the Poaceae or Brassicaceae, and uniformly produced by RNA-sequencing technology is expected to yield more robust inferences regarding the evolution of global gene regulation networks in plants.
Different Causations of Rapid Evolution of Male Reproductive Genes in Animals and Plants
In this study, we found that genes specifically expressed in stamen in plants (i.e., stamen, anthers, or pollen) are relatively rapidly evolving, consistent with the discovery in animals that testis-specific genes tend to evolve at the fastest rate in the genome. However, further analysis revealed different causes of rapid evolution in the two kingdoms, namely, positive selection in animals versus relaxed functional constraints (= decreased negative selection) in plants. The distinct forces driving the evolution of male reproductive genes in plants and animals may be interpreted by their fundamentally different reproductive behaviors. For example, most plants are hermaphroditic and a third party is usually required to achieve pollination without any involvement of sentient influence from female’s choice. This means that sexual selection in plants tends to be weaker than that in animals (Moore and Pannell, 2011). Furthermore, we did not observe substantial differences across the seven organs in terms of the total DNA mutations (Figure 4A). This finding suggests that the frequencies of spontaneous mutation occur almost equally in the seven groups of organ-specific genes, ruling out the hypothesis that reproductive genes have higher mutation rates than vegetative organs. However, the rate of nonsynonymous mutation and the proportion of fast-evolving genes in sexual organs are substantially above those in vegetative organs (Figures 4B and 4C). The best explanation for this pattern is that a higher degree of sequence divergence of proteins related to reproduction occurs in plants, possibly because of relaxed functional constraints in sexual organs.
To infer the evolutionary mechanisms behind this pattern, we examined the functions of the stamen-specific genes with the highest protein divergence rates. Among these genes, we observed a significant enrichment of genes involved in “plant-type cell wall modification (P value = 7.3E-06)” and “pollen tube growth (P value = 1.6E-04)” (see Supplemental Data Set 2 online). Many of the genes under these two GO categories encode pectin methylesterases, which have been found essential for the development of pollen tubes and interactions between pollen tubes and female floral tissues through the modification of cell walls (Jiang et al., 2005). Studies have shown that mutation in one member of this gene family, VANGUARD1 (VGD1), can cause shorter siliques with fewer seeds and substantially reduced fertility in Arabidopsis compared with wild-type plants (Jiang et al., 2005). However, vgd1 mutation did not produce abnormal phenotypes in floral organs and vegetative parts or affect the normal growth of the mutant plants. Therefore, our analysis supports the hypothesis that highly diverged reproductive proteins, especially for those involved in sperm–egg interaction, may be responsible for establishing prefertilization barriers to interspecific hybridization and consequently driving the process of speciation (Swanson and Vacquier, 2002). We want to point out that, although our analysis indicated that relaxed functional constraints may be the primary force causing the accelerated evolution of stamen-specific genes in plants, we cannot rule out any effect of positive selection in stamen-expressed genes. As a matter of fact, there is evidence that a few reproductive genes, such as the S-LOCUS RECEPTOR KINASE and S-LOCUS GLYCOPROTEIN genes for self-incompatibility in Arabidopsis, may be under positive selection (Clark et al., 2006). Although a clear signature of positive selection was exhibited in S-LOCUS, a high diversity of protein sequences was also maintained within the species (Richman and Kohn, 2000). This phenomenon may be explained by the hypothesis raised by Wagner (2008) that previous accumulation of neutral or near-neutral mutations due to decreased negative selection may provide raw genetic materials for later positive selection to explore the fitness landscape.
Relationship of Gene Function and Gene Evolution
The positive correlation found between expression divergence and sequence divergence at the organ level suggests that the evolution of gene sequences and expression patterns occur in parallel, with different evolutionary rates associated with different organ types. This result is consistent with the studies in animals hypothesizing that different tissues may have different degrees of functional constraint influencing the coevolution of genome and transcriptome (Khaitovich et al., 2005b; Gu and Su, 2007). Inspired by Gu and Su’s tissue-driven hypothesis that stresses the role of tissue factor in gene evolution and Khaitovich et al.’s proposition that functional constraints may accumulate from multiple tissues to influence gene evolution, we developed a new measure of eFC based on gene coregulation modules inferred from expression profiles. The information from gene coregulation modules, including the expression breadth of a gene and the genes with which it may interact, along with the predicted module functions, may indicate the biological processes in which a gene participates to contribute to a tissue phenotype. The idea of a tissue factor may be more explicitly interpreted using eFC, which in fact associates with the specific physiological function of a tissue. For instance, the stamen-specific genes mostly are related to pollen tube growth, cell wall biogenesis, etc., and are among the most rapidly evolved genes in the genome, the root-specific genes functioning in ion transportation evolve at relatively slower rates, while the leaf-specific genes involved in a broader range of pathways, such as photosynthesis, secondary metabolic process, and responses to biotic stress, etc., evolve with an intermediate rate between stamen and root (see Supplemental Data Set 2 online). This result implies that the different degree of evolutionary constraint at the tissue level, which is referred to as the “tissue factor” by Gu et al., is actually a reflection of the importance of the tissue’s primary physiological function.
The negative correlation established between eFCs of genes and the evolutionary rates of protein sequences may also support the gene pleiotropy hypothesis proposed by Gu and his colleagues based on complicated mathematical deduction using population genetics and genotype-phenotype mapping data (Gu, 2007; Zeng and Gu, 2010). Their theory suggests that genes capable of affecting multiple phenotypes or involved in multiple biological functions are more conserved in evolution than genes with single function. To an extent, the eFC derived from expression profiles may be considered a measure of the pleiotropy (or multifunctionality) of a gene since the number of biological processes requiring the gene’s function can be quantitatively inferred from the ISA modules. Thus, the eFC in essence combines the concepts of tissue factor and gene pleiotropy: While the former stresses the role of function specificity in gene evolution (e.g., genes specifically expressed in root may be more evolutionarily constrained than genes specifically expressed in stamen), the latter stresses the role of multifunctionality in gene evolution (e.g., genes expressed in multiple tissues may be more evolutionarily constrained than genes expressed in single tissue).
In the past decades, many kinds of association studies have been performed to interpret the relationships between gene evolution and so-called genome factors that include gene expression levels (Pál et al., 2001; Hunt et al., 2011), protein structures (Kim et al., 2006), molecular interactions (Liao et al., 2006), topological characteristics of a gene in a network (Kim et al., 2007; Jovelin and Phillips, 2009), and so on. The list of these factors may be endlessly extended with the availability of new forms of biological data (Zeng and Gu, 2010). In summary, we believe that the eFC inferred from expression profiles can be used as a general metric that may summarize all of these functional characteristics of genes to elucidate the relationship between gene function and gene evolution.
METHODS
Tissue Specificity Score
We developed a scoring algorithm to compute a TS metric to quantify the contribution of a gene’s expression to an organ. Each gene was assigned seven TS scores associated with the seven organs. For organ O, the TS score for gene g is defined as 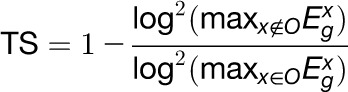 , where
, where 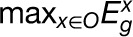 and
and 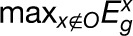 denote the maximum expression levels of gene g in the organ O and in the other six organs, respectively. Thus, the higher the TS score of a gene in an organ, the more likely this gene contributes to the species-specific phenotype of this organ to a greater extent. The relative contribution of a particular gene’s expression to an organ can then be ranked according to the TS scores of all genes.
denote the maximum expression levels of gene g in the organ O and in the other six organs, respectively. Thus, the higher the TS score of a gene in an organ, the more likely this gene contributes to the species-specific phenotype of this organ to a greater extent. The relative contribution of a particular gene’s expression to an organ can then be ranked according to the TS scores of all genes.
Divergence Rate of Protein Evolution
The putative one-to-one ortholog groups in Arabidopsis thaliana, rice (Oryza sativa), and maize (Zea mays) were determined by all-against-all comparisons of the protein sequences in each pair of the species with the BLASTP program. The hits in any comparison pair with an E-value < 1E-6 and an alignment length covering at least two-thirds of both query and subject sequences were selected to construct the ortholog expression matrix. The protein sequences of a group of orthologs were then aligned using the ClustalW program (Thompson et al., 1994). With the codeml program (seqtype = 2 and runmode = −2) in the PAML package, the Poisson-corrected distance was calculated for every pair of orthologs (Yang, 1997); the average of this distances of three pairs of protein alignments represents the relative evolutionary rate of the corresponding genes because they share the same evolutionary history since the recent common ancestor of the maize, rice, and Arabidopsis.
The ISA
We used the ISA developed by Bergmann et al. (2003) to decompose the interspecific expression matrix into coregulation modules. Specifically, we first conducted a near-exhaustive search of the modules with a large number of seeds and a wide combination of thresholds using the eisa software package (Csárdi et al., 2010). To derive the optimal parameters, 56 combinations of the gene thresholds from 2.1 to 3.5 and the condition thresholds from 1.3 to 2.5, both in increments of 0.2, were tested. To reduce the redundancy of the ISA modules, we used a merging parameter of 0.8 to combine the modules containing a similar group of genes. Subsequently, the identified 1183 modules were annotated by GO and KEGG pathway enrichment analysis in the eisa package. The 1181 ISA modules assigned clear functions based on GO analysis are publicly accessible at http://www.cmbb.arizona.edu/ISA.
eFC
Based on the ISA modules, we developed an additive model to quantify the functional constraint acting on a gene based on considerations of the number of species, tissues, and modules in which it participates and the number of genes with which it is coregulated. Given that gene i participates in n modules, i.e., 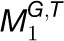 ,
,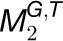 ,
, 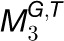 ,… and,
,… and, 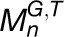 , where G and T denote the gene set and tissue set in the modules, respectively, we first calculated the functional constraint of gene i in an individual module
, where G and T denote the gene set and tissue set in the modules, respectively, we first calculated the functional constraint of gene i in an individual module 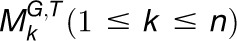 . For simplicity and without ambiguity, we denote this constraint as IFCk to omit the subscript i. We assumed that gene i in
. For simplicity and without ambiguity, we denote this constraint as IFCk to omit the subscript i. We assumed that gene i in 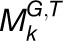 is concordantly regulated in j organs, i.e., Tk,1, Tk,2, Tk,3,…, and Tk,j, where Tk,m(1 ≤ m ≤ j ≤ 7) is one of the organs from root, leaf, seedling, stem, flower, stamen, and seed. We then assigned a coefficient Sk,m to each Tk,m according to the equation
is concordantly regulated in j organs, i.e., Tk,1, Tk,2, Tk,3,…, and Tk,j, where Tk,m(1 ≤ m ≤ j ≤ 7) is one of the organs from root, leaf, seedling, stem, flower, stamen, and seed. We then assigned a coefficient Sk,m to each Tk,m according to the equation 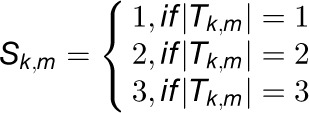 , where |Tk,m| represents the number of species in the module
, where |Tk,m| represents the number of species in the module 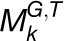 functioning in an organ Tk,m. Using the average number of genes (
functioning in an organ Tk,m. Using the average number of genes (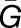 ) and the average number of organs (
) and the average number of organs (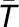 ) derived from all the modules as two scaling factors, the functional constraint of the individual module
) derived from all the modules as two scaling factors, the functional constraint of the individual module 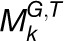 on gene i was then formulated as
on gene i was then formulated as
where |G| denotes the number of genes in gene set G and 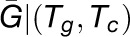 denotes the average number of genes affiliated with a set of modules identified under a specific combination of thresholds (Tg, Tc). Finally, the total functional constraint of gene i functioning in all the participated modules was defined as the sum of the functional constraint from each module, computed by
denotes the average number of genes affiliated with a set of modules identified under a specific combination of thresholds (Tg, Tc). Finally, the total functional constraint of gene i functioning in all the participated modules was defined as the sum of the functional constraint from each module, computed by 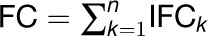 . To balance the opposite effects of the redundancy of similar modules and the coverage for genes, 117 ISA modules (Tg = 2.7 or 2.9 and Tc = 1.7 or 1.9) with overrepresented functional signatures were selected for the calculation of eFC.
. To balance the opposite effects of the redundancy of similar modules and the coverage for genes, 117 ISA modules (Tg = 2.7 or 2.9 and Tc = 1.7 or 1.9) with overrepresented functional signatures were selected for the calculation of eFC.
Accession Numbers
Accession numbers for samples used in this work can be found in Supplemental Data Set 1 online.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure 1. Global Relationships of Tissue and Organ Expression
Supplemental Figure 2. Dendrogram Tree of the 46 Tissue Groups Using Hierarchical Clustering on the 4117 Orthologs.
Supplemental Table 1. Categorization of Organ Types and Tissue Groups.
Supplemental Data Set 1. Descriptions of the Samples from the Arabidopsis, Rice, and Maize Microarray Experiments.
Supplemental Data Set 2. GO Enrichment Analysis on the Top 200 Tissue-Specific Genes in the Seven Organs.
AUTHOR CONTRIBUTIONS
R.Y. and X.W. designed the research, performed the analyses, and wrote the article.
Glossary
- eFC
expression-based functional constraint
- PCA
principal component analysis
- NJ
neighbor-joining
- TS
tissue specificity
- GO
Gene Ontology
- SNP
single nucleotide polymorphism
- ISA
iterative signature algorithm
- MYA
million years ago
References
- Bergmann, S., Ihmels, J., and Barkai, N. (2003). Iterative signature algorithm for the analysis of large-scale gene expression data. Phys. Rev. E Stat Nonlin. Soft Matter Phys. 67: 031902. [DOI] [PubMed]
- Brawand D., et al. (2011). The evolution of gene expression levels in mammalian organs. Nature 478: 343–348 [DOI] [PubMed] [Google Scholar]
- Cao J., et al. (2011). Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat. Genet. 43: 956–963 [DOI] [PubMed] [Google Scholar]
- Clark N.L., Aagaard J.E., Swanson W.J. (2006). Evolution of reproductive proteins from animals and plants. Reproduction 131: 11–22 [DOI] [PubMed] [Google Scholar]
- Csárdi G., Kutalik Z., Bergmann S. (2010). Modular analysis of gene expression data with R. Bioinformatics 26: 1376–1377 [DOI] [PubMed] [Google Scholar]
- Dorus S., Evans P.D., Wyckoff G.J., Choi S.S., Lahn B.T. (2004). Rate of molecular evolution of the seminal protein gene SEMG2 correlates with levels of female promiscuity. Nat. Genet. 36: 1326–1329 [DOI] [PubMed] [Google Scholar]
- Good J.M., Hayden C.A., Wheeler T.J. (2006). Adaptive protein evolution and regulatory divergence in Drosophila. Mol. Biol. Evol. 23: 1101–1103 [DOI] [PubMed] [Google Scholar]
- Gu X. (2007). Evolutionary framework for protein sequence evolution and gene pleiotropy. Genetics 175: 1813–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X., Su Z.X. (2007). Tissue-driven hypothesis of genomic evolution and sequence-expression correlations. Proc. Natl. Acad. Sci. USA 104: 2779–2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt B.G., Ometto L., Wurm Y., Shoemaker D., Yi S.V., Keller L., Goodisman M.A. (2011). Relaxed selection is a precursor to the evolution of phenotypic plasticity. Proc. Natl. Acad. Sci. USA 108: 15936–15941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L.X., Yang S.L., Xie L.F., Puah C.S., Zhang X.Q., Yang W.C., Sundaresan V., Ye D. (2005). VANGUARD1 encodes a pectin methylesterase that enhances pollen tube growth in the Arabidopsis style and transmitting tract. Plant Cell 17: 584–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovelin R., Phillips P.C. (2009). Evolutionary rates and centrality in the yeast gene regulatory network. Genome Biol. 10: R35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitovich P., Enard W., Lachmann M., Pääbo S. (2006). Evolution of primate gene expression. Nat. Rev. Genet. 7: 693–702 [DOI] [PubMed] [Google Scholar]
- Khaitovich P., Hellmann I., Enard W., Nowick K., Leinweber M., Franz H., Weiss G., Lachmann M., Pääbo S. (2005b). Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science 309: 1850–1854 [DOI] [PubMed] [Google Scholar]
- Khaitovich P., Pääbo S., Weiss G. (2005a). Toward a neutral evolutionary model of gene expression. Genetics 170: 929–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitovich P., Weiss G., Lachmann M., Hellmann I., Enard W., Muetzel B., Wirkner U., Ansorge W., Pääbo S. (2004). A neutral model of transcriptome evolution. PLoS Biol. 2: E132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim P.M., Korbel J.O., Gerstein M.B. (2007). Positive selection at the protein network periphery: Evaluation in terms of structural constraints and cellular context. Proc. Natl. Acad. Sci. USA 104: 20274–20279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim P.M., Lu L.J., Xia Y., Gerstein M.B. (2006). Relating three-dimensional structures to protein networks provides evolutionary insights. Science 314: 1938–1941 [DOI] [PubMed] [Google Scholar]
- Kimura, M. (1983). The Neutral Theory of Molecular Evolution. (Cambridge, UK: Cambridge University Press). [Google Scholar]
- King M.C., Wilson A.C. (1975). Evolution at two levels in humans and chimpanzees. Science 188: 107–116 [DOI] [PubMed] [Google Scholar]
- Liao B.Y., Scott N.M., Zhang J. (2006). Impacts of gene essentiality, expression pattern, and gene compactness on the evolutionary rate of mammalian proteins. Mol. Biol. Evol. 23: 2072–2080 [DOI] [PubMed] [Google Scholar]
- Moore J.C., Pannell J.R. (2011). Sexual selection in plants. Curr. Biol. 21: R176–R182 [DOI] [PubMed] [Google Scholar]
- Pál C., Papp B., Hurst L.D. (2001). Highly expressed genes in yeast evolve slowly. Genetics 158: 927–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman A.D., Kohn J.R. (2000). Evolutionary genetics of self-incompatibility in the Solanaceae. Plant Mol. Biol. 42: 169–179 [PubMed] [Google Scholar]
- Saitou N., Nei M. (1987). The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4: 406–425 [DOI] [PubMed] [Google Scholar]
- Schmid M., Davison T.S., Henz S.R., Pape U.J., Demar M., Vingron M., Schölkopf B., Weigel D., Lohmann J.U. (2005). A gene expression map of Arabidopsis thaliana development. Nat. Genet. 37: 501–506 [DOI] [PubMed] [Google Scholar]
- Sekhon R.S., Lin H., Childs K.L., Hansey C.N., Buell C.R., de Leon N., Kaeppler S.M. (2011). Genome-wide atlas of transcription during maize development. Plant J. 66: 553–563 [DOI] [PubMed] [Google Scholar]
- Swanson W.J., Vacquier V.D. (2002). The rapid evolution of reproductive proteins. Nat. Rev. Genet. 3: 137–144 [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Higgins D.G., Gibson T.J. (1994). CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I., Barkai N. (2008). Evolution of gene sequence and gene expression are not correlated in yeast. Trends Genet. 24: 109–113 [DOI] [PubMed] [Google Scholar]
- Wagner A. (2008). Neutralism and selectionism: A network-based reconciliation. Nat. Rev. Genet. 9: 965–974 [DOI] [PubMed] [Google Scholar]
- Wagner A. (2000). Decoupled evolution of coding region and mRNA expression patterns after gene duplication: Implications for the neutralist-selectionist debate. Proc. Natl. Acad. Sci. USA 97: 6579–6584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Xie W., Chen Y., Tang W., Yang J., Ye R., Liu L., Lin Y., Xu C., Xiao J., Zhang Q. (2010). A dynamic gene expression atlas covering the entire life cycle of rice. Plant J. 61: 752–766 [DOI] [PubMed] [Google Scholar]
- Wittkopp P.J., Kalay G. (2012). Cis-regulatory elements: Molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet. 13: 59–69 [DOI] [PubMed] [Google Scholar]
- Yang Z. (1997). PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13: 555–556 [DOI] [PubMed] [Google Scholar]
- Zeng Y.W., Gu X. (2010). Genome factor and gene pleiotropy hypotheses in protein evolution. Biology Direct 5: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]