

Abstract
Neurofibromatosis type 1 (NF1) is a common autosomal dominant genetic disorder caused by mutation of the NF1 tumor suppressor gene. Spinal deformities are common skeletal manifestations in patients with NF1. To date, the mechanism of vertebral abnormalities remains unclear because of the lack of appropriate animal models for the skeletal manifestations of NF1. In the present study, we report a novel murine NF1 model, Nf1flox/−;Col2.3Cre+ mice. These mice display short vertebral segments. In addition, a significant reduction in cortical and trabecular bone mass of the vertebrae was observed in Nf1flox/−;Col2.3Cre+ mice as measured by dual-energy X-ray absorptiometry (DEXA) and peripheral quantitative computed tomography (pQCT). Peak stress and peak load were also significantly reduced in Nf1flox/−;Col2.3Cre+ mice as compared to controls. Furthermore, the lumbar vertebrae showed enlargement of the inter-vertebral canal, a characteristic feature of lumbar vertebrae in NF1 patients. Finally, histologic analysis demonstrated increased numbers of osteoclasts and decreased numbers of osteoblasts in the vertebrae of Nf1flox/−;Col2.3Cre+ mice in comparison to controls. In summary, Nf1flox/−;Col2.3Cre+ mice demonstrate multiple structural and functional abnormalities in the lumbar vertebrae which recapitulate the dystrophic vertebral changes in NF1 patients. This novel murine model provides a platform to understand the cellular and molecular mechanisms underlying the pathogenesis of spinal deficits in NF1 patients.
Keywords: Neurofibromatosis type 1, Spinal deformity, Osteopathy, Osteoclast, Osteoblast
Introduction
NF1 is a common genetic disorder affecting 1 in every 3000–4000 individuals [1-5]. Inheritance is autosomal dominant, although approximately 50% of cases arise sporadically as new mutations [6]. Patients with NF1 present with a wide variety of malignant and non-malignant clinical manifestations, including plexiform neurofibromas, malignant peripheral nerve sheath tumors, and learning deficits [2-5,7-12]. Almost one century ago, both Gould and Weiss called attention to the high incidence of spinal deformities in patients with NF1 [13,14]. However, the true prevalence of spinal deformities in NF1 remains unknown, with values in the literature ranging from 2% to 69% [15-27]. Conversely, 2–3% of all scoliotic patients with significant curves have NF1 [28].
Spinal deformities in NF1 include dystrophic and nondystrophic vertebral body changes [29]. The radiographic appearance of nondystrophic deformities is no different from idiopathic deformities. Dystrophic changes include scalloping of the posterior vertebral margins, severe rotation of the apical vertebrae, vertebral wedging, short vertebral segments, widening of the spinal canal, enlargement of the neural foramina, osteoporosis, widened interpediculate distance, defective pedicles, presence of a paraspinal mass, spindling of the transverse process, and rotation of the ribs [30]. Erosive lesions have been described in the thoracic spine in NF1 cases with dysplastic meninges without evidence of associated tumors [13,14]. Dystrophic changes appearing in the adjacent bony elements could then be triggered by a locally active biochemical substance or hormone produced by neurofibromas.
Until now, little is known about vertebral biology in patients with NF1, due in part to a lack of relevant animal models. The Nf1 null mouse cannot be used for bone studies because embryos die before 13 days, because of a disruption in heart development associated with excessive proliferation and premature apoptosis of the endocardium [31]. Although several murine models of NF1 exhibit certain skeletal pathologies analogous to human NF1 patients [32-35], no relevant mouse model has been reported to show spinal deformities, a major skeletal defect in humans.
In the present study, we report a murine model that develops multiple spinal deficiencies associated with reduced localized bone mass. This murine model provides a foundation for investigating the cellular and molecular mechanism(s) underlying spinal deformities in NF1.
Materials and methods
Animals
Nf1+/− mice were obtained from Dr. Tyler Jacks at the Massachusetts Institute of Technology (Cambridge, MA) [31]. Nf1flox/flox mice were provided by Dr. Luis Parada at the University of Texas Southwestern Medical Center [7]. Col2.3Cre transgenic mice were generated as described elsewhere [36]. In this model, the 2.3 α1(I) Collagen promoter is used to selectively drive Cre expression in the osteoblast lineage. As further validation of this model, Col2.3Cre transgenic mice were crossed with ROSA26flox/flox reporter mice. LacZ staining was performed to assess Cre recombination in skeletal tissues. Abundant β-gal expression was observed in the long bones and vertebrae of ROSA26flox/flox;Col2.3Cre+ mice but absent in ROSA26flox/flox;Col2.3Cre− controls (Supplementary Fig. 1).
Four different genotypes of mice were generated with the intercross. (1) Nf1flox/flox;Col2.3Cre− mice (systemically wildtype, WT); (2) Nf1flox/−;Col2.3Cre− mice (Nf1+/− systemically); (3) Nf1flox/flox;Col2.3Cre+ mice (WT in hematopoietic lineages and Nf1−/− in osteoblasts); and (4) Nf1flox/−;Col2.3Cre+ mice (Nf1+/− in hematopoietic lineages and Nf1−/− in osteoblasts). PCR based genotyping was accomplished utilizing tail DNA and primers as described elsewhere (Supplementary Fig. 2) [31,37,38]. The successful deletion of neurofibromin in osteoblasts was confirmed by western blot with optimal exposure (top panel) and overexposure (middle panel) (Supplementary Fig. 3). All studies were approved by the Institutional Animal Care and Use Committee.
Longitudinal in vivo X-ray and peripheral dual-energy X-ray absorptiometry (pDEXA)
X-rays were taken using a piXarray 100 DSR System with Biopix Software (Version No: 1.0.6., Bioptics, Inc. Tucson, AZ). The axial bone was dissected after the mice were euthanized. Whole spine anterior–posterior radiographs were taken. To avoid the possible influence of transition of L6 in the lumbosacral region, the height of each L1–L5 vertebral body was measured between the cranial and caudal ends. To evaluate the bone mass in each genotype of mice, bone mineral density (BMD) of the axial bone was evaluated in vivo using pDEXA (PIXImus II; GE-Lunar Corp., Madison, WI) [39]. The mice were placed in a prone position on a specimen tray and scanned. The head was excluded from total body scans. The region of interest included the center of the whole axial bone and the L1–L5 lumbar region.
Peripheral quantitative computed tomography
Whole axial bone was placed in the gantry of a XCT Research SA + peripheral quantitative computed tomography (pQCT) scanner (Stratec Medizintechnik GmbH, Pforzheim, Germany) with Software Version 5.50. A voxel size of 70 μm was used for analysis [40]. A scout view was obtained and the anatomical reference line was positioned at the midpoint of the rostral/caudal margin of the L5 pedicle. Individual slices were obtained from each vertebra.
The centrum of the vertebra was selected as a region of interest (ROI). Volumetric bone mineral density (vBMD; mg/cm3) of the cortical and trabecular bone was measured. The ratio of spinal canal area/vertebral body area was derived by measuring the area of the L5 vertebral body and the spinal intervertebral canal from the pQCT images.
Biomechanical testing
To evaluate the structural integrity of L5 vertebrae, biomechanical testing was conducted utilizing Testresouse R systems with MtestwR software version: R 1.4.2 (TestResources Inc. Shakopee, MN). After removing all tissue, L5 vertebral bodies were isolated and prepared with flat and parallel cranial and caudal ends by removing only the soft cartilage (intervertebral disk) and exposing the bone surface. These vertebral bodies were subjected to mechanical testing in compression using a servo-controlled mechanical testing machine. All biomechanical tests were conducted at room temperature (RT) and at a rate of 20 mm/min. Peak load was defined as the maximum load that the specimen can withstand before fracturing. The cross sectional area of each vertebra was measured on a medial and lateral (transverse) plane. The peak stress was calculated by the equation: peak stress=peak load/cross sectional area of vertebra.
Isolation of osteoblast-like cells from lumbar vertebrae and osteoblast differentiation assays
Osteoblast-like cells were cultured from vertebrae as previously reported with minor modifications [41,42]. Briefly, one-month-old mice were sacrificed and vertebrae were harvested by depleting muscle and connective tissue. Individual vertebrae (L1–L5) were collected, minced, and marrow cells were collected by digestion of the minced tissue using type III collagenase (0.1 mg/mL), 2.5% trypsin, and shaking in a hot water bath at 37 °C for 40 min. The reaction was stopped by addition of 10 volumes of Dulbecco’s Modified Eagle Media (DMEM) supplemented with 10% fetal bovine serum (FBS). The cells were then washed once with DMEM, and aggregates were removed from the cell suspension by sieving through a 70 μm strainer. Cell number was determined by counting trypan blue negative cells.
To examine the frequency of osteoblast progenitors in the marrow of Nf1flox/−;Col2.3Cre+ lumbar vertebrae, colony forming units of osteoblasts (CFU-OBL) were assessed following ALP staining as previously described [43]. To evaluate the osteoblast differentiation in more detail, Alizarin staining was performed on the osteoblast differentiation cultures. In brief, mononuclear cells from lumbar vertebrae were cultured at 2×106/mL in osteogenic differentiation medium (MesenCult PLUS supplemented with 10−8 mol/L dexamethasone, 5 μg/mL ascorbic acid 2-phosphate, and 10 mmol/L β-glycerophosphate). After two weeks of culture, cells were washed with PBS and stained with 2% Alizarin Red S solution (pH=4.2) for 1 h. Then the cells were washed with distilled water and incubated with 500 μL of 1% hydrochloric acid in 70% ethanol and shaken on a plate rotator for 30 min. The extracted supernatant was collected and the optical density was measured at a wavelength of 450 nm. For each sample, measurements were performed in triplicate.
Von Kossa staining was used to evaluate the osteogenic differentiation of marrow cells from the vertebrae [44]. Briefly, after 4 weeks culture in osteoblast differentiation media in 6-well tissue culture plates, the cells were fixed with 10% neutral formalin. Freshly prepared 5% silver nitrate (2 mL) was added to each well, and then incubated under UV light for 60 min. The dishes were then rinsed with distilled water and fixed with 5% sodium thiosulfate for 2 min. The wells were photographed using a phase-contrast microscope, and the number of mineralized bone nodules was recorded.
Histological analysis and biohistochemistry
Histological analysis was conducted to evaluate the osteoclast and osteoblast distribution and frequency [45]. Briefly, 6 month old mice of each genotype were sacrificed. Lumbar vertebrae were harvested from each animal and fixed in 4% formaldehyde buffered solution at RT. The vertebrae were then decalcified in 10% ethylenediamine-tetraacetic acid (EDTA) in 4% formaldehyde buffered solution for 4 days. The tissues were then dehydrated in graded alcohols and embedded in paraffin, and longitudinal sections of the L5 vertebrae were obtained and processed for tartrate resistant acid phosphatase (TRACP) staining and MacNeal’s staining. To evaluate the osteoclasts in the vertebral sections, TRACP staining was performed. Briefly, vertebrae were fixed in 10% formaldehyde and processed for paraffin embedding. 5 μm thick sections were cut using a microtome (Nussloch GmbH, Germany) and the slides were incubated in 50 mL 0.2 M acetate buffer for 20 min at RT, transferred from pre-incubation to incubation media which contained 0.2 M acetate buffer (pH=5.0), AS-MX phosphate 0.5 mg/mL, and fast red TR Salt 1.1 mg/mL as previously described [46]. To evaluate the morphology and number of osteoblasts, the slides were stained in a 2% tetrachrome solution for 10 min. Osteoblasts in the L5 vertebrae were quantified following MacNeal’s staining. The quantification of osteoclast and osteoblast cell number was normalized to the bone surface in millimeters (mm), to control for the total amount of endosteal bone surface (cancellous bone surface + endocortical bone surface) within each histological section. To determine the neurofibromin expression in the osteoblasts at the protein level, western blot was performed [43].
Statistical analysis
Two-tailed Student’s t test and analysis of variance (ANOVA) were used to evaluate statistical difference. P values less than 0.05 were considered significant.
Results
Nf1flox/−;Col2.3Cre+ mice have short vertebrae
Shortened vertebral segments are one of the dystrophic features of NF1 related spinal deformities. To determine if Nf1flox/−;Col2.3Cre+ mice exhibited similar pathological findings, the height of the lumbar vertebrae was evaluated by X-ray. The average height of L5 was significantly less in Nf1flox/−;Col2.3Cre+ mice than in WT, Nf1+/−, and Nf1flox/flox;Col2.3Cre+ mice (Figs. 1A, B, *p<0.05), while there was no significant difference in the mean height of L1 to L5 between all genotypes of mice (data not shown). This result indicates that Nf1flox/−;Col2.3Cre+ mice recapitulate the shortened vertebral segment phenotype in NF1 patients.
Fig. 1.
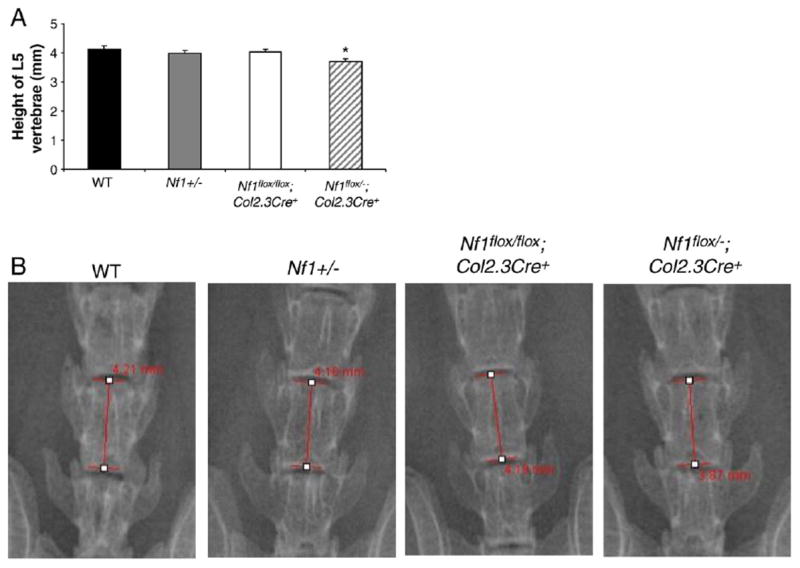
Short segmentation in Nf1flox/−;Col2.3Cre+ mice. (A) Average height (mm) of L5 is shown. Data represent the mean ± SEM of 9 mice in each genotype group. *p<0.01 for comparing the height of Nf1flox/−;Col2.3Cre+ with the other three groups of mice as measured by ANOVA and post-hoc t-tests corrected for multiple comparisons between Nf1flox/−;Col2.3Cre+ and the other genotypes. (B) Representative photographs of L5 vertebrae of four genotypes are shown.
Reduced BMD in the L1–L5 vertebrae of Nf1flox/−;Col2.3Cre+ mice
It has been reported that pediatric NF1 patients have reduced bone mass in the lumbar region [47]. To examine whether bone mass was altered in the experimental mice, DEXA was performed to assess the BMD in the spines of these mice. The muscle and soft tissue were removed from the axial bone to avoid the influence of surrounding tissue to the BMD. The BMD of both the entire axial bone and segments L1–L5 were evaluated separately in all four genotypes of mice (Fig. 2A). Although no difference in BMD was seen in the whole axial bone between the four genotypes of mice (Fig. 2B), a significant reduction in BMD was observed in the L1–L5 region of Nf1flox/−;Col2.3Cre+ mice (Figs. 2C–D, *p<0.05) as analyzed by ANOVA and post-hoc t-tests with correction for multiple comparisons. This result is consistent with the report by Kuorilehto and colleagues that the lowest local values of bone mineral density are located in the load-carrying parts of the body in patients with NF1 [17].
Fig. 2.
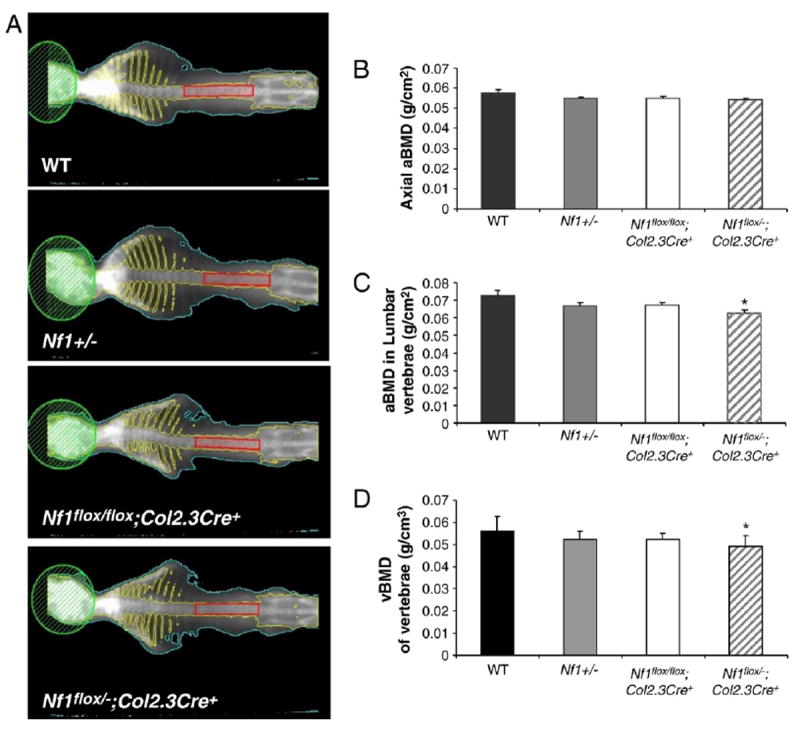
Reduced bone mineral density (BMD) in the L1–L5 vertebrae of Nf1flox/−;Col2.3Cre+ mice. (A) Representative photographs of DEXA. Skull area was excluded from the total axial bone scan. The region enclosed by the red rectangle includes the L1–L5 vertebrae. (B) Total axial BMD (g/cm2). Data represent the mean ± SEM of 9 mice in each genotype group. (C) BMD of the L1–L5 region. Data are the mean ± SEM of 9 mice in each genotype group. *p<0.05 for Nf1flox/−;Col2.3Cre+ mice vs. WT controls as determined by ANOVA and multiple-comparison post-hoc t-tests. (D) Lumbar vBMD of L1–L5 region. Data are the mean ± SEM of 9 mice in each genotype group. *p<0.05 for Nf1flox/−;Col2.3Cre+ mice vs. WT controls as determined by ANOVA and multiple-comparison post-hoc t-tests.
Nf1flox/−;Col2.3Cre+ mice have reduced cortical bone area and bone density
Given that the lumbar vertebrae are commonly affected in NF1 patients suffering from spinal deficits, and the L5 vertebrae of Nf1flox/−;Col2.3Cre+ mice was short and had reduced BMD, L5 was used to check the cortical bone density by pQCT. Since BMD varies according to the region of the vertebrae examined, the midpoint of the rostral/caudal pedicular margin was selected to insure that all pQCT images acquired were at the same level of the vertebrae for all samples. As shown in Fig. 3A, reduced cortical bone density was observed in L5 of the Nf1flox/flox;Col2.3Cre+ and Nf1flox/−;Col2.3Cre+ mice, while no change in the cortical bone mass was seen in the Nf1+/− mice. Interestingly, cortical bone area was significantly decreased in the L5 vertebrae of not only Nf1flox/−;Col2.3Cre+ mice, but also Nf1flox/flox;Col2.3Cre+ mice as compared to WT (Fig. 3B). In addition, a significant reduction in vBMD of trabecular bone was observed (Figs. 3C and D).
Fig. 3.
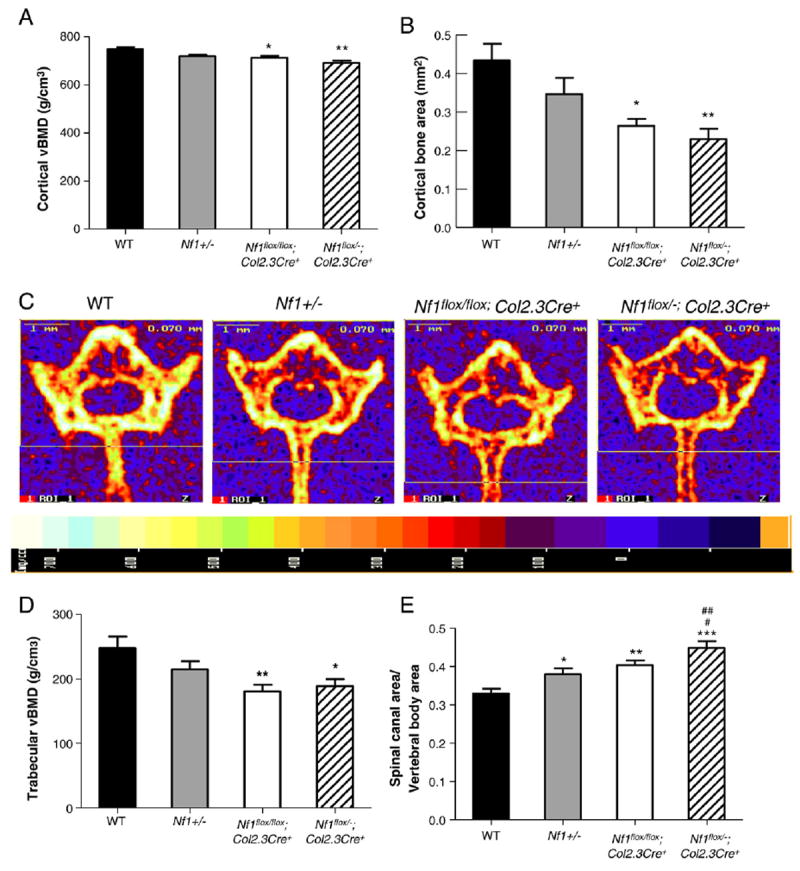
Nf1flox/−;Col2.3Cre+ mice have reduced cortical bone area and bone mineral density. (A) Cortical bone mineral density of L5 at the midpoint between the rostral/caudal margin of the pedicle in four different genotypes is shown. Data represent mean ± SEM of 9–10 mice per genotype. *p<0.05 Nf1flox/flox;Col2.3Cre+ vs WT. **p<0.001 Nf1flox/−;Col2.3Cre+ vs WT. (B). The cortical bone area in the L5 vertebral body was compared between the 4 groups of mice. Data represent mean ± SEM of 9–10 mice per genotype. *p<0.01 Nf1flox/flox;Col2.3Cre+ vs WT. **p<0.01 Nf1flox/−;Col2.3Cre+ vs WT. (C) Representative pQCT cross sections from L5 of four genotypes of mice. The relative intensity of bone is shown in the bottom panel. (D) The trabecular vBMD in the L5 vertebral body was compared between the 4 groups of mice. Data represent mean ± SEM of 9–10 mice per group. **p<0.01 Nf1flox/flox;Col2.3Cre+ vs WT. *p<0.05 Nf1flox/−;Col2.3Cre+ vs WT. (E) The ratio of spinal canal area/vertebral body area was compared between the 4 groups of mice. Data represented mean ± SEM of 9–10 mice per genotype. *p<0.05 Nf1+/− vs WT. **p<0.01 Nf1flox/flox;Col2.3Cre+ vs WT. ***p<0.001 Nf1flox/−;Col2.3Cre+ vs WT. #p<0.05 Nf1flox/flox;Col2.3Cre+ vs Nf1flox/−;Col2.3Cre−. ##p<0.01 Nf1flox/−;Col2.3Cre+ vs Nf1+/−.
One of the characteristics of NF1 related spinal deformities is the enlargement of the spinal canal. To examine whether the spinal canal was enlarged in vertebrae of Nf1flox/−;Col2.3Cre+ mice, the ratio of spinal canal area (C)/vertebral body area (V) (C/V ratio) was measured by pQCT (Fig. 3E). An increased C/V ratio indicates a comparatively larger area of the spinal canal. No significant difference in vertebral body area (V) was observed between the four groups of mice (data not shown). In contrast, a significant increase in the C/V ratio was found in Nf1+/−, Nf1flox/flox;Col2.3Cre+, and Nf1flox/−;Col2.3Cre+ mice as compared with WT mice (Fig. 3E). A further increase in the C/V ratio of L5 vertebrae from Nf1flox/−;Col2.3Cre+ mice was observed as compared to Nf1+/− and Nf1flox/flox;Col2.3Cre+ mice. This finding may be related to compression mediated by the cerebrospinal fluid pressure, which pushes outward against the weakened trabecular bone of the Nf1flox/−;Col2.3Cre+ vertebral body, leading to the enlargement of the spinal canal in vivo. The enlargement of the canal in the Nf1flox/−;Col2.3Cre+ mice is consistent with the observation that NF1 patients have enlarged spinal canals.
Nf1flox/−;Col2.3Cre+ vertebrae have reduced load bearing capacity
It has been reported that reduced bone mineral density in NF1 patients is most apparent in the load-carrying parts of the body, including the lumbar spine [17]. Since the L5 vertebrae of Nf1flox/−;Col2.3Cre+ mice showed reduced bone mass, we sought to evaluate whether the load bearing capacity of this segment was also affected by performing mechanical loading experiments to determine peak load to failure. Peak load to failure in the L5 vertebrae of Nf1+/−, Nf1flox/flox;Col2.3Cre+, and Nf1flox/−;Col2.3Cre+ mice was significantly reduced in comparison to WT mice (Fig. 4A, *p<0.05, **p<0.01). A similar pattern was observed in a peak stress to failure assay (Fig. 4B). Peak stress to failure of the L5 vertebra was lowest in Nf1flox/−;Col2.3Cre+ mice as compared to all other experimental groups, although L5 of both Nf1+/− mice and Nf1flox/flox;Col2.3Cre+ mice showed significantly reduced peak stress to failure than WT mice (*p<0.05). The greatest reduction in L5 peak stress observed in the Nf1flox/−;Col2.3Cre+ mice may be associated with the fact that these mice also exhibit the most profound reduction in L5 BMD as described above.
Fig. 4.
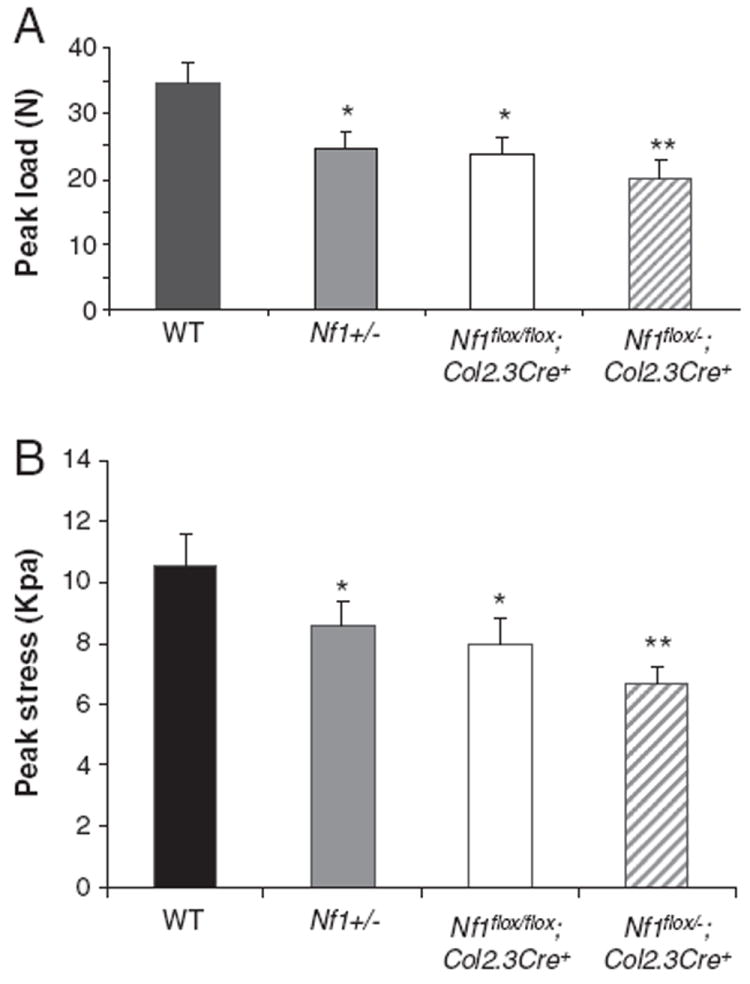
Quantitative evaluation of L5 vertebral peak load. (A) Comparison of peak load to failure (N) in L5 vertebra between four genotypes. *p<0.01 for comparing Nf1+/− or Nf1flox/flox;Col2.3Cre+ mice with WT controls. **p<0.001 for comparing Nf1flox/−;Col2.3Cre+ mice with WT mice. (B) Comparison of peak stress (Kpa) of L5 vertebrae between four genotypes. *p<0.01 for comparing Nf1+/− or Nf1flox/flox;Col2.3Cre+ mice with WT controls. **p<0.001 for comparing Nf1flox/−;Col2.3Cre+ mice with WT mice.
Histological analysis of L5
Osteoclasts and osteoblasts are the two major cell lineages responsible for maintaining skeletal homeostasis. The number and morphology of these lineages was evaluated quantitatively by histological analysis. In contrast to bipeds, quadrupeds, including mice, bear stress loading with the upper facet process. Thus, we chose the upper facet of L5 as the area of interest for histological examination. Significantly increased osteoclast number per mm of bone surface was observed in the upper facet process of Nf1+/− and Nf1flox/−;Col2.3Cre+ mice compared with WT mice and Nf1flox/flox;Col2.3Cre+ mice (Figs. 5A, B). In addition, significantly reduced osteoblast number per mm of bone surface was observed in the upper facet process of Nf1flox/−;Col2.3Cre+ mice than in the other three genotypes of mice (Figs. 5C, D). The histological changes in the upper facet process of L5 are consistent with the reduced BMD of this region.
Fig. 5.
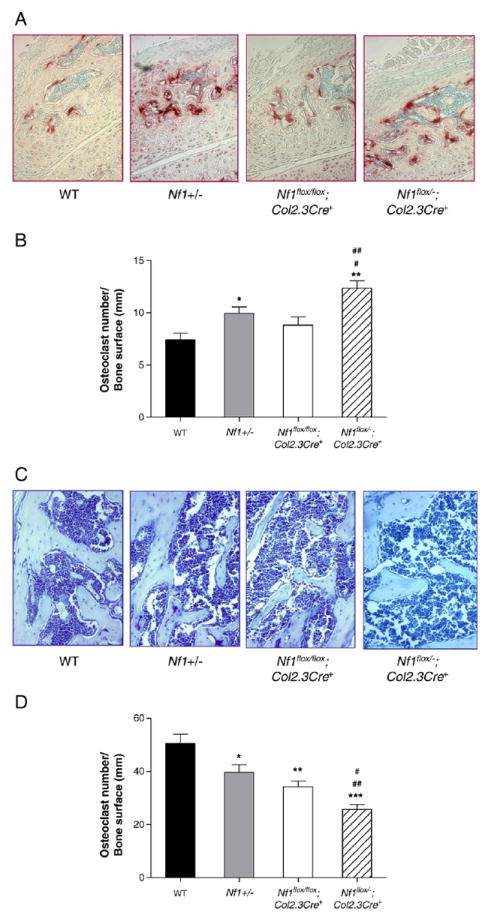
Nf1flox/−;Col2.3Cre+ mice have increased osteoclast number and reduced osteoblast number per mm of bone surface in the upper facet processes. (A) Representative photomicrographs (magnification, 200×) of the upper facet processes of four genotypes of mice following TRACP staining. (B) Mean osteoclast number per mm of bone surface was quantified per 200× field. Data represent mean ± SEM of 5 fields for each genotype. 5 mice were used for each group. *p<0.01 Nf1+/− vs WT. **p<0.001 Nf1flox/−;Col2.3Cre+ vs WT. #p<0.05 Nf1flox/−;Col2.3Cre+ vs Nf1+/−. ##p<0.01 Nf1flox/−;Col2.3Cre+ vs Nf1flox/flox;Col2.3Cre+. (C) Representative photomicrographs (magnification, 200×) of the upper facet processes of four genotypes of mice following MacNeal’s staining. (D) Quantitative evaluation of the osteoblast number per mm of bone surface. Data represent mean ± SEM of 5 high power fields per mouse and 5 mice were used in each group. *p<0.01 Nf1+/− vs WT. **p<0.01 Nf1flox/flox;Col2.3Cre+ vs WT. ***p<0.001 Nf1flox/−;Col2.3Cre+ vs WT. #p<0.05 Nf1flox/flox;Col2.3Cre+ vs Nf1flox/−;Col2.3Cre−. ##p<0.01 Nf1flox/−;Col2.3Cre+ vs Nf1+/−.
Nullizygosity of Nf1 impairs osteoblast differentiation in vitro
Previous studies have shown that Nf1 haploinsufficiency (Nf1+/−) results in reduced osteoblast differentiation as measured by alkaline phosphate activity [39,43]. To examine the frequency of pre-osteoblasts in the marrow of vertebrae, one-month-old vertebrae were harvested and flushed thoroughly with culture media. The bone marrow mononuclear cells were plated on plastic tissue culture dishes and supplemented with osteogenic medium for the CFU-OBL assay. BMMNCs cultured from Nf1flox/−;Col2.3Cre+ mice demonstrated significantly reduced CFU-OBL forming capacity as compared to controls (Fig. 6A). To examine if osteoblast mineral deposition was deficient in vitro, osteoblasts were harvested from the vertebrae and osteoblast differentiation was evaluated by Alizarin and von Kossa staining. Cultures collected from WT vertebrae displayed strong Alizarin staining, whereas reduced Alizarin staining was observed in Nf1+/−, Nf1flox/flox;Col2.3Cre+, as well as in Nf1flox/−;Col2.3Cre+ cultures in a gene dosage dependent fashion (Fig. 6B). Von Kossa staining was also used to measure mineral deposition in vitro. A significant reduction in the number of mineralized bone nodules was observed in osteoblasts cultured from Nf1+/−, Nf1flox/flox;Col2.3Cre+, as well as Nf1flox/−;Col2.3Cre+ mice as compared to WT mice (Fig. 6C). Consistent with previous studies [39,43], a reduced osteoblast differentiation was observed in Nf1+/− cultures. Furthermore, a markedly impaired osteoblast differentiation was shown in the Nf1flox/flox;Col2.3Cre+ and Nf1flox/−;Col2.3Cre+ cultures, suggesting a gene dosage dependent reduction in osteoblast differentiation.
Fig. 6.
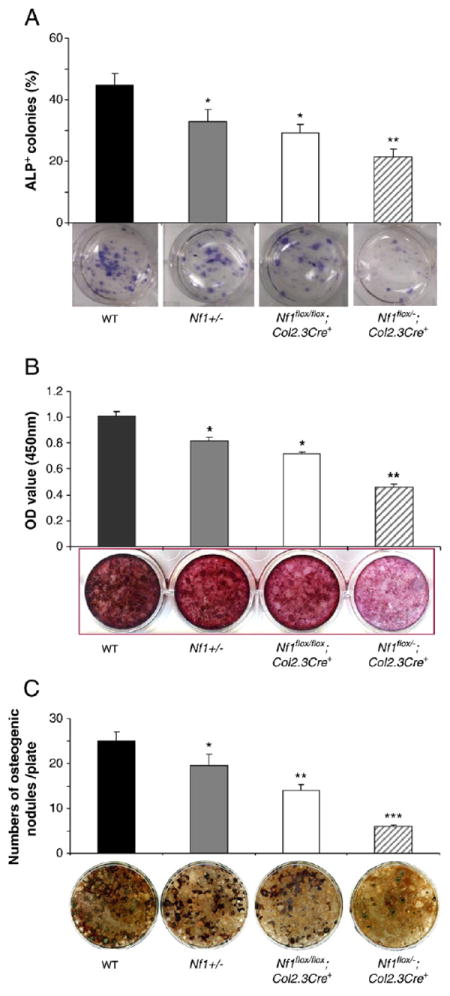
Impaired osteoblast differentiation in Nf1 deficient mice in vitro. (A) Quantitative evaluation of ALP+ colony formation in primary osteoblast cultures from four genotypes of mice. Data represent mean ± SEM of triplicate cultures from each mouse. Three mice were used for each experiment per genotype. Three independent experiments were conducted with similar results. *p<0.05 for comparing Nf1+/− vs. WT mice, or Nf1flox/flox;Col2.3Cre+ mice vs. WT mice, and **p<0.01 for comparing Nf1flox/−;Col2.3Cre+ mice vs. three other groups of mice. The lower panel shows representative photographs of each culture. (B) Quantitative evaluation of osteoblast differentiation following Alizarin staining. Data represent the mean ± SEM of triplicate cultures per mouse and three mice were used per condition per genotype. Three independent experiments were conducted. *p<0.05 for comparing Nf1+/− with WT mice, or Nf1flox/flox;Col2.3Cre+ mice vs. WT mice, and **p<0.01 for comparing Nf1flox/−;Col2.3Cre+ mice vs. the other three groups of mice. (C) Quantitative evaluation of osteogenic nodules of four genotypes of mice following von Kossa staining. Data represent the mean ± SEM of triplicate cultures, and three mice were used in each group. Three independent experiments were conducted with similar results. *p<0.05 for comparing Nf1+/− vs. WT mice, **p<0.01 for comparing Nf1flox/flox;Col2.3Cre+ mice vs. WT mice, and ***p<0.001 for comparing Nf1flox/−;Col2.3Cre+ mice vs. the other three groups of mice. Representative photomicrographs of osteogenic nodules are shown in the lower panel.
To determine the neurofibromin expression in osteoblasts at the protein level, western blot was performed. As shown in Supplemental Fig. 3, neurofibromin is strongly expressed in wildtype osteoblasts. In contrast, neurofibromin expression was undetectable in Nf1flox/− and Nf1flox/flox;Col2.3Cre+ osteoblasts. The reduced osteoblast differentiation in Nf1flox/flox;Col2.3Cre+ cultures differs from the results of a previous study [32]. In that study, no difference in osteoblast differentiation was observed in the Nf1flox/flox;Col2.3Cre+ cultures established from calvaria. These disparate results from two independent laboratories may be explained by the different tissue sources of the osteoblast cultures. The most deficient osteoblast differentiation was observed in Nf1flox/−;Col2.3Cre+ cultures and may reflect the in vivo impact of the WT vs. Nf1+/− environment.
Discussion
Spinal deformities, common osseous deficits in NF1, have traditionally been classified as either dystrophic deformities, characterized by a rapid course of progression, or non-dystrophic deformities [48,49]. Among them, vertebral deformities are the most frequent dystrophic features, including anterior, lateral and posterior scalloping, wedging, shortened vertebral segments, osteoporosis and severe rotation of the apical vertebrae [29]. However, no animal model for spinal deformity in NF1 has been reported. In the present study, utilizing a genetic intercross approach, we have established that Nf1flox/−;Col2.3Cre+ mice carrying both Nf1 nullizygous (Nf1−/−) osteoblasts and Nf1 haploinsufficient (Nf1+/−) osteoclasts acquire multiple vertebral anomalies, such as shortened vertebral segments, reduced bone mass in the lumbar vertebrae, widening of the spinal canal, and impaired mechanical integrity including reduced peak load and peak stress.
Currently, the pathogenesis of vertebral osteopathy in NF1 remains unclear. Clinically, it is known that the severity and progression of spinal deformities is associated with the characteristic morphologic changes of vertebrae [13,14,50]. However, it is debated whether spinal deformities in NF1 are primary osseous defects or secondary to the pressure of an adjacent paraspinal tumor [51,52]. In the present study, we show that Nf1flox/−;Col2.3Cre+ mice display reduced bone mass, increased osteoclast formation and reduced osteoblast numbers in the vertebrae, thereby indicating that a primary osseous deficit in the spinal vertebrae exists. Given the fact that Col2.3Cre mediated recombination occurs specifically in osteoblasts, the Nf1flox/−;Col2.3Cre+ mice have no undue influences of increased tumor formation such as that seen in Nf1flox/−;Krox20Cre mice [7].
It has been previously reported that haploinsufficiency in non-tumorigenic lineages is required for neurofibroma development [7]. We also reported that haploinsufficiency of Nf1 in the hematopoietic microenvironment plays a critical role in plexiform neurofibroma formation [53]. In the present study, we report that a combination of Nf1 nullizygous osteoblastic cells and Nf1 haploinsufficient cells (including osteoclasts) are sufficient for the development of vertebral anomalies in the NF1 murine model, Nf1flox/−;Col2.34Cre+ mice. While homozygous loss of the Nf1 tumor suppressor gene together with haploinsufficiency of Nf1 in the microenvironment has recently been shown to be required for the malignant manifestations of NF1 [7,53-55], our present study provides evidence that a similar paradigm exists in the development of NF1-related vertebral anomalies. Given our previous report that haploinsufficiency of Nf1 results in increased bone resorptive activity by osteoclasts [32,46,56,57], it is likely that this lineage is the key effector of the microenvironment. Further investigation is ongoing to dissect the specific lineage(s) in the microenvironment that contribute to the skeletal deficits.
The vertebrae in quadrupedal animals do not receive an axial load to maintain balance of the torso, which is the major loading site for bipedal human locomotion [58]. As the mechanical circumstance is quite different in the bipedal condition from the quadrupedal condition, it is reasonable that the animal model does not display some spinal deformities, including characteristic scalloping of vertebrae and kyphoscoliosis, which are major spinal deformities in NF1 patients [30]. Despite such limitations, Nf1flox/−;Col2.3Cre+ mice still exhibit reduced cortical and trabecular bone mass, and a relatively larger L5 spinal canal relative to controls. Furthermore, Nf1flox/−;Col2.3Cre+ mice showed dramatically reduced peak load and peak stress to failure in the L5 vertebrae as compared to control mice. The alteration in mechanical load bearing capacity is likely a direct consequence of the intrinsic pathological Nf1flox/−;Col2.3Cre+ vertebral architecture. Casselman and colleagues speculated that NF1 bone is intrinsically more susceptible to the distorting effects of an adjacent tumor [59]. In fact, patients with intraspinal tumors who are not affected with NF1 do not exhibit similar dystrophic skeletal abnormalities [60]. Our study suggests that the dystrophic vertebral changes in humans may be secondary to different mechanical circumstances.
The remodeling of mammalian bone is an active, continuous process that requires a balance between osteoclasts and osteoblasts in order to maintain homeostasis. It remains controversial whether these bony changes represent primary osseous dysplasia or secondary responses to neurofibromas within the intervertebral foramina or adjacent tissues. Here, we show that deficiency of Nf1 results in increased osteoclast formation and decreased osteoblast development in the spine both in vivo and in vitro. The primary pathological defects in these cell lineages may explain the rapid development of spinal deformities, high complications of instrumentation and progression after operation, and high rate of pseudoarthrosis after spinal fusion, which are the specific clinical phenotypic changes in NF1 [61,62].
Conclusion
In summary, our present NF1 murine model provides strong evidence that primary cellular defects exist in NF1, which may be the cause for multiple spinal deformities in NF1 patients. This model provides a tool to further investigate the molecular mechanism(s) underlying the primary osteopathy in NF1 vertebrae.
Supplementary Material
Acknowledgments
We thank Susan Stanley for administrative support. This work was supported in part by the Department of Defense (DOD) NF043032 (FCY), NF073112 (FCY) and NIH/NCI/5R01CA074177 (FCY and DWC). SD Rhodes was supported in part by an Indiana CTSI Career Development Award (PHS (NCCR) Grant No: 5TL1RR025759-03).
Abbreviations
- NF1
Neurofibromatosis type 1
- PDEXA
Peripheral dual-energy X-ray absorptiometry
- BMD
Bone mineral density
- pQCT
Peripheral quantitative computed tomography
- ROI
Region of interest
- vBMD
Volumetric bone mineral density
- L
Lumbar
- CFU-OBL
Colony forming units of osteoblast
- BMMNCs
Bone marrow mononuclear cells
- DMEM
Dulbecco’s Modified Eagle Media
- ALP
Alkaline phosphatase
- UV
Ultraviolet
- EDTA
Ethylene diamine tetraacetic acid
- TRACP
Tartrate resistant acid phosphatase
- RT
Room temperature
- WT
Wild type
- C/V ratio
Spinal canal area (C)/vertebral body area (V)
- ANOVA
Analysis of Variance
Footnotes
Supplementary materials related to this article can be found online at doi:10.1016/j.bone.2011.03.760.
References
- 1.Wallace MR, Marchuk DA, Andersen LB, Letcher R, Odeh HM, Saulino AM, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249:181–6. doi: 10.1126/science.2134734. [DOI] [PubMed] [Google Scholar]
- 2.Atit RP, Mitchell K, Nguyen L, Warshawsky D, Ratner N. The neurofibromatosis type 1 (Nf1) tumor suppressor is a modifier of carcinogen-induced pigmentation and papilloma formation in C57BL/6 mice. J Invest Dermatol. 2000;114:1093–100. doi: 10.1046/j.1523-1747.2000.00994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atit RP, Crowe MJ, Greenhalgh DG, Wenstrup RJ, Ratner N. The Nf1 tumor suppressor regulates mouse skin wound healing, fibroblast proliferation, and collagen deposited by fibroblasts. J Invest Dermatol. 1999;112:835–42. doi: 10.1046/j.1523-1747.1999.00609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bajenaru ML, Hernandez MR, Perry A, Zhu Y, Parada LF, Garbow JR, et al. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. 2003;63:8573–7. [PubMed] [Google Scholar]
- 5.Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell. 2001;104:593–604. doi: 10.1016/s0092-8674(01)00245-8. [DOI] [PubMed] [Google Scholar]
- 6.Cawthon RM, Weiss R, Xu GF, Viskochil D, Culver M, Stevens J, et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell. 1990;62:193–201. doi: 10.1016/0092-8674(90)90253-b. [DOI] [PubMed] [Google Scholar]
- 7.Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002;296:920–2. doi: 10.1126/science.1068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ajuebor MN, Hogaboam CM, Kunkel SL, Proudfoot AE, Wallace JL. The chemokine RANTES is a crucial mediator of the progression from acute to chronic colitis in the rat. J Immunol. 2001;166:552–8. doi: 10.4049/jimmunol.166.1.552. [DOI] [PubMed] [Google Scholar]
- 9.Rutkowski JL, Wu K, Gutmann DH, Boyer PJ, Legius E. Genetic and cellular defects contributing to benign tumor formation in neurofibromatosis type 1. Hum Mol Genet. 2000;9:1059–66. doi: 10.1093/hmg/9.7.1059. [DOI] [PubMed] [Google Scholar]
- 10.Ingram DA, Yang FC, Travers JB, Wenning MJ, Hiatt K, New S, et al. Genetic and biochemical evidence that haploinsufficiency of the Nf1 tumor suppressor gene modulates melanocyte and mast cell fates in vivo. J Exp Med. 2000;191:181–8. doi: 10.1084/jem.191.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang FC, Ingram DA, Chen S, Hingtgen CM, Ratner N, Monk KR, et al. Neurofibromin-deficient Schwann cells secrete a potent migratory stimulus for Nf1+/− mast cells. J Clin Invest. 2003;112:1851–61. doi: 10.1172/JCI19195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci U S A. 2005;102:8573–8. doi: 10.1073/pnas.0503224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gould E. The bone changes occuring in Von Recklinghausen’s disease. Q J Med. 1918;11:221–8. [Google Scholar]
- 14.Weiss R. (A) Von Recklinghausen’s disease in the Negro; (B) curvature of the spine in von Recklinghausen’s disease. Arch Dermatol Syphilol. 1921;3:144–51. [Google Scholar]
- 15.Kuorilehto T, Kinnunen P, Nissinen M, Alanne M, Leskela HV, Lehenkari P, et al. Vasculopathy in two cases of NF1-related congenital pseudarthrosis. Pathol Res Pract. 2006;202:687–90. doi: 10.1016/j.prp.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 16.Kuorilehto T, Nissinen M, Koivunen J, Benson MD, Peltonen J. NF1 tumor suppressor protein and mRNA in skeletal tissues of developing and adult normal mouse and NF1-deficient embryos. J Bone Miner Res. 2004;19:983–9. doi: 10.1359/JBMR.040130. [DOI] [PubMed] [Google Scholar]
- 17.Kuorilehto T, Poyhonen M, Bloigu R, Heikkinen J, Vaananen K, Peltonen J. Decreased bone mineral density and content in neurofibromatosis type 1: lowest local values are located in the load-carrying parts of the body. Osteoporos Int. 2005;16:928–36. doi: 10.1007/s00198-004-1801-4. [DOI] [PubMed] [Google Scholar]
- 18.Lammert M, Kappler M, Mautner VF, Lammert K, Storkel S, Friedman JM, et al. Decreased bone mineral density in patients with neurofibromatosis 1. Osteoporos Int. 2005;16:1161–6. doi: 10.1007/s00198-005-1940-2. [DOI] [PubMed] [Google Scholar]
- 19.Jacques C, Dietemann JL. Imaging features of neurofibromatosis type 1. J Neuroradiol. 2005;32:180–97. doi: 10.1016/s0150-9861(05)83136-0. [DOI] [PubMed] [Google Scholar]
- 20.Illes T, Halmai V, de Jonge T, Dubousset J. Decreased bone mineral density in neurofibromatosis-1 patients with spinal deformities. Osteoporos Int. 2001;12:823–7. doi: 10.1007/s001980170032. [DOI] [PubMed] [Google Scholar]
- 21.Heuze Y, Piot B, Mercier J. Difficult surgical management of facial neurofibromatosis type I or von Recklinghausen disease in children. Rev Stomatol Chir Maxillofac. 2002;103:105–13. [PubMed] [Google Scholar]
- 22.Stevenson DA, Moyer-Mileur LJ, Murray M, Slater H, Sheng X, Carey JC, et al. Bone mineral density in children and adolescents with neurofibromatosis type 1. J Pediatr. 2007;150:83–8. doi: 10.1016/j.jpeds.2006.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stevenson DA, Zhou H, Ashrafi S, Messiaen LM, Carey JC, D’Astous JL, et al. Double inactivation of NF1 in tibial pseudarthrosis. Am J Hum Genet. 2006;79:143–8. doi: 10.1086/504441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riccardi VM. Von Recklinghausen neurofibromatosis. N Engl J Med. 1981;305:1617–27. doi: 10.1056/NEJM198112313052704. [DOI] [PubMed] [Google Scholar]
- 25.Crawford AH, Jr, Bagamery N. Osseous manifestations of neurofibromatosis in childhood. J Pediatr Orthop. 1986;6:72–88. doi: 10.1097/01241398-198601000-00015. [DOI] [PubMed] [Google Scholar]
- 26.Crawford AH. Pitfalls of spinal deformities associated with neurofibromatosis in children. Clin Orthop Relat Res. 1989:29–42. [PubMed] [Google Scholar]
- 27.Overweg-Plandsoen WC, Weersink RG, Sillevis Smitt JH, Fleury P, van Asperen CJ. Neurofibromatosis type 1: a survey of 195 patients. Ned Tijdschr Geneeskd. 1997;141:624–9. [PubMed] [Google Scholar]
- 28.Garfin SR. Cervical Spine Research Society Presidential Address: a 25-year correlation with history. Spine (Phila Pa 1976) 1997;22:2589–97. doi: 10.1097/00007632-199711150-00001. [DOI] [PubMed] [Google Scholar]
- 29.Tsirikos AI, Ramachandran M, Lee J, Saifuddin A. Assessment of vertebral scalloping in neurofibromatosis type 1 with plain radiography and MRI. Clin Radiol. 2004;59:1009–17. doi: 10.1016/j.crad.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 30.Alwan S, Armstrong L, Joe H, Birch PH, Szudek J, Friedman JM. Associations of osseous abnormalities in Neurofibromatosis 1. Am J Med Genet A. 2007;143A:1326–33. doi: 10.1002/ajmg.a.31754. [DOI] [PubMed] [Google Scholar]
- 31.Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet. 1994;7:353–61. doi: 10.1038/ng0794-353. [DOI] [PubMed] [Google Scholar]
- 32.Elefteriou F, Benson MD, Sowa H, Starbuck M, Liu X, Ron D, et al. ATF4 mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasiae. Cell Metab. 2006;4:441–51. doi: 10.1016/j.cmet.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kolanczyk M, Kossler N, Kuhnisch J, Lavitas L, Stricker S, Wilkening U, et al. Multiple roles for neurofibromin in skeletal development and growth. Hum Mol Genet. 2007;16:874–86. doi: 10.1093/hmg/ddm032. [DOI] [PubMed] [Google Scholar]
- 34.Kuorilehto T, Ekholm E, Nissinen M, Hietaniemi K, Hiltunen A, Paavolainen P, et al. NF1 gene expression in mouse fracture healing and in experimental rat pseudarthrosis. J Histochem Cytochem. 2006;54:363–70. doi: 10.1369/jhc.5A6784.2005. [DOI] [PubMed] [Google Scholar]
- 35.Schindeler A, Ramachandran M, Godfrey C, Morse A, McDonald M, Mikulec K, et al. Modeling bone morphogenetic protein and bisphosphonate combination therapy in wild-type and Nf1 haploinsufficient mice. J Orthop Res. 2008;26:65–74. doi: 10.1002/jor.20481. [DOI] [PubMed] [Google Scholar]
- 36.Dacquin R, Starbuck M, Schinke T, Karsenty G. Mouse alpha1(I)-collagen promoter is the best known promoter to drive efficient Cre recombinase expression in osteoblast. Dev Dyn. 2002;224:245–51. doi: 10.1002/dvdy.10100. [DOI] [PubMed] [Google Scholar]
- 37.Liu F, Woitge HW, Braut A, Kronenberg MS, Lichtler AC, Mina M, et al. Expression and activity of osteoblast-targeted Cre recombinase transgenes in murine skeletal tissues. Int J Dev Biol. 2004;48:645–53. doi: 10.1387/ijdb.041816fl. [DOI] [PubMed] [Google Scholar]
- 38.Kwon CH, Zhao D, Chen J, Alcantara S, Li Y, Burns DK, et al. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res. 2008;68:3286–94. doi: 10.1158/0008-5472.CAN-07-6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu X, Chen S, Potter OL, Murthy SM, Li J, Pulcini JM, et al. Neurofibromin and its inactivation of Ras are prerequisites for osteoblast functioning. Bone. 2005;36:793–802. doi: 10.1016/j.bone.2005.01.022. [DOI] [PubMed] [Google Scholar]
- 40.Koller DL, Liu L, Alam I, Sun Q, Econs MJ, Foroud T, et al. Epistasis between QTLs for bone density variation in Copenhagen x dark agouti F2 rats. Mamm Genome. 2009;20:180–6. doi: 10.1007/s00335-008-9161-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Milne M, Kang MI, Quail JM, Baran DT. Thyroid hormone excess increases insulin-like growth factor I transcripts in bone marrow cell cultures: divergent effects on vertebral and femoral cell cultures. Endocrinology. 1998;139:2527–34. doi: 10.1210/endo.139.5.5992. [DOI] [PubMed] [Google Scholar]
- 42.Ishida Y, Heersche JN. Progesterone- and dexamethasone-dependent osteoprogenitors in bone cell populations derived from rat vertebrae are different and distinct. Endocrinology. 1999;140:3210–8. doi: 10.1210/endo.140.7.6850. [DOI] [PubMed] [Google Scholar]
- 43.Wu X, Estwick SA, Chen S, Yu M, Ming W, Nebesio TD, et al. Neurofibromin plays a critical role in modulating osteoblast differentiation of mesenchymal stem/progenitor cells. Hum Mol Genet. 2006;15:2837–45. doi: 10.1093/hmg/ddl208. [DOI] [PubMed] [Google Scholar]
- 44.Ge Y, Chen D, Xie L, Zhang R. Enhancing effect of daidzein on the differentiation and mineralization in mouse osteoblast-like MC3T3-E1 cells. Yakugaku Zasshi. 2006;126:651–6. doi: 10.1248/yakushi.126.651. [DOI] [PubMed] [Google Scholar]
- 45.Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- 46.Yang FC, Chen S, Robling AG, Yu X, Nebesio TD, Yan J, et al. Hyperactivation of p21 and PI3K cooperate to alter murine and human neurofibromatosis type 1-haploinsufficient osteoclast functions. J Clin Invest. 2006;116:2880–91. doi: 10.1172/JCI29092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yilmaz K, Ozmen M, Bora Goksan S, Eskiyurt N. Bone mineral density in children with neurofibromatosis 1. Acta Paediatr. 2007;96:1220–2. doi: 10.1111/j.1651-2227.2007.00401.x. [DOI] [PubMed] [Google Scholar]
- 48.Crawford AH, Herrera-Soto J. Scoliosis associated with neurofibromatosis. Orthop Clin North Am. 2007;38:553–62. vii. doi: 10.1016/j.ocl.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 49.Crawford AH, Schorry EK. Neurofibromatosis update. J Pediatr Orthop. 2006;26:413–23. doi: 10.1097/01.bpo.0000217719.10728.39. [DOI] [PubMed] [Google Scholar]
- 50.Kwok ES, Sawatzky B, Birch P, Friedman JM, Tredwell SJ. Vertebral scalloping in neurofibromatosis type 1: a quantitative approach. Can J Surg. 2002;45:181–4. [PMC free article] [PubMed] [Google Scholar]
- 51.Vitale MG, Guha A, Skaggs DL. Orthopaedic manifestations of neurofibromatosis in children: an update. Clin Orthop Relat Res. 2002:107–18. doi: 10.1097/00003086-200208000-00013. [DOI] [PubMed] [Google Scholar]
- 52.Funasaki H, Winter RB, Lonstein JB, Denis F. Pathophysiology of spinal deformities in neurofibromatosis. An analysis of seventy-one patients who had curves associated with dystrophic changes. J Bone Joint Surg Am. 1994;76:692–700. doi: 10.2106/00004623-199405000-00010. [DOI] [PubMed] [Google Scholar]
- 53.Yang FC, Ingram DA, Chen S, Zhu Y, Yuan J, Li X, et al. Nf1-dependent tumors require a microenvironment containing Nf1+/−- and c-kit-dependent bone marrow. Cell. 2008;135:437–48. doi: 10.1016/j.cell.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Daginakatte GC, Gianino SM, Zhao NW, Parsadanian AS, Gutmann DH. Increased c-Jun-NH2-kinase signaling in neurofibromatosis-1 heterozygous microglia drives microglia activation and promotes optic glioma proliferation. Cancer Res. 2008;68:10358–66. doi: 10.1158/0008-5472.CAN-08-2506. [DOI] [PubMed] [Google Scholar]
- 55.Daginakatte GC, Gutmann DH. Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1-deficient astrocyte and glioma growth. Hum Mol Genet. 2007;16:1098–112. doi: 10.1093/hmg/ddm059. [DOI] [PubMed] [Google Scholar]
- 56.Yan J, Chen S, Zhang Y, Li X, Li Y, Wu X, et al. Rac1 mediates the osteoclast gains-in-function induced by haploinsufficiency of Nf1. Hum Mol Genet. 2008;17:936–48. doi: 10.1093/hmg/ddm366. [DOI] [PubMed] [Google Scholar]
- 57.Li H, Liu Y, Zhang Q, Jing Y, Chen S, Song Z, et al. Ras dependent paracrine secretion of osteopontin by Nf1+/− osteoblasts promote osteoclast activation in a neurofibromatosis type I murine model. Pediatr Res. 2009 doi: 10.1203/PDR.0b013e3181a1c607. [DOI] [PubMed] [Google Scholar]
- 58.Goff CW, Landmesser W. Bipedal rats and mice; laboratory animals for orthopaedic research. J Bone Joint Surg Am. 1957;39-A:616–22. [PubMed] [Google Scholar]
- 59.Casselman ES, Mandell GA. Vertebral scalloping in neurofibromatosis. Radiology. 1979;131:89–94. doi: 10.1148/131.1.89. [DOI] [PubMed] [Google Scholar]
- 60.Alwan S, Tredwell SJ, Friedman JM. Is osseous dysplasia a primary feature of neurofibromatosis 1 (NF1)? Clin Genet. 2005;67:378–90. doi: 10.1111/j.1399-0004.2005.00410.x. [DOI] [PubMed] [Google Scholar]
- 61.Abdel-Wanis ME, Kawahara N. The role of neurofibromin and melatonin in pathogenesis of pseudarthrosis after spinal fusion for neurofibromatous scoliosis. Med Hypotheses. 2002;58:395–8. doi: 10.1054/mehy.2001.1467. [DOI] [PubMed] [Google Scholar]
- 62.Ramachandran M, Tsirikos AI, Lee J, Saifuddin A. Whole-spine magnetic resonance imaging in patients with neurofibromatosis type 1 and spinal deformity. J Spinal Disord Tech. 2004;17:483–91. doi: 10.1097/01.bsd.0000133466.97241.50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.