

Abstract
Background
Signaling defects in the Toll-like receptor (TLR) pathway, such as interleukin-1 receptor–associated kinase 4 deficiency, highlight the prominence of TLR signaling in the defense against bacterial disease. Because myeloid differentiation primary response gene 88 (MyD88) can transduce signals from almost all TLRs, we studied its role in otitis media (OM), the most common upper respiratory tract bacterial infectious disease in young children.
Methods
The middle ears (MEs) of wild-type (WT) and MyD88−/− mice were inoculated with nontypeable Haemophilus influenzae (NTHi). ME infection and inflammation were monitored for 21 days after surgery. Bone marrow–derived macrophages from WT and MyD88−/− mice were infected with NTHi in vitro to assess their interaction with bacteria.
Results
In WT mice, MyD88 expression was detected in the ME stroma at baseline. MyD88−/− mice displayed prolonged ME mucosal thickening and delayed recruitment of neutrophils and macrophages. Although WT mice cleared NTHi within 5 days, viable NTHi were isolated for up to 21 days in MyD88−/− mice. The interaction between macrophages and NTHi was significantly altered in MyD88−/− mice.
Conclusions
In this mouse model, MyD88-mediated signaling was important for clearance of infection and resolution of inflammation in acute OM due to NTHi. The role played by innate signaling in children susceptible to chronic or recurrent OM deserves further study.
Innate immune mechanisms are critical for host responses to specific pathogens. The Toll-like receptor (TLR) pathway recognizes pathogen-associated molecular patterns of bacteria and viruses to initiate rapid innate immune responses. TLRs are present on multiple cell types, particularly those that interact with the external environment, such as surface epithelial, mast, and dendritic cells, which are prominent at mucosal surfaces. The activation of this pathway leads to the elaboration of multiple cytokines and also primes a targeted adaptive immune response.
TLRs employ adapter proteins to activate intracellular signaling cascades. All TLRs other than TLR3 can use the adapter myeloid differentiation primary response gene 88 (MyD88) to induce NF-κB and/or mitogen-activated protein (MAP) kinase–dependent proinflammatory gene expression [1]. MyD88 is also an adapter for the interleukin (IL)–1β receptor, another activator of NF-κB.
Innate immune responses appear to be particularly important in childhood pyogenic infections. One study found that children with IL-1 receptor (IL-1R)–associated kinase 4 deficiency have impaired TLR/IL-1R immunity, predisposing them to severe pneumoccocal infections [2]. However, death from invasive bacterial disease was not reported if these patients survived into adolescence, suggesting less dependence on innate immunity with maturation of the adaptive immune system. Upstream defects in TLR signaling have also been associated with disease susceptibility, such as dominant-negative TLR3 mutations in otherwise healthy children with Herpes simplex encephalitis [3].
The contributions of TLR signaling to the pathophysiology of one of the most common infectious diseases of childhood, otitis media (OM), is not well defined. Mucin production, a component of innate immune responses that prevents pathogen adherence and forms part of the mucociliary clearance apparatus, was up-regulated by live nontypeable Haemophilus influenzae (NTHi) lipoprotein P6 via a TLR2-MyD88–dependent p38 MAP kinase pathway in a middle ear (ME) epithelial cell line [4]. In a mouse model of OM, TLR4 deficiency was recently noted to promote the persistence of live NTHi in the ME up to 72 h after NTHi inoculation [5].
The present study was designed to evaluate the role played by MyD88 in a well-characterized murine model of OM induced by NTHi, one of the most common pathogens associated with OM. We hypothesized that, without signaling through MyD88, resolution of acute OM would be impaired because of delayed inflammatory cell recruitment to the ME after NTHi challenge.
METHODS
Ethics
All experiments were performed according to National Institutes of Health guidelines on the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee of the San Diego VA Medical Center.
Bacterial strain and culture conditions
H. influenzae strain 3655 (nontypeable, biotype II; originally isolated from the ME of a child with OM) was used at a concentration of 1 × 105–1 × 106 bacteria/mL to induce an inflammatory response in the ME. Inocula were prepared as described by Melhus and Ryan [6].
Histology
Ninety-six mice were divided into 2 experimental groups: 48 MyD88−/− mice and 48 wild-type (WT) control mice (C57BL/6). MyD88−/− mice were originally generated by Adachi et al. [7] and were crossed >6 times onto a C57BL/6 background. Age-matched WT C57BL/6 mice were purchased from Jackson Laboratories. Each group was subdivided into sets of 6 mice, 1 for each of 8 time points. Three mice per set were used for histology, and 3 mice per set were used for ME bacterial cultures.
WT and MyD88−/− mice were anesthetized with an intraperitoneal injection of rodent cocktail (0.1–0.2 mL/25–30 g of body weight, 13.3 mg/mL ketamine hydrochloride, 1.3 mg/mL xylazine, and 0.25 mg/mL acepromazine). The MEs were exposed bilaterally by a ventral approach through a midline neck incision. The ME bulla was fenestrated using a 25-gauge needle. Approximately 5 μL of NTHi (500–5000 cfu) was injected into the ME via a 30-gauge needle. Excess fluid was absorbed with a sterile cotton swab, the wound was closed by replacing soft tissue, and the skin incision was stapled. After surgery, the mice received a subcutaneous injection of buprenorphine and lactated Ringer’s solution.
Mice used for histology were killed under general anesthesia by intracardiac perfusion with PBS followed by 4% paraformal-dehyde (PFA) at baseline (0 h), 6 and 12 h after inoculation, and 1, 2, 3, 5, 10, 14, and 21 days after inoculation. For all experiments, the 0-h time point represents untreated MEs. The MEs were dissected, postfixed (4% PFA) overnight, and decalcified (8% EDTA and 4% PFA) for 14 days. Paraffin sections of the ME were stained with hematoxylineosin. Sections from the same region of each ME, in the largest part of the ME cavity, were digitally recorded, and mucosal thickness and the percentage area of the ME lumen occupied by inflammatory cells were determined at 6 standard locations, as described elsewhere [8].
For immunohistochemistry, sections were deparaffinized and rehydrated, fixed with acetone for 20 min, washed with PBS, and blocked with Power Block (BioGenex) for 1 h at room temperature. Sections were incubated with primary antibody overnight at 4°C (anti-MyD88 at 1:100 [Cell Sciences] and anti-pancytokeratin at 1:100 [Novocastra]). Sections were washed with PBS and incubated with secondary antibodies at 1:200 for 1 h at room temperature (Alexa 405–conjugated goat anti-mouse and Alexa 488 – conjugated goat anti-rabbit antibodies [both from Invitrogen]). Slides from MyD88−/− mice served as negative controls. Sections were washed with PBS and visualized with a Nikon C1Si confocal microscope. Images were processed using EZ-C1 FreeViewer software (version 3; Nikon).
Bacterial clearance
MEs were dissected from 3 WT and 3 MyD88−/− mice for each time point. MEs were opened, and a 1-μL loop sample was obtained from the ME lumen for NTHi culture on chocolate agar plates. Each loop was streaked onto 3 quadrants of each plate, followed by streaking onto a fourth quadrant without touching the other quadrants. The plates were evaluated 24 h after streaking. The presence of NTHi was verified by Gram staining of colony-forming units and negative cultures on blood agar plates versus chocolate agar plates.
A semiquantitative analysis of colony-forming units obtained from ME cultures was performed, using the following scoring system to classify the degree of colonization of each plate: 0 indicates no colony-forming units, 1 indicates colony-forming units in 1 quadrant, 2 indicates colony-forming units in 2 quadrants, 3 indicates colony-forming units in 3 quadrants, and 4 indicates colony-forming units in all 4 quadrants.
Inflammatory cell recruitment
The numbers of neutrophils and macrophages comprising ME cellular infiltrates were assessed by 2 independent observers who manually counted cell types in 5 randomly selected clusters of cellular ME effusions for each ME in a ×400 high-power field. The numbers were then averaged by computer.
Isolation and culture of bone marrow–derived macrophages
Bone marrow was isolated from 3 WT and 3 MyD88−/− mice. Macrophages were isolated and cultured as described elsewhere [9]. Macrophages were seeded onto 48-well tissue culture plates at ~5 × 105 cells/well and incubated overnight at 37°C. Macrophage viability was assessed with phase-contrast microscopy. No differences in cellular morphology were noted throughout the duration of the assay.
In vitro macrophage/NTHi interaction
Macrophage function was evaluated using an in vitro assay [10, 11], in which phagocytes are incubated with viable bacteria to allow internalization. Extracellular bacteria are then removed by rinsing and/or killed by antibiotic exposure. The cells are lysed at intervals, and the lysates are cultured to quantify viable intracellular bacteria, which is the final measure.
NTHi strain 3655 was stored at −80°C in brain-heart infusion broth with 20% glycerol. For preparation of inocula, bacteria were streaked from frozen aliquots onto a chocolate agar plate and incubated overnight at 37°C. Midexponential-phase bacteria were harvested from freshly growing cultures in brain-heart infusion broth with Fildes enrichment (shaken at 37°C), centrifuged at 1400 g for 10 min, and resuspended in PBS. Ten micro-liters (5 × 107 bacteria) were added to each well to yield an MOI of ~100:1.
Tissue culture plates were spun in a room temperature centrifuge at 200 g for 5 min to enhance contact between the bacteria and macrophages. Infected monolayers were then incubated for 1 h at 37°C. After this period, monolayers were washed with warm Dulbecco’s modified Eagle medium (DMEM) to remove extracellular bacteria. Fresh DMEM containing 10% fetal calf serum, macrophage colony-stimulating factor, and 50 μg/mL gentamicin was added to each well to rapidly kill any organisms not yet internalized by the macrophages. The minimum concentration of gentamicin required to kill extracellular NTHi had been determined to be 1 μg/mL. Incubation was continued for 1, 2, 5, 24, or 48 h after the addition of gentamicin. Six wells were used per time point. Intracellular NTHi were recovered by cell lysis in 0.5 mL of pyrogen-free water followed by aspiration of the lysate 5 times through a 23-gauge syringe. Supernatants and cell lysates were plated on chocolate agar, incubated overnight at 37°C in serial dilutions of 1:1 to 1:105, and evaluated by counting colony-forming units.
To estimate the elimination of internalized NTHi by WT and MyD88−/− macrophages, a mean killing index was calculated for WT and MyD88−/− macrophages, as follows: (mean colony-forming units 1 h after addition of gentamicin-mean colony-forming units 2 or 5 h after addition of gentamicin)/mean colony-forming units 1 h after addition of gentamicin.
Statistics
A 2-tailed t test comparing WT and MyD88−/− mice was performed at each time point on measures of mucosal thickness and ME inflammatory cells; tests were performed with the Bonferroni correction, using StatView software (version 5.0; JMP-SAS Institute). Differences between experimental groups were considered significant at P < .05. The 2 ears in each mouse had previously been found to be independent of each other and were therefore analyzed independently, which yielded 6 ears per time point and treatment (as discussed in detail by Ebmeyer et al. [8]). Descriptive statistics alone (i.e., means) were used for bacterial load from ME cultures, because this parameter was evaluated using semiquantitative measures.
For the macrophage assay, the Wilcoxon rank sum test was performed on NTHi lysate counts, owing to the lack of a normal distribution of these data points. Results were recorded as inter-quartile ranges, medians, and means.
RESULTS
Expression of MyD88 in the ME mucosa of C57BL/6 mice
The location of MyD88 expression was defined at baseline in the ME mucosa and during the early stages of NTHi infection. MyD88 was detected by immunohistochemical analysis in the stromal layer of the mucosa at baseline in WT mice (figure 1C and 1D). By 2 days after NTHi inoculation, MyD88 was detected throughout the mucosa: in some epithelial cells in the stromal layer and in inflammatory cells traversing the mucosa to enter the ME cavity (figure 1E and 1F). MyD88 was expressed in virtually all inflammatory cells comprising ME exudates (figure 1G and 1H).
Figure 1.
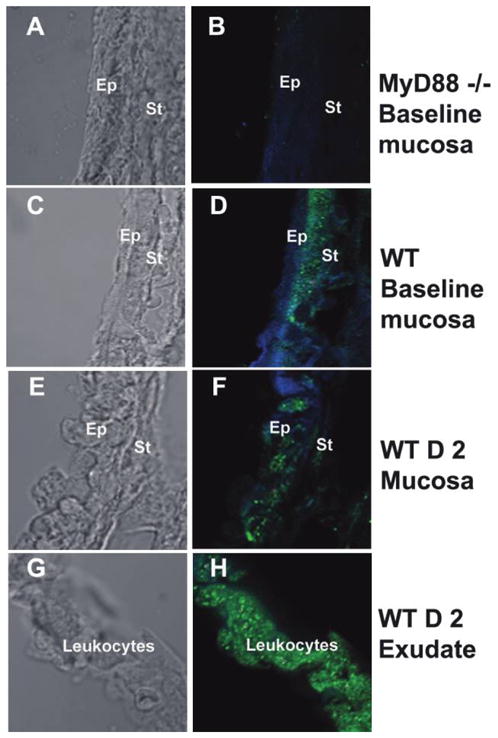
Expression of myeloid differentiation primary response gene 88 (MyD88) in the middle ear (ME). The left column shows light microscopy, and the right column shows immunohistochemical staining with fluorescence. Blue represents cytokeratin, and green represents MyD88. At baseline, MyD88 was not detected in the ME mucosa of MyD88−/− mice (A and B), as at any time point after inoculation with nontypeable Haemophilus influenzae (NTHi). In C57BL/6mice, MyD88 was detected in the stromal layer at baseline (C and D). The boundary between the epithelium and stromal layers is blurred as leukocytes traverse the ME mucosa (E and F). MyD88 was detected throughout the ME mucosa within 2 days after NTHi inoculation in wild-type (WT) mice. MyD88 was strongly detected in leukocytes (identified from adjacent hematoxylineosin–stained sections [data not shown]) comprising the ME exudate (G and H). D 2, day 2; Ep, epithelium; St, stroma.
Histology of the ME during OM
The ME undergoes characteristic inflammatory changes during bacterial infection with NTHi. The epithelial and stromal layers of the mucosa expand substantially, and an exudate of inflammatory cells is noted in the ME cavity. In WT mice, these changes are most prominent 2–3 days after NTHi inoculation (figure 2B). The ME cavity resembles its baseline appearance by 10–14 days after infection (figure 2C and 2D). The ears of MyD88−/− mice also experience mucosal thickening and an infiltrate of inflammatory cells by days 2–3 (figure 2F). However, 14 days after NTHi inoculation, the mucosa remains thickened and an exudate is still present (figure 2G and 2H).
Figure 2.
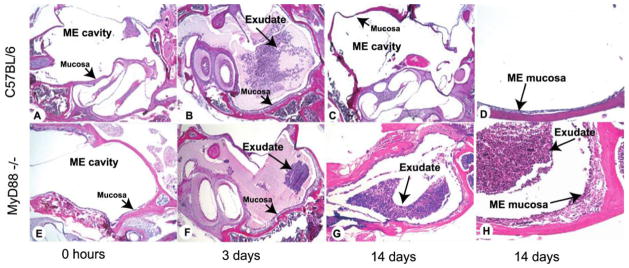
Middle ear (ME) response to nontypeable Haemophilus influenzae (NTHi) in C57BL/6 mice (A–D) and myeloid differentiation primary response gene 88 (MyD88)−/− mice (E–H). The MEs of C57BL/6 mice before NTHi infection demonstrated a very thin mucosal layer and no cellular infiltrate in the ME cavity (A). By day 3 after infection with NTHi, the ME cavities of C57BL/6 mice were filled with inflammatory cells and transudated fluid (B). By 14 days, no inflammatory cells were evident in the ME cavity of C57BL/6 mice; the mucosal layer had remodeled to its baseline appearance (C and D). In MyD88−/− mice, the morphology at 0 h (untreated) was similar to that in wild-type mice (E). By 3 days after NTHi infection, MyD88−/− mice had fewer inflammatory cells evident in the ME cavity (F). At 14 days after infection, the MEs of MyD88−/− mice showed a persistent inflammatory cell infiltrate and increased mucosal thickness compared with C57BL/6 mice (G and H). Original magnifications, ×40 (A–C and E–G) and ×100 (D and H).
Prolonging of ME mucosal thickening due to deficiency of MyD88
The degree of inflammation in the ME can be quantified by mucosal thickness and the percentage of the ME cavity covered by inflammatory cells [12]. In WT mice, mucosal thickness peaked 2–3 days after infection with NTHi (figure 3A), remodeling to its baseline dimensions by days 10–14. In MyD88−/− mice, mucosal thickness was similar to that in WT mice through 24 h after NTHi inoculation (figure 3A). Although the mucosal thickness in WT mice exceeded that in MyD88−/− mice at 2 days (P < .05), at 3 days the 2 groups of mice were again equivalent. Thereafter, mucosal thickness in MyD88−/− mice failed to recover, and it remained significantly greater than in WT mice from 5 to 21 days (P < .05) (figure 3A).
Figure 3.
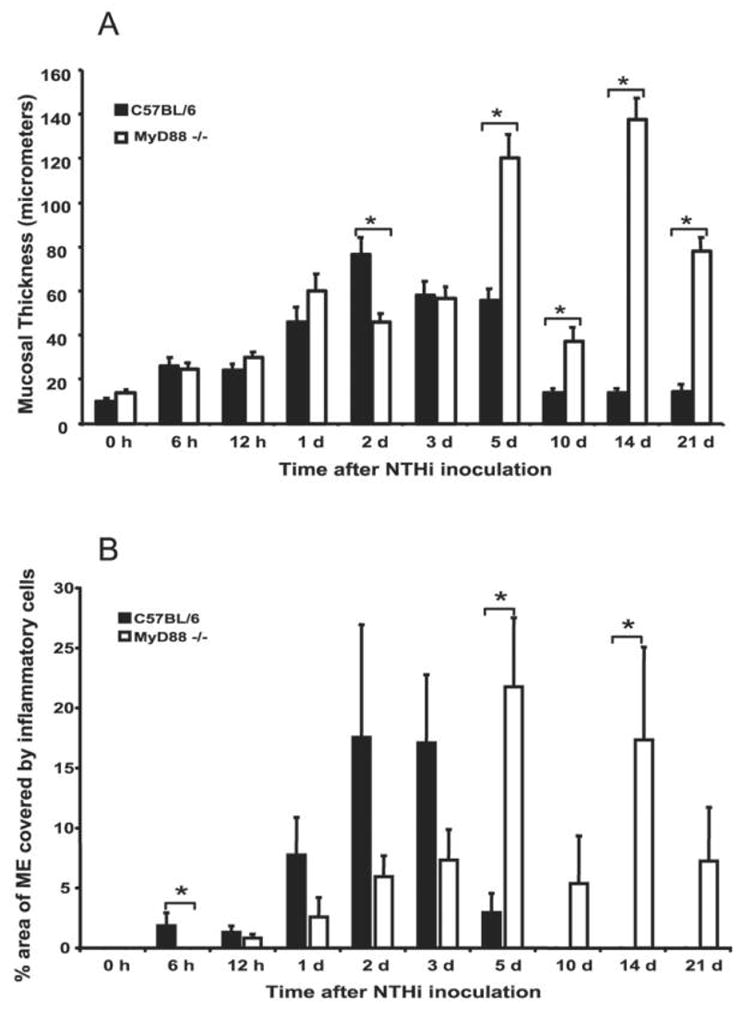
Markers of middle ear (ME) inflammation in C57BL/6 and myeloid differentiation primary response gene 88 (MyD88)−/− mice. A, Mucosal thickness. MEs of C57BL/6 mice and MyD88−/− mice showed similar degrees of mucosal thickness through 1 day after infection with nontypeable Haemophilus influenzae (NTHi), with thicker mucosa evident on day 2 in C57BL/6 mice. The degrees of mucosal thickness diverged after day 5, when the ME mucosae of MyD88−/− mice were persistently thickened but those of C57BL/6 mice had remodeled to their baseline thickness. B, Coverage of inflammatory cells. A greater percentage of the ME was occupied by inflammatory cells in C57BL/6 mice than in MyD88−/− mice through 3 days after NTHi inoculation. Inflammatory cells were recruited later in MyD88−/− mice, peaked at 5 days after NTHi inoculation, and persisted through day 21 (n = 6 ears per time point; bars and error bars represent means and SEs). *P < .05.
Delay in neutrophil and macrophage recruitment to the site of ME infection in the absence of signaling through MyD88
Inflammatory cell recruitment was noted as early as 6 h in WT mice (figure 3B). The inflammatory exudate peaked 2–3 days after NTHi inoculation and was cleared by 10 days (figure 3B). In MyD88−/− mice, recruitment of inflammatory cells was delayed, with significantly fewer leukocytes at 6 h (P < .05) and a later peak. The inflammatory exudate exceeded that of WT mice at 5 and 14 days (P < .05) and persisted throughout the entire period of observation (figure 3B).
We further assessed the ME infiltrate by quantifying the participation of neutrophils versus macrophages. In WT mice, neutrophils comprised the majority of ME cellular infiltrates 1 and 2 days after NTHi inoculation (figure 4A and 4E). Macrophages were subsequently recruited and were the primary inflammatory cells at later time points (figure 4B and 4F). No neutrophils or macrophages were evident by 10 days. MyD88−/− mice had impaired influx of neutrophils on days 1 and 2 after infection with NTHi (figure 4C), with peaks in neutrophil numbers delayed until 5 days (figure 4E). Macrophage recruitment was also delayed, with only a few macrophages noted on day 3 but high macrophage numbers at 10–21 days (figure 4F).
Figure 4.
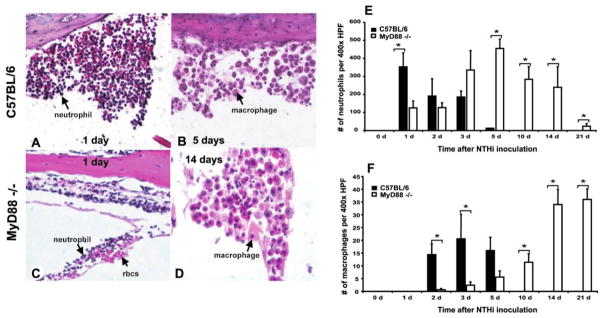
Cells composing the middle ear (ME) cell infiltrate after nontypeable Haemophilus influenzae (NTHi) infection. A large infiltrate of neutrophils was seen on day 1 after NTHi infection in C57BL/6 mice (A). Macrophages were the primary cell type present on day 5 in the MEs of C57BL/6 mice (B). In myeloid differentiation primary response gene 88 (MyD88)−/− mice, fewer neutrophils were noted in the ME cavity 1 day after NTHi infection (C). Macrophages did not become the predominant cell type in the ME infiltrate until day 14 after NTHi inoculation (D). C57BL/6 mice showed peak neutrophil numbers 1 day after infection with NTHi, and no neutrophils were evident by day 10 after infection; neutrophil infiltration peaked 5 days after NTHi infection in MyD88−/− mice, and some neutrophils were still present 21 days after NTHi inoculation (E). Macrophages were recruited to the ME by 2 days after NTHi infection in C57BL/6 mice, with no macrophages noted by day 10 after infection; macrophage recruitment to the ME was delayed in MyD88−/− mice (F). Like neutrophils, they persisted in the ME cavity through 21 days after inoculation with NTHi and were the primary cell type in the ME cavity at that time point (n = 6 ears per time point; bars and error bars represent means and SEs). rbcs, red blood cells. *P < .05.
Requirement of MyD88 for bacterial clearance after NTHi infection
Because of its central role as an adapter protein in innate immune signaling and its role in bacterial clearance at other sites, MyD88 was hypothesized to participate in the clearance of NTHi infection from the ME. NTHi were not cultured from WT mice 5 days after NTHi inoculation (table 1), consistent with the resolution of the ME inflammation and effusion. In contrast, NTHi were isolated from MyD88−/− mice throughout the 21-day observation period.
Table 1.
Bacterial clearance after nontypeable Haemophilus influenzae (NTHi) inoculation of the middle ear (ME) cavity.
| Time after NTHi inoculation | C57BL/6 mice
|
MyD88−/− mice
|
||
|---|---|---|---|---|
| CPPs, proportion | Bacterial colonization of CPPs,a mean | CPPs, proportion | Bacterial colonization of CPPs,a mean | |
| Day 1 | 4/6 | 4 | 6/6 | 4 |
|
| ||||
| Day 2 | 6/6 | 3 | 6/6 | 4 |
|
| ||||
| Day 3 | 3/6 | 1 | 6/6 | 4 |
|
| ||||
| Day 5 | 0/6 | 0 | 6/6 | 4 |
|
| ||||
| Day 10 | 0/6 | 0 | 6/6 | 4 |
|
| ||||
| Day 14 | 0/6 | 0 | 6/6 | 2.7 |
NOTE. No colony-forming units were detected by day 5 after NTHi inoculation in C57BL/6 (wild-type [WT]) mice. Bacterial clearance was impaired in myeloid differentiation primary response gene 88 (MyD88)−/− mice; NTHi were isolated from more than half of the MEs of MyD88−/− mice through 21 days after inoculation. CPPs, culture-positive plates.
For the semiquantitative analysis of bacterial colonization of the MEs of WT vs. MyD88−/− mice after NTHi inoculation, 0 indicates no colony-forming units, 1 indicates colony-forming units in 1 quadrant, 2 indicates colony-forming units in 2 quadrants, 3 indicates colony-forming units in 3 quadrants, and 4 indicates colony-forming units in all 4 quadrants.
Decrease in the initial recovery of viable NTHi and in the reduction of intracellular bacterial numbers in MyD88−/− mice
Although leukocyte recruitment was delayed in MyD88−/− mice, neutrophil and macrophage numbers eventually peaked at levels equal to or greater than those noted at earlier time points in WT mice (figure 4E and 4F). However, viable NTHi were still isolated from ME cultures after these inflammatory cells were present in the ME cavity (table 1). Therefore, we hypothesized that leukocytes from MyD88−/− mice were less efficient in clearing NTHi.
After 1 h of bone marrow–derived macrophage incubation with NTHi followed by an additional 1 h of gentamicin treatment (and at later time points), cultures of supernatants showed no colony-forming units, confirming that extracellular bacteria were killed by the gentamicin. After the 1-h exposure to gentamicin, lysates of macrophages from WT mice contained significantly more viable NTHi (mean, 2.9 × 106 cfu; median, 2.5 × 106 cfu) than those from MyD88−/− mice (mean, 6.1 × 105 cfu; median, 3.75 × 105 cfu) (P = .015) (figure 5). After 2 h of gentamicin exposure, the mean number of colony-forming units recovered from WT macrophages had decreased dramatically (mean, 2 × 105 cfu; median, 6.0 × 104 cfu) (P < .01). In contrast, essentially the same numbers of bacteria (mean, 5.12 × 105 cfu; median, 2.5 × 105 cfu) (P = .18) were isolated from MyD88−/− macrophages at 2 h and at 1 h. To quantify the presumed capacity of macrophages to kill internalized NTHi, we used a mean killing index (as described in Methods). Comparing the time points 1 and 2 h after gentamicin treatment, WT macrophages eliminated an average of 93% of internalized NTHi in 1 h (P = .022), whereas MyD88−/− macrophages eradicated only 16% of internalized NTHi during the same period (P = .70). Both WT and MyD88−/− macrophages had eliminated virtually all intracellular NTHi at 5 h after gentamicin treatment (for WT, 375 cfu; for MyD88−/−, 916 cfu), and no bacteria could be isolated from either group by 24 h (data not shown).
Figure 5.
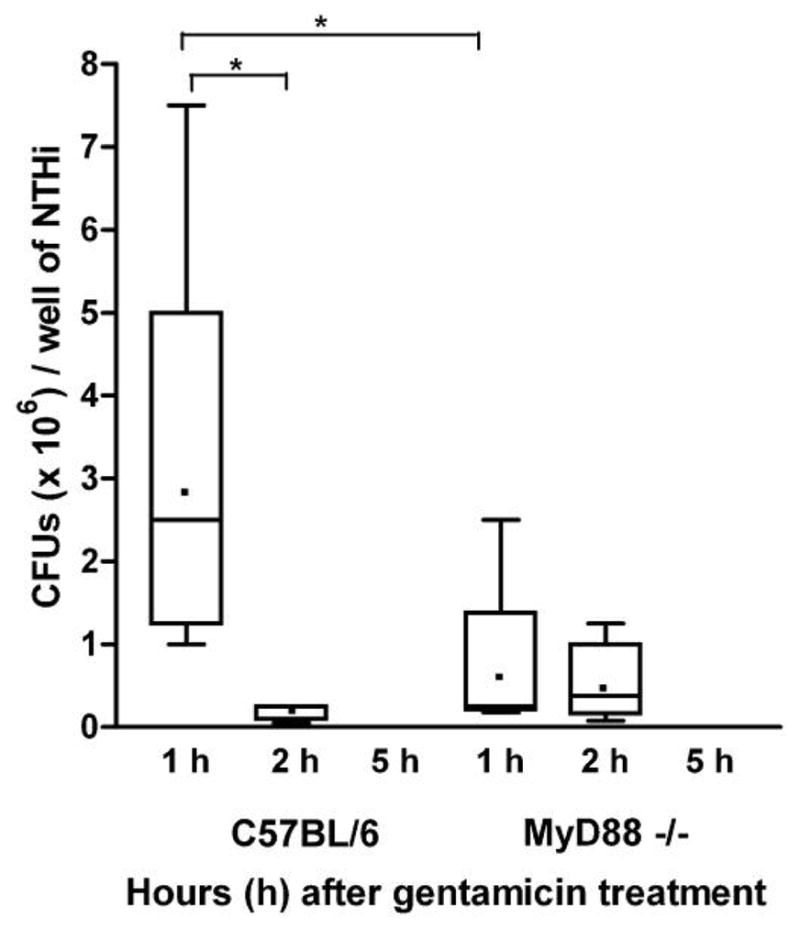
In vitro assay of macrophage interaction with nontypeable Haemophilus influenzae (NTHi). Bone marrow–derived macrophages from C57BL/6 and myeloid differentiation primary response gene 88 (MyD88)−/− mice were incubated with NTHi for 1 h, after which they were treated with a high concentration of gentamicin that killed all extracellular bacteria. Recovery of viable internalized bacteria from cell lysates was then assessed after an additional 1, 2, or 5 h of culture with gentamicin (n = 6 wells per time point). Results are shown as box plots encompassing the interquartile range; horizontal lines represent median values, dots represent mean nos. of colony-forming units per well, and error bars represent extremes of the data. After 1 h of gentamicin treatment, macrophages from C57BL/6 mice contained significantly more NTHi than did macrophages from MyD88−/− mice (P < .02). After 2 h of gentamicin treatment, the no. of colony-forming units recovered from wild-type (WT) macrophages had declined dramatically (P < .01), whereas recovery from MyD88−/− macrophages was unchanged (P = .18). By 5 h, most bacteria recovered from WT and MyD88−/− macrophages were nonviable. Comparing the 1- and 2-h time points, C57BL/6 macrophages killed significantly more internalized NTHi (P < .03) than did MyD88−/− macrophages (P = .70).
DISCUSSION
Our group has shown that signaling through MyD88 is essential for early neutrophil and macrophage recruitment to the ME after NTHi infection. We have also found that, once neutrophils and macrophages encounter this pathogen, their ability to eradicate it is dependent on MyD88. In the absence of MyD88, normally acute OM becomes chronic.
In the absence of MyD88, the delayed activation of endogenous and pathogen-associated molecular pattern signals could permit microorganisms to evade immune detection for longer periods, delaying compensatory responses and promoting further pathogen replication and the perpetuation of tissue damage. MyD88 has been shown to promote the rapid production of molecules important for leukocyte recruitment in response to NTHi, such as tumor necrosis factor (TNF)–α and IL-1β, products of NF-κB activation that are normally seen during the early stages of NTHi infection [13]. Failure to recruit neutrophils and macrophages to sites of infection is expected to promote enhanced bacterial replication and tissue damage.
After entering the MEs of MyD88−/− mice, macrophages and neutrophils coexisted with NTHi in the ME cavity for a prolonged period without effectively clearing the bacteria, suggesting that these cells were not able to properly phagocytose or kill NTHi. Our data show that the ability of macrophages to challenge NTHi infection is compromised, but not eliminated, by a lack of MyD88.
Using a macrophage cell culture inoculation assay, we obtained evidence that macrophages from MyD88−/− mice demonstrated decreased internalization of viable NTHi and a diminished rate of intracellular killing (figure 5). Much higher numbers of viable intracellular bacteria were observed inside WT macrophages 1 h after the addition of gentamicin to the cultures, compared with those for MyD88-deficient macrophages. Bacterial recovery at this 1-h time point can be interpreted as being dominated by the phagocytic activity of the macrophages, although some intracellular killing undoubtedly also occurs during this initial period. Within these constraints, MyD88−/− macrophages seem to be significantly deficient in phagocytosis. An alternative interpretation is that MyD88−/− macrophages internalized an equivalent number of viable bacteria but were more effective at intracellular killing. This explanation is unlikely given that MyD88−/− cells showed very little reduction in intracellular bacterial numbers between 1 and 2 h after the addition of gentamicin. It should be noted that the use of techniques that assess phagocytic capacity without regard to bacterial viability would address differences in that capacity more definitively [14].
The difference between the numbers of viable bacteria 1 h after the addition of gentamicin (during which time extracellular bacteria are presumed to be dead) and the numbers at the 2-h time point and later is a less ambiguous measurement of intracellular killing. Therefore, the lack of significant reduction in internalized bacteria in MyD88−/− macrophages between 1 and 2 h, compared with the >90% reduction seen in WT macrophages (P < .02), is consistent with compromised intracellular killing in the absence of MyD88.
Although viable NTHi were recovered from the MEs of MyD88−/− mice throughout the 21-day observation period, NTHi numbers decreased at 14 and 21 days, and some mice were able to eliminate NTHi at 21 days, implicating MyD88-independent mechanisms. NTHi express lipoproteins that are known or potential TLR2 ligands on its surface, such as the outer membrane lipoprotein P6 [15]. NTHi also express lipooligosaccharide, which is closely related to the known TLR4 ligand lipopolysaccharide [16]. Although TLR2 signaling is wholly dependent on MyD88, TLR4 can use both MyD88 and Toll/IL-1R domain–containing adapter-inducing interferon-β (TRIF) as adapter molecules in mediating downstream signaling. Signaling through TRIF can mediate the production of TNF-α and interferons [17]. In experiments involving NTHi-induced OM in TLR2−/− and TLR4−/− mice, we found that bacterial clearance was significantly impaired in both groups, although in neither group to the degree seen in MyD88−/− mice. This finding suggests that both TLRs contribute to ME bacterial clearance and that signaling via TLR4 and TRIF could operate independently of MyD88. However, Wieland et al. [18] found that MyD88, but not TRIF, contributed to NTHi clearance from the lungs at 48 h. Studies to clarify the participation of TRIF in NTHi-induced OM are currently under way. Of course, other innate immune signaling mechanisms, such as the NOD-like receptors, could also mediate ME bacterial clearance independently of MyD88.
It should be noted that we used only 1 strain of NTHi to evaluate OM in MyD88−/− mice. There can be significant heterogeneity in NTHi strains, and therefore the results of the present study do not represent the full spectrum of ME disease caused by this organism. However, the response of the mouse ME to this strain has been well characterized and is similar to that seen with other NTHi isolates [5, 6, 8].
Our study identifies a previously unrecognized role for MyD88-dependent innate immune signaling in the control of acute NTHi OM in mice. MyD88 is critical for the timely recruitment of leukocytes to the site of infection as well as for the effective resolution of infection by these leukocytes. Because routine studies of immune competence are often unrevealing in children with frequent or recurrent OM, our data suggest new avenues to explore for disease susceptibility to specific bacteria in humans. In this regard, it has recently been reported that polymorphisms in the genes encoding TLR4 and TNF-α are associated with recurrent OM in children [19].
Acknowledgments
We thank Drs. Shizuo Akira and Eyal Raz for the MyD88−/− mice, Eduardo Chavez for mouse colony maintenance, and Missy Brighton for assistance with immunohistochemistry.
Financial support: National Institute on Deafness and Other Communication Disorders, National Institutes of Health (grants DC006279 and DC000129); German Research Foundation (grant EU 120/1-1); Research Service of the Veterans Administration.
Footnotes
Potential conflicts of interest: none reported.
Presented in part: 2006 American Academy of Allergy, Asthma, and Immunology Annual Meeting, Miami, 3–7 March 2006; 2007 American Academy of Allergy, Asthma, and Immunology Annual Meeting, San Diego, 23–27 February 2007.
References
- 1.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 2.Ku CL, von Bernuth H, Picard C, et al. Selective predisposition to bacterial infections in IRAK-4-deficient children: IRAK-4-dependent TLRs are otherwise redundant in protective immunity. J Exp Med. 2007;204:2407–22. doi: 10.1084/jem.20070628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang SY, Jouanguy E, Ugolini S, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–7. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 4.Chen R, Lim JH, Jono H, et al. Nontypeable Haemophilus influenzae lipoprotein P6 induces MUC5AC mucin transcription via TLR2-TAK1-dependent p38 MAPK-AP1 and IKKbeta-IkappaBalpha-NF-kappaB signaling pathways. Biochem Biophys Res Commun. 2004;324:1087–94. doi: 10.1016/j.bbrc.2004.09.157. [DOI] [PubMed] [Google Scholar]
- 5.Hirano T, Kodama S, Fujita K, Maeda K, Suzuki M. Role of Toll-like receptor 4 in innate immune responses in a mouse model of acute otitis media. FEMS Immunol Med Microbiol. 2007;49:75–83. doi: 10.1111/j.1574-695X.2006.00186.x. [DOI] [PubMed] [Google Scholar]
- 6.Melhus A, Ryan AF. A mouse model for acute otitis media. APMIS. 2003;111:989–94. doi: 10.1034/j.1600-0463.2003.1111012.x. [DOI] [PubMed] [Google Scholar]
- 7.Adachi O, Kawai T, Takeda K, et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–50. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 8.Ebmeyer J, Furukawa M, Pak K, et al. Role of mast cells in otitis media. J Allergy Clin Immunol. 2005;116:1129–35. doi: 10.1016/j.jaci.2005.07.026. [DOI] [PubMed] [Google Scholar]
- 9.Obonyo M, Sabet M, Cole SP, et al. Deficiencies of myeloid differentiation factor 88, Toll-like receptor 2 (TLR2), or TLR4 produce specific defects in macrophage cytokine secretion induced by Helicobacter pylori. Infect Immun. 2007;75:2408–14. doi: 10.1128/IAI.01794-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang TJ, Fenton MJ, Weiner MA, et al. Murine macrophages kill the vegetative form of Bacillus anthracis. Infect Immun. 2005;73:7495–501. doi: 10.1128/IAI.73.11.7495-7501.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Barke RA, Charboneau R, Schwendener R, Roy S. Morphine induces defects in early response of alveolar macrophages to Streptococcus pneumoniae by modulating TLR9-NF-kappaB signaling. J Immunol. 2008;180:3594– 600. doi: 10.4049/jimmunol.180.5.3594. [DOI] [PubMed] [Google Scholar]
- 12.Ryan AF, Ebmeyer J, Furukawa M, et al. Mouse models of induced otitis media. Brain Res. 2006;1091:3–8. doi: 10.1016/j.brainres.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Melhus A, Ryan AF. Expression of cytokine genes during pneumococcal and nontypeable Haemophilus influenzae acute otitis media in the rat. Infect Immun. 2000;68:4024–31. doi: 10.1128/iai.68.7.4024-4031.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berenson CS, Garlipp MA, Grove LJ, Maloney J, Sethi S. Impaired phagocytosis of nontypeable Haemophilus influenzae by human alveolar macrophages in chronic obstructive pulmonary disease. J Infect Dis. 2006;194:1375–84. doi: 10.1086/508428. [DOI] [PubMed] [Google Scholar]
- 15.Shuto T, Xu H, Wang B, et al. Activation of NF-kappa B by nontypeable Hemophilus influenzae is mediated by toll-like receptor 2-TAK1-dependent NIK-IKK alpha /beta-I kappa B alpha and MKK3/6-p38 MAP kinase signaling pathways in epithelial cells. Proc Natl Acad Sci USA. 2001;98:8774–9. doi: 10.1073/pnas.151236098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hajjar AM, Ernst RK, Tsai JH, Wilson CB, Miller SI. Human Toll-like receptor 4 recognizes host-specific LPS modifications. Nat Immunol. 2002;3:354–9. doi: 10.1038/ni777. [DOI] [PubMed] [Google Scholar]
- 17.Hoebe K, Du X, Georgel P, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signaling. Nature. 2003;424:743–8. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 18.Wieland CW, Florquin S, Maris NA, et al. The MyD88-dependent, but not the MyD88-independent, pathway of TLR4 signaling is important in clearing nontypeable Haemophilus influenzae from the mouse lung. J Immunol. 2005;175:6042–9. doi: 10.4049/jimmunol.175.9.6042. [DOI] [PubMed] [Google Scholar]
- 19.Emonts M, Veenhoven RH, Wiertsema SP, et al. Genetic polymorphisms in immunoresponse genes TNFA, IL6, IL10, and TLR4 are associated with recurrent acute otitis media. Pediatrics. 2007;120:814–23. doi: 10.1542/peds.2007-0524. [DOI] [PubMed] [Google Scholar]