

Abstract
Background
Breast cancer is one of the most common cancers in the world, and the identification of biomarkers for the early detection of breast cancer is a relevant target. The present study aims to determine serum peptidome patterns for screening of breast cancer.
Methods
The present work focused on the serum proteomic analysis of 36 healthy volunteers and 37 breast cancer patients using a ClinProt Kit combined with mass spectrometry (MS). This approach allows the determination of peptidome patterns that are able to differentiate the studied populations. An independent group of sera (36 healthy volunteers and 37 breast cancer patients) was used to verify the diagnostic capabilities of the peptidome patterns blindly. An immunoassay method was used to determine the serum mucin 1 (CA15-3) of validation group samples.
Results
Support Vector Machine (SVM) Algorithm was used to construct the peptidome patterns for the identification of breast cancer from the healthy volunteers. Three of the identified peaks at m/z 698, 720 and 1866 were used to construct the peptidome patterns with 91.78% accuracy. Furthermore, the peptidome patterns could differentiate the validation group achieving a sensitivity of 91.89% (34/37) and a specitivity of 91.67% (33/36) (> CA 15–3, P < 0.05).
Conclusions
These results suggest that the ClinProt Kit combined with MS shows great potentiality for the diagnosis of breast cancer.
Virtual slides
The virtual slide(s) for this article can be found here: http://www.diagnosticpathology.diagnomx.eu/vs/1501556838687844
Keywords: Breast neoplasms, Diagnosis, Proteomics, Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry
Background
Breast cancer is a leading cause of mortality and morbidity for women [1]. Surgery, chemotherapy, and radiation treatments can be effective, depending on stage of cancer and other factors [2,3]. At present, the best available tool for the early detection of breast cancer is mammography[4]. Data acquisition, processing and visualization techniques of medical images facilitate diagnosis and improve their functionalities [5]. However, it is well established that mammography is better able to detect certain types of breast cancer (such as ductal carcinomas) than other types (such as poor prognosis estrogen receptor (ER)-negative tumors)[6-9]. Considering ER status, interval detected tumors are 1.8 to 2.6-fold more likely to be ER-compared to screen detected tumors [8,9]. Combining findings derived from both cytology and histology best allows for the proper management of patients suffering from breast cancer [10,11]. However, cytology and histology testes are invasive, which are not suitable for screening of breast cancer. Continued improvements in our ability to detect breast cancer early offer the promise of further reducing the burden of this disease, as breast cancer detected at an earlier stage is much more curable than is metastatic disease.
Proteomics, which concerns comprehensive protein profile changes caused by multiple gene alterations, is currently considered the most powerful tool for the global evaluation of protein expression [12]. Human serum contains thousands of proteolytically derived peptides called peptidomes, which may provide a robust correlation with the physiologic and pathologic processes in the entire body [13,14]. Preliminary studies have shown that great interest has been focused on the low-molecular-weight region, particularly on peptides smaller than 20 kDa, which may provide a novel means of diagnosing cancer and other diseases [14-16].
Advances in mass spectrometry (MS) now permit the display of hundreds of small- to medium-sized peptides using only microliters of serum [17,18]. Matrix-assisted laser desorption/ionization time-of-flight MS (MALDI-TOF MS) can detect peptides with low molecular weights with the necessary sensitivity and resolution, which makes it a useful technique for serum peptide profiling. Furthermore, for accurate MS analysis, the peptidome fractionation procedure and the preanalytical conditions of peptidome mapping must be carefully assessed [19]. Magnetic beads (MBs), based on nanomaterials, have been developed and considered as a promising material for convenient and efficient enrichment of peptides and proteins in biological samples [20,21]. The combination of MALDI-TOF MS and MBs enables the high throughput and sensitive investigation of peptides and proteins.
In the current study, a novel technology platform called ClinProt (BrukerDaltonics, Ettlingen, Germany) was used. It comprises a immobilized affinity copper ions MB (IMAC-MB)-based sample separation, MALDI-TOF MS for peptide profiling, and a bioinformatics package for inspection and comparison of data sets to create “disease-specific” peptidome pattern models, which could serve as a powerful tool for breast cancer diagnosis [22-24]. The diagnostic model, which consists of three differentially expressed peptides, was established and validated by the Support Vector Machine (SVM) Algorithm, by which different groups were effectively discriminated. Then, the diagnostic model was further verified using blinded samples from breast cancer and healthy volunteers.
Patients and methods
Reagents and instruments
The AutoFlex III MALDI-TOF mass spectrometer, MTP 384 target plate polished steel, α-cyano-hydroxycinnamic acid (CHCA), MB-IMAC kit, and peptide calibration standard were purchased from BrukerDaltonics (Leipzig, Germany). The trifluoroacetic acid (TFA) and acetonitrile (ACN), and mucin 1 (CA15-3) diagnostic kit (ELISA) were purchased from Alfa Aesar (Ward Hill, MA, USA), Sigma (St. Louis, MO, USA), and Roche Diagnostics GmbH (SandhoferStrasse, Germany), respectively.
Patients and sample collection
With their consent, 72 healthy volunteers and 74 breast cancer patients (TNM I 26, II48) were enrolled into the study, from whom blood samples were collected. Serum samples were prepared by collecting blood in a vacuum tube and allowing it to clot for 30 min at room temperature. About 1 mL of serum was obtained after centrifugation at 2000 rpm for 10 min and stored in small aliquots at −80°C until analysis.
Study design
The data set, including 72 health subject and 74 breast cancer patients, was randomly split into model construction group and external evaluation group. Model construction group (36 healthy volunteers and 37 breast cancer patients) was used for the identification of signals related to peptides expressed differentially among breast cancer patients compared with healthy volunteers. The group was also used for the pattern recognition. The external evaluation group (36 healthy volunteers and 37 breast cancer patients) was used for the blind independent pattern validation of the cluster. The accuracy of the peptidome model was compared with that of CA 15–3. The mean ages (years, means ± SD) of the healthy volunteers and breast cancer patients were 56.35 ± 2.58 and 58.57 ± 9.55, respectively. The difference of ages between the healthy volunteers in the model construction group and those in the external evaluation group were not significant. No significant differences were also observed for the ages of the breast cancer patients and healthy volunteers, as well as for the TNM stages of the breast cancer patients in the model construction group and external evaluation group.
Sample purification
IMAC-MBs were used for the peptidome separation of samples following the manufacturer's standard protocol [25]. First, 50 μL of IMAC-MB binding solution and 5 μL of IMAC beads were combined in a 0.5 mL microfuge tube after thoroughly vortexing both reagents. The microfuge tubes were then placed in an MB separator (MBS) and agitated 10 times. The beads were collected from the tube walls 1 min later. Then resuspend the MB in 20 μL of IMAC-MB binding solution. Second, 5 μL of serum sample was added and mixed by pipetting up and down. The microfuge tubes were then placed in an MBS and agitated 10 times. The beads were collected from the tube walls 1 min later and the supernate was carefully removed using a pipette. Third, 100 μL of IMAC-MB wash buffer was added into tubes, which were again agitated 10 times in the MBS. The beads were then collected from the tube walls, and the supernate was carefully removed using a pipette. After three times washing, 10 μL of the IMAC-MB elution buffer was added to disperse the beads in tubes by pipetting up and down. The beads were collected from the tube walls after 5 min, and the clear supernate was transferred into fresh tubes. The supernate was then ready for spotting onto MALDI-TOF MS targets and measurement. Finally, prior to the MALDI-TOF MS analysis, the targets were prepared by spotting 1 μL of the proteome fraction on the polished steel target (BrukerDaltonics, Bremen, Germany). After air-drying, 1 μL of 3 mg/mL CHCA in 50% ACN and 50% Milli-Q with 2% TFA was applied onto each spot, and then, the target was air-dried again (cocrystallization). The peptide calibration standard (1 pmol/μL peptide mixture) was applied for machine calibration.
MS analysis
For proteome analysis, a linear Autoflex III MALDI-TOF mass spectrometer was used with the following settings: ion source 1, 20.00 kV; ion source 2, 18.60 kV; lens, 6.60 kV; and pulsed ion extraction, 120 ns. Ionization was achieved via irradiation with a crystal laser operating at 200 Hz. For the matrix suppression, a high gating factor with signal suppression up to 600 Da was used. The mass spectra were recorded in linear positive mode. Mass calibration was performed using the calibration mixture of the peptides and proteins in the mass range of 1–18 kDa. Three MALDI preparations (MALDI spots) were measured for each MB fraction. For each MALDI spot, 1600 spectra were quantified (200 laser shots at eight different spot positions). The spectra were recorded automatically using the Autoflex Analysis software (BrukerDaltonics, Bremen, Germany) for the fuzzy-controlled adjustment of the critical instrument settings to generate raw data with optimized quality.
Bioinformatics and statistical analysis
The ClinProt Tools software 2.2 (BrukerDaltonics) was used to analyze all serum sample data derived from either the patients or the normal health subjects. The data analysis began with raw-data pretreatment, including baseline subtraction of spectra, normalization of a set of spectra, internal peak alignment using prominent peaks, and a peak-picking procedure. The pretreated data were then used for visualization and statistical analysis in ClinProt Tools.
Statistically significant differences in peptide quantity were determined using Welch’s t-tests. The significance was set at P < 0.05. The class prediction model was set up using the SVM Algorithm. Then, the classified peptidome patterns were constructed. To determine the accuracy of the class prediction, a cross-validation was first implemented. Twenty percent of the samples from the model construction group were randomly selected as a test set and the remaining samples were taken as a training set in the class predictor algorithm. Second, by designing a double-blind test, the samples of external evaluation group were classified using the classified peptidome patterns constructed by the SVM Algorithm.
Detection of CA15-3
The serum CA15-3 levels of the 37 breast cancer patients and 36 healthy volunteers in the evaluation group were measured using an electrochemiluminescence immunoassay following the manufacturer’s standard protocols (the methods were omitted). The samples were diagnosed as breast cancer (≥ 50 U/mL) or healthy (< 50 U/mL).
Statistical methods and evaluation of assay precision
Each spectrum recorded using the MALDI-TOF MS was analyzed with Autoflex Analysis to detect the peak intensities of interest and with ClinProt™ software (BrukerDaltonics) to compile the peaks across the spectra recorded from all samples. This setup allowed differentiation between the cancer and the health subject samples. To evaluate the precision of the assay, the within- and between-run variations were determined using multiple analyses of bead fractionation and MS for two plasma samples. For the within- and between-run variations, three peaks with various intensities were examined. The within-run imprecision was determined by evaluating the coefficient variations (CVs) for each sample, using eight assays within a run, and then the between-run imprecision was determined by performing eight different assays over a period of seven days. SPSS 16.0 was used to analyze the clinical characteristics of the volunteers using a χ2 test or a t-test. The significance was set at P < 0.05. In addition, SPSS 16.0 was used to compare the accuracies of the peptidome models and the CA15-3 determination.
Results
For the reproducibility of the protein profiling, the within- and between-run reproducibility of two samples was determined via IMAC-MB fractionation and MALDI-TOF MS analysis. In each profile, three peaks with different molecular masses were selected to evaluate assay precision. Despite varying peptide masses and spectral intensities, the peak CVs were all <3% and <9% in the within- and between-run assays, respectively. These values were consistent with the reproducibility data for the Protein Biology System reported by BrukerDaltonics.
In the pilot study, the differences between the serum proteome profiles of breast cancer patients and healthy volunteers were evaluated. The mass spectra from 600 Da to 18 kDa were obtained using MALDI-TOF MS in linear mode. The representative mass spectra of the prefractionated sera of the model construction group are reported in Figure 1. On average, 70 signals common to the two groups have been detected in this mass range and 24 were identified by the ClinProt software with a statistically different area (P < 0.05 using the Wilcoxon analysis) in the model construction population, including 15 upregulated and 9 downregulated peptides, respectively (Table 1).
Figure 1 .
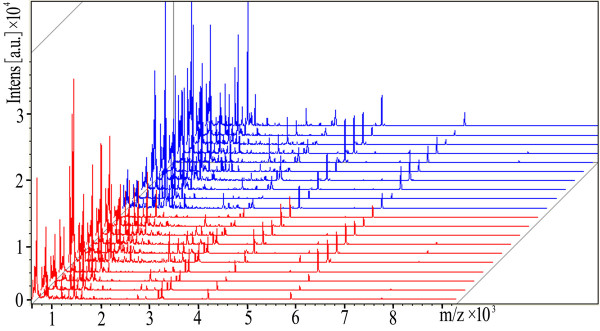
View of the aligned mass spectra of the serum protein profiles of the model construction group (red represents healthy volunteers, and blue denotes breast cancer patients) obtained using MALDI-TOF after purification with IMAC-MBs.
Table 1.
Statistics of the 24 candidate biomarkers for identifying breast cancer patients from healthy individuals
|
Mass |
Breast cancer |
Health |
Regulation in breast cancer |
P
★
|
||
|---|---|---|---|---|---|---|
| Means ▲ | SD | Means ▲ | SD | |||
| 622.48 |
48.9 |
24.43 |
34.29 |
18.28 |
↑ |
0.0454 |
| 622.97 |
48.26 |
24.67 |
30.41 |
14.76 |
↑ |
0.013 |
| 654.73 |
27.08 |
9.66 |
18.02 |
5.6 |
↑ |
0.0048 |
| 655.23 |
23.48 |
8.61 |
16.35 |
4.46 |
↑ |
0.00827 |
| 666.78 |
13.96 |
6.64 |
20.73 |
6.29 |
↓ |
0.00821 |
| 667.12 |
13.07 |
6.56 |
18.81 |
5.77 |
↓ |
0.0146 |
| 676.83 |
39.48 |
25.42 |
15.69 |
15.75 |
↑ |
1.98E-05 |
| 698.4 |
116.2 |
35.99 |
76.07 |
27.99 |
↑ |
0.0048 |
| 698.81§ |
114.32 |
35.83 |
70.97 |
23.34 |
↑ |
0.00138 |
| 720.8§ |
13.59 |
6.5 |
23.41 |
8.61 |
↓ |
0.00199 |
| 721.4 |
13.31 |
6.39 |
19.92 |
7.96 |
↓ |
0.0165 |
| 858.11 |
13.28 |
6.03 |
8.11 |
3.76 |
↑ |
0.0182 |
| 887.23 |
10.15 |
3.38 |
8.33 |
5.93 |
↑ |
0.0222 |
| 893.99 |
12.58 |
7.56 |
21.87 |
8.11 |
↓ |
0.00218 |
| 909.06 |
27.78 |
15.1 |
40.15 |
14.33 |
↓ |
0.0127 |
| 909.75 |
26.61 |
15.1 |
37.89 |
14.06 |
↓ |
0.0222 |
| 1618.46 |
11.12 |
8.56 |
21.88 |
16.05 |
↓ |
0.017 |
| 1866.64§ |
6.66 |
5.23 |
12.97 |
7.21 |
↓ |
0.00199 |
| 2770.34 |
6.64 |
5.11 |
2.26 |
1.07 |
↑ |
0.00123 |
| 2771.86 |
5.73 |
4.78 |
1.48 |
0.68 |
↑ |
0.000427 |
| 2933.97 |
3.69 |
2.06 |
1.86 |
0.85 |
↑ |
0.0103 |
| 5963.91 |
0.71 |
0.76 |
0.2 |
0.24 |
↑ |
0.0222 |
| 7772.04 |
0.73 |
0.94 |
0.25 |
0.46 |
↑ |
0.0176 |
| 7777.16 | 0.8 | 1.01 | 0.3 | 0.51 | ↑ | 0.0406 |
§The peptide selected for model construction.
▲Peak area.
★P value calculated with the Wilcoxon test; values lower than 0.05 suggest statistical relevance.
Classification models were developed to classify between the breast cancer and healthy volunteers samples of model construction group. The use of individual peaks as diagnostic biomarkers for breast cancer was addressed using SVM algorithm analysis. First, the breast cancer patients and healthy volunteers were compared. Second, all detected peaks were analyzed using ClinProt 2.2 to generate the cross-validated classification models. The optimized model resulted in the following correct sample classification. Three peptide ion signatures (m/z 698, 720 and 1866) were provided as a class prediction for a cross-validation set to discriminate the breast cancer patients from healthy volunteers, which achieved 91.78% recognition and 91.78% cross-validation accuracy. The regions of the mass spectra obtained at 800 resolution are reported in Figure 2.
Figure 2 .
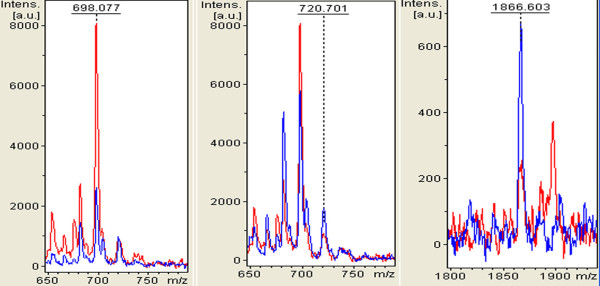
Zoom of the mass range for the three signals (MALDI-TOF linear mode) used in the cluster to differentiate BREAST CANCER from healthy (H) individuals. Red represents breast cancer patient, and blue represents healthy volunteers.
The preliminary statistical analysis was performed for each single marker and signal cluster using the receiver operating characteristic curve analysis. The area under curve (AUCs) of receiver operator characteristic (ROC)of peak A at m/z698, peak B at m/z 720 and peak C at m/z 1866 were 0.85, 0.83 and 0.83, respectively (Figure 3). Moreover, the areas of these peaks in the spectra of breast cancer patients were statistically different from those of the healthy volunteers (Figure 2). A combination of these three peaks yielded 88.89% (32/36) specificity and 94.59% (35/37) sensitivity for the breast cancer samples (Table 2).
Figure 3 .
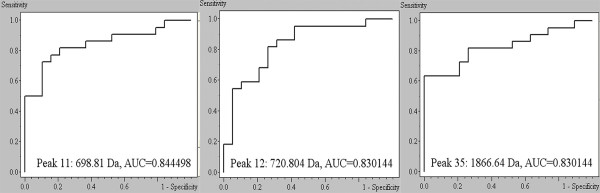
The receiver operating characteristic (ROC) curves of the three signals selected for the diagnostic model. AUC: Area under the receiver operating characteristic curve.
Table 2.
The predicted results of peptidome pattern distinguishing breast cancer patients from health volunteers
|
Group |
Cases |
Sensitivity (%) |
Specificity (%) |
PV
+
|
PV
−
|
YI |
|
|---|---|---|---|---|---|---|---|
| Breast cancer | Health | ||||||
| Model construction |
37 |
36 |
94.59 |
88.89 |
89.74 |
94.12 |
0.83 |
| External validation | 37 | 36 | 91.89 | 91.67 | 91.89 | 91.67 | 0.83 |
Abbreviations: PV+, Positive predict value; PV−, Negective predict value; YI, Youden index.
To verify the accuracy of the established SVM classification model with the adopted peptides, the samples of external evaluation group was introduced (not used in the model construction), which consisted of 37 breast cancer patients and 36 healthy subjects. As a result, the model correctly classified 91.89% (34/37) of the breast cancer (sensitivity) and 91.67% (33/36) of the healthy (specificity) samples, which surpassed the results of CA15-3 (41.67% (15/36) specificity and 43.24% (16/37) sensitivity) (Table 2).
Discussion
The usefulness of multiple markers for diagnosis, prognosis, and prediction of the risk of developing diseases or their complications is now widely recognized [13,26]. Various proteomic approaches have been applied to biomarker discovery using biological fluids. Interestingly, low-molecular-weight peptides, such as S100A8 and fibrinogen, have been recognized to play important roles in physiologic and pathologic processes and could be used as relevant biomarker candidates [27,28]. Recently, the mass spectrum that directly detects and differentiates short peptides has offered a promising approach for peptidomic biomarker discovery [14,15,29-31].
MS instrumentation and analysis tools have continued to rapidly evolve and improve our ability to detect lessabundant serum proteins. Until now, the most commonly used instrument was the SELDI-TOF MS[32-35]. However, SELDI-TOF MS does not allow a direct identification of the discriminatory proteins and the debate about the reproducibility has been particularly strong[36]. Alternative approaches for measuring polypeptides, such as the surface-enhanced laser desorption and ionization, recently reported by several groups, have several disadvantages, such as low resolution and the loss of most proteins and peptides [37-39]. MALDI is a soft ionization technique used in MS that allows the analysis of biomolecules such as proteins, peptide sugars, and large organic molecules. As a powerful tool for surveying the complex patterns of biologically informative molecules, MALDI-TOF MS protein/peptide profiling has been applied in proteomics biomarker research and has become a promising tool in cancer biomarker research [29,40,41].
In the present study, by integrating short peptide purification with IMAC-MBs, peak intensity detection with MALDI-TOF MS, and profile analysis with ClinProt Tools software 2.2, a series of differentially expressed short peptides in the sera of breast cancer patients has been successfully detected. A comparative case control analysis between breast cancer and healthy volunteers was performed. Peptidomic maps associated with the disease were drawn. The results show that compared with the healthy volunteers, the breast cancer patients share 24 significantly differentiated peptides, including 15 upregulated and 9 downregulated peptides. Genomic and proteomic technologies will further help us understand the intracellular signaling and gene transcription systems, as well as the protein pathways that connect the extracellular microenvironment to the serum or plasma macroenvironment of cancer [42]. These 24 interesting significantly differentiated peptides may provide further evidence for understanding the occurrence and progress of breast cancer.
Using SVM algorithm analysis, classification models were developed to classify samples between healthy volunteers and breast cancer. A cluster of three peptides at m/z 698, 720 and 1866 achieved a recognition capacity and a cross-validation of 91.78% to discriminate breast cancer from healthy volunteers. The blinded verification of the SVM classification model proved the correct classification of 91.89% (34/37) of the breast cancer (sensitivity) and 91.67% (33/36) of the healthy volunteers(specificity). To our knowledge, this is the first study to screen for breast cancer-related short peptides in sera by combining IMAC-MBs and MALDI-TOF MS. The classification model that we have built up has potential applications in providing alternatives for breast cancer diagnosis and may provide a better understanding of breast cancer pathogenesis or help in tailoring the use of chemotherapy for each patient, finally resulting in improved patient outcomes.
In conclusion, peptidome patterns from IMAC-MB-purified serum samples were directly profiled with MALDI-TOF MS and a peptidome model that differentiated breast cancer from the healthy volunteers was constructed with high sensitivity and specificity. Despite the high sensitivity and specificity, the number of specimens analyzed in this study was relatively small, which may limit the validity of the results. The next step in our study will be to analyze larger patient cohorts and to run blinded samples to confirm the usefulness of the currently identified peptides for breast cancer diagnosis. After this confirmation, the biomarkers of the interest will then be isolated and identified and their biological role in breast cancer pathogenesis will be studied.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
N-JF carried out magnetic bead separation and mass spectrometry analysis, participated in the design of the study, and drafted the manuscript. C-FG conceived of the study, and participated in its design and coordination and helped to draft the manuscript. GZ participated in mass spectrometry analysis and performed the statistical analysis. X-LW carried out the clinical sample and data collection. Q-YL carried out the immunoassays. All authors read and approved the final manuscript.
Contributor Information
Nai-Jun Fan, Email: fandoctor123@163.com.
Chun-Fang Gao, Email: chunfanggao@126.com.
Guang Zhao, Email: macroguang@163.com.
Xiu-Li Wang, Email: xiuli150@163.com.
Qing-Yin Liu, Email: qingyinliu@163.com.
Acknowledgment
This research was supported by The Medical Science Research Project of The Chinese People’s Liberation Army (No. 08Z006).
References
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Elmore JG, Armstrong K, Lehman CD, Fletcher SW. Screening for breast cancer. Jama. 2005;293:1245–1256. doi: 10.1001/jama.293.10.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duric VM, Stockler MR, Heritier S, Boyle F, Beith J, Sullivan A, Wilcken N, Coates AS, Simes RJ. Patients' preferences for adjuvant chemotherapy in early breast cancer: what makes AC and CMF worthwhile now? Ann Oncol. 2005;16:1786–1794. doi: 10.1093/annonc/mdi370. [DOI] [PubMed] [Google Scholar]
- Geiser WR, Haygood TM, Santiago L, Stephens T, Thames D, Whitman GJ. Challenges in mammography: part 1, artifacts in digital mammography. AJR Am J Roentgenol. 2011;197:W1023–1030. doi: 10.2214/AJR.10.7246. [DOI] [PubMed] [Google Scholar]
- Balafar MA. Spatial based expectation maximizing (EM) Diagn Pathol. 2011;6:103. doi: 10.1186/1746-1596-6-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JM, Anderson TJ, Page DL, Lee D, Duffy SW. Infiltrating lobular carcinoma of the breast. Histopathology. 1982;6:149–161. doi: 10.1111/j.1365-2559.1982.tb02712.x. [DOI] [PubMed] [Google Scholar]
- Silverstein MJ, Lewinsky BS, Waisman JR, Gierson ED, Colburn WJ, Senofsky GM, Gamagami P. Infiltrating lobular carcinoma. Is it different from infiltrating duct carcinoma? Cancer. 1994;73:1673–1677. doi: 10.1002/1097-0142(19940315)73:6<1673::AID-CNCR2820730620>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Porter PL, El-Bastawissi AY, Mandelson MT, Lin MG, Khalid N, Watney EA, Cousens L, White D, Taplin S, White E. Breast tumor characteristics as predictors of mammographic detection: comparison of interval- and screen-detected cancers. J Natl Cancer Inst. 1999;91:2020–2028. doi: 10.1093/jnci/91.23.2020. [DOI] [PubMed] [Google Scholar]
- Collett K, Stefansson IM, Eide J, Braaten A, Wang H, Eide GE, Thoresen SO, Foulkes WD, Akslen LA. A basal epithelial phenotype is more frequent in interval breast cancers compared with screen detected tumors. Cancer Epidemiol Biomarkers Prev. 2005;14:1108–1112. doi: 10.1158/1055-9965.EPI-04-0394. [DOI] [PubMed] [Google Scholar]
- Howard JH, Bland KI. Current management and treatment strategies for breast cancer. Curr Opin Obstet Gynecol. 2012;24:44–48. doi: 10.1097/GCO.0b013e32834da4b1. [DOI] [PubMed] [Google Scholar]
- Lale S, Kure K, Lingamfelter D. Challenges to diagnose metaplastic carcinoma of the breast through cytologic methods: an eight-case series. Diagn Pathol. 2011;6:7. doi: 10.1186/1746-1596-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky E, Bastiaens PI, Zhu H, Snyder M, Fields S. Protein analysis on a proteomic scale. Nature. 2003;422:208–215. doi: 10.1038/nature01512. [DOI] [PubMed] [Google Scholar]
- Issaq HJ, Xiao Z, Veenstra TD. Serum and plasma proteomics. Chem Rev. 2007;107:3601–3620. doi: 10.1021/cr068287r. [DOI] [PubMed] [Google Scholar]
- Dai Y, Hu C, Wang L, Huang Y, Zhang L, Xiao X, Tan Y. Serum peptidome patterns of human systemic lupus erythematosus based on magnetic bead separation and MALDI-TOF mass spectrometry analysis. Scand J Rheumatol. 2010;39:240–246. doi: 10.3109/03009740903456292. [DOI] [PubMed] [Google Scholar]
- Liu LH, Shan BE, Tian ZQ, Sang MX, Ai J, Zhang ZF, Meng J, Zhu H, Wang SJ. Potential biomarkers for esophageal carcinoma detected by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Clin Chem Lab Med. 2010;48:855–861. doi: 10.1515/CCLM.2010.138. [DOI] [PubMed] [Google Scholar]
- Sui W, Huang L, Dai Y, Chen J, Yan Q, Huang H. Proteomic profiling of renal allograft rejection in serum using magnetic bead-based sample fractionation and MALDI-TOF MS. Clin Exp Med. 2010;10:259–268. doi: 10.1007/s10238-010-0094-5. [DOI] [PubMed] [Google Scholar]
- Koomen JM, Li D, Xiao LC, Liu TC, Coombes KR, Abbruzzese J, Kobayashi R. Direct tandem mass spectrometry reveals limitations in protein profiling experiments for plasma biomarker discovery. J Proteome Res. 2005;4:972–981. doi: 10.1021/pr050046x. [DOI] [PubMed] [Google Scholar]
- Villanueva J, Philip J, Entenberg D, Chaparro CA, Tanwar MK, Holland EC, Tempst P. Serum peptide profiling by magnetic particle-assisted, automated sample processing and MALDI-TOF mass spectrometry. Anal Chem. 2004;76:1560–1570. doi: 10.1021/ac0352171. [DOI] [PubMed] [Google Scholar]
- Omenn GS. The human proteome organization plasma proteome project pilot phase: reference specimens, technology platform comparisons, and standardized data submissions and analyses. Proteomics. 2004;4:1235–1240. doi: 10.1002/pmic.200300686. [DOI] [PubMed] [Google Scholar]
- Yao N, Chen H, Lin H, Deng C, Zhang X. Enrichment of peptides in serum by C(8)-functionalized magnetic nanoparticles for direct matrix-assisted laser desorption/ionization time-of-flight mass spectrometry analysis. J Chromatogr A. 2008;1185:93–101. doi: 10.1016/j.chroma.2008.01.047. [DOI] [PubMed] [Google Scholar]
- Whiteaker JR, Zhao L, Zhang HY, Feng LC, Piening BD, Anderson L, Paulovich AG. Antibody-based enrichment of peptides on magnetic beads for mass-spectrometry-based quantification of serum biomarkers. Anal Biochem. 2007;362:44–54. doi: 10.1016/j.ab.2006.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West-Norager M, Kelstrup CD, Schou C, Hogdall EV, Hogdall CK, Heegaard NH. Unravelling in vitro variables of major importance for the outcome of mass spectrometry-based serum proteomics. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;847:30–37. doi: 10.1016/j.jchromb.2006.09.048. [DOI] [PubMed] [Google Scholar]
- Shin S, Cazares L, Schneider H, Mitchell S, Laronga C, Semmes OJ, Perry RR, Drake RR. Serum biomarkers to differentiate benign and malignant mammographic lesions. J Am Coll Surg. 2007;204:1065–1071. doi: 10.1016/j.jamcollsurg.2007.01.036. discussion 1071–1063. [DOI] [PubMed] [Google Scholar]
- Baumann S, Ceglarek U, Fiedler GM, Lembcke J, Leichtle A, Thiery J. Standardized approach to proteome profiling of human serum based on magnetic bead separation and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Clin Chem. 2005;51:973–980. doi: 10.1373/clinchem.2004.047308. [DOI] [PubMed] [Google Scholar]
- Ketterlinus R, Hsieh SY, Teng SH, Lee H, Pusch W. Fishing for biomarkers: analyzing mass spectrometry data with the new ClinProTools software. Biotechniques. 2005. pp. 37–40. [DOI] [PubMed]
- Zethelius B, Berglund L, Sundstrom J, Ingelsson E, Basu S, Larsson A, Venge P, Arnlov J. Use of multiple biomarkers to improve the prediction of death from cardiovascular causes. N Engl J Med. 2008;358:2107–2116. doi: 10.1056/NEJMoa0707064. [DOI] [PubMed] [Google Scholar]
- Tolson JP, Flad T, Gnau V, Dihazi H, Hennenlotter J, Beck A, Mueller GA, Kuczyk M, Mueller CA. Differential detection of S100A8 in transitional cell carcinoma of the bladder by pair wise tissue proteomic and immunohistochemical analysis. Proteomics. 2006;6:697–708. doi: 10.1002/pmic.200500033. [DOI] [PubMed] [Google Scholar]
- Cheng AJ, Chen LC, Chien KY, Chen YJ, Chang JT, Wang HM, Liao CT, Chen IH. Oral cancer plasma tumor marker identified with bead-based affinity-fractionated proteomic technology. Clin Chem. 2005;51:2236–2244. doi: 10.1373/clinchem.2005.052324. [DOI] [PubMed] [Google Scholar]
- Alagaratnam S, Mertens BJ, Dalebout JC, Deelder AM, van Ommen GJ, den Dunnen JT, t Hoen PA. Serum protein profiling in mice: identification of Factor XIIIa as a potential biomarker for muscular dystrophy. Proteomics. 2008;8:1552–1563. doi: 10.1002/pmic.200700857. [DOI] [PubMed] [Google Scholar]
- Chinello C, Gianazza E, Zoppis I, Mainini V, Galbusera C, Picozzi S, Rocco F, Galasso G, Bosari S, Ferrero S. et al. Serum biomarkers of renal cell carcinoma assessed using a protein profiling approach based on ClinProt technique. Urology. 2010;75:842–847. doi: 10.1016/j.urology.2009.09.050. [DOI] [PubMed] [Google Scholar]
- Du J, Yang S, Lin X, Bu L, Nan Y, Huo S, Shang W. Use of anchorchip-time-of-flight spectrometry technology to screen tumor biomarker proteins in serum for small cell lung cancer. Diagn Pathol. 2010;5:60. doi: 10.1186/1746-1596-5-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gast MC, Zapatka M, van Tinteren H, Bontenbal M, Span PN, Tjan-Heijnen VC, Knol JC, Jimenez CR, Schellens JH, Beijnen JH. Postoperative serum proteomic profiles may predict recurrence-free survival in high-risk primary breast cancer. J Cancer Res Clin Oncol. 2011;137:1773–1783. doi: 10.1007/s00432-011-1055-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. The application of SELDI-TOF-MS in clinical diagnosis of cancers. J Biomed Biotechnol. 2011;2011:245821. doi: 10.1155/2011/245821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooker BC, Newman LS, Bowler RP, Karjalainen A, Oksa P, Vainio H, Pukkala E, Brandt-Rauf PW. Proteomic detection of cancer in asbestosis patients using SELDI-TOF discovered serum protein biomarkers. Biomarkers. 2011;16:181–191. doi: 10.3109/1354750X.2010.543289. [DOI] [PubMed] [Google Scholar]
- Fan NJ, Gao CF, Wang XL. Identification of Regional Lymph Node Involvement of Colorectal Cancer by Serum SELDI Proteomic Patterns. Gastroenterol Res Pract. 2011;2011:784967. doi: 10.1155/2011/784967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engwegen JY, Gast MC, Schellens JH, Beijnen JH. Clinical proteomics: searching for better tumour markers with SELDI-TOF mass spectrometry. Trends Pharmacol Sci. 2006;27:251–259. doi: 10.1016/j.tips.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Petricoin EF, Ardekani AM, Hitt BA, Levine PJ, Fusaro VA, Steinberg SM, Mills GB, Simone C, Fishman DA, Kohn EC, Liotta LA. Use of proteomic patterns in serum to identify ovarian cancer. Lancet. 2002;359:572–577. doi: 10.1016/S0140-6736(02)07746-2. [DOI] [PubMed] [Google Scholar]
- Chen YD, Zheng S, Yu JK, Hu X. Artificial neural networks analysis of surface-enhanced laser desorption/ionization mass spectra of serum protein pattern distinguishes colorectal cancer from healthy population. Clin Cancer Res. 2004;10:8380–8385. doi: 10.1158/1078-0432.CCR-1162-03. [DOI] [PubMed] [Google Scholar]
- Liu XP, Shen J, Li ZF, Yan L, Gu J. A serum proteomic pattern for the detection of colorectal adenocarcinoma using surface enhanced laser desorption and ionization mass spectrometry. Cancer Invest. 2006;24:747–753. doi: 10.1080/07357900601063873. [DOI] [PubMed] [Google Scholar]
- Hortin GL. The MALDI-TOF mass spectrometric view of the plasma proteome and peptidome. Clin Chem. 2006;52:1223–1237. doi: 10.1373/clinchem.2006.069252. [DOI] [PubMed] [Google Scholar]
- Navare A, Zhou M, McDonald J, Noriega FG, Sullards MC, Fernandez FM. Serum biomarker profiling by solid-phase extraction with particle-embedded micro tips and matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun Mass Spectrom. 2008;22:997–1008. doi: 10.1002/rcm.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho WC. Contribution of oncoproteomics to cancer biomarker discovery. Mol Cancer. 2007;6:25. doi: 10.1186/1476-4598-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]