

Summary
Diabetes results in vascular changes and dysfunction, and vascular complications are the leading cause of morbidity and mortality in diabetic patients. There has been a continual increase in the number of diabetic nephropathy patients and epidemic increases in the number of patients progressing to end-stage renal diseases. To identify targets for therapeutic intervention, most studies have focused on understanding how abnormal levels of glucose metabolites cause diabetic nephropathy, which is of paramount importance in devising strategies to combat the development and progression of diabetic nephropathy. However, less studied than the systemic toxic mechanisms, hyperglycemia and dyslipidemia might inhibit the endogenous vascular protective factors such as insulin, vascular endothelial growth factor, and platelet-derived growth factor. In this review, we highlight the importance of enhancing endogenous protective factors to prevent or delay diabetic nephropathy.
Keywords: Hyperglycemia, protein kinase C (PKC)β, advanced glycation end products (AGEs), platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF)
Diabetic nephropathy (DN) is the leading cause of end-stage renal disease in the world. Despite the many advances in the clinical management of DN, the number of cases of diabetic nephropathy continues to increase, in part, owing to a pandemic increase of people with diabetes. Two large studies, the Diabetes Control and Complications Trial in type 1 diabetes and the United Kingdom Prospective Diabetes Study in type 2 diabetes, showed that intensive blood glucose control delays the onset and the progression of diabetic microvascular complications including nephropathy.1,2 These clinical intervention studies strongly suggest that hyperglycemia is the major factor responsible for the pathogenesis of DN. In addition to hyperglycemia, enhanced activation of the renin-angiotensin-aldosterone system is thought to contribute significantly to the pathogenesis of DN, as evidenced by numerous studies showing that renin-angiotensin-aldosterone system blockade by angiotensin-converting enzyme inhibitors (ACEI) and AT1 receptor blockers (ARB) delay the progression of DN, although recent studies suggest that ACEI and ARB do not stop the progression of DN.3
The focus of research on the pathogenesis of DN has been on the risk factor or the mechanism by which hyperglycemia is causing glomerular or tubular pathologies. However, the lack of success in obtaining therapies based on mechanisms of hyperglycemia’s adverse production of toxic metabolites suggest that effective therapy may require a change in approach. Studies from our laboratory called the Joslin 50-year Medalist Study examined people who have had type 1 diabetes for more than 50 years and revealed that 87% of these diabetic patients did not show clinically significant DN that was confirmed by renal pathologic examinations.4,5 This study suggested that endogenous protective factors such as antioxidant enzymes, insulin, and vascular endothelial growth factor (VEGF) can neutralize the adverse effects of hyperglycemia4 (Fig. 1). In addition, we also reported that hyperglycemia can cause retinal vascular cell apoptosis by dual mechanisms of increasing toxic metabolites such as oxidants and inhibition of protective growth factor action such as platelet-derived growth factors (PDGF) receptors6 (Fig. 1). For DN, we are proposing that hyperglycemia via AGEs and oxidants can directly activate protein kinase C (PKC)α, β, and δ, stress mitogen-activated protein (MAP) kinase, and oxidases to cause fibrosis of the glomeruli and tubules. The fibrosis is caused by overexpression of transforming growth factor-β (TGF-β) and connective tissue growth factor (CTGF). This process is accelerated by angiotensin and inflammation and decreased by activated protein C (APC), insulin, and VEGF. Because PKC activation also can inhibit the activation of insulin and the actions of VEGF, the protective affects of these cytokines are reduced in hyperglycemia, resulting in proteinuria, mesangium expansion, nephromegaly, and, finally, tubular-glomerular sclerotic lesions (Fig. 2). In this review, we survey the results of clinical trials of various therapeutics related to the toxic metabolites of hyperglycemia. Later in this review, we will discuss some of the potential protective factors that could be involved in preventing renal pathologies of diabetes.
Figure 1.
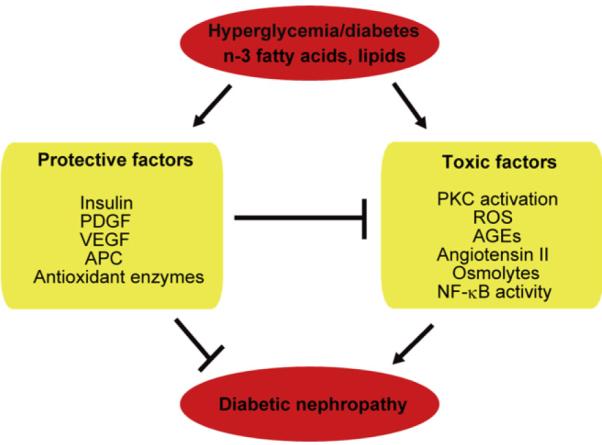
Diabetes induces an imbalance between toxic and protective factors to induce diabetic nephropathy. ROS, reactive oxygen species; NF-κB, nuclear factor-κB.
Figure 2.
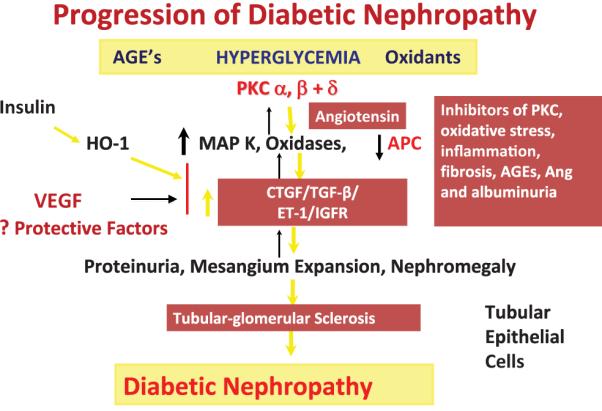
Schematic diagram on the progression of diabetic nephropathy with both pathogenic and protective factors. Ang, angiotensin; HO-1, heme-oxygenase-1; ET-1, endothelin-1; IGFR, insulin-like growth factor receptor.
NEUTRALIZING AGENTS AGAINST HYPERGLYCEMIA’S TOXIC METABOLITES
Advanced Glycation End Product Inhibitors
The receptor for advanced glycation end products (AGEs) transduces intracellular signaling and involves the generation of reactive oxygen species, which then activates transcription factors such as nuclear factor-κB.7 AGEs have been reported to increase growth factors and fibrotic factors including TGF-β and CTGF.8 Inhibitors of AGE formation or its receptor have been tested in clinical trials.
Aminoguanidine
A clinical study examining the renoprotective effects of aminoguanidine was performed in diabetic patients with DN (Table 1). In this study, patients treated with aminoguanidine showed significant decreases in proteinuria and maintaining a stable level of estimated glomerular filtration rate (eGFR).9 However, significant side effects were observed such as decreases in nitric oxide (NO)10 and increases in DNA damage through pro-oxidant activity.11
Table 1.
Clinical Trials of Potential New Therapeutic Agents for Diabetic Nephropathy
| Study (drug) | Patient Numbers | Treatment Plan | Outcome |
|---|---|---|---|
| Tuttle et al65 (RBX; 2005) | RBX, 61; placebo, 62 | RBX 32 mg/d (12 months) versus placebo |
Significant decreases in UAE in treatment versus placebo (−24% versus −9%; P = .02) |
| ACTION I9 (aminoguanidine; 2004) |
Low-dose, 229; high-dose, 225; placebo, 236 |
150 or 300 mg/d (2.49 y) versus placebo |
SCr doubled in fewer patients than placebo group (20% versus 26%; P = .099) |
| Williams et al14 (pyridoxamine; 2007) |
Pyridoxamine, 122; placebo, 90 |
Pyridoxamine 50 or 250 mg/d (6 mo) versus placebo |
Significant decreases in the increase in SCr (0.20 versus 0.76 mg/dL/y; P = .01) |
| Passariello et al16 (tolrestat; 1993) |
Tolrestat, 20 | Tolrestat 200 mg/d (6 mo) versus placebo |
Significant decreases in albuminuria (219 to 58.6 μg/ min; P < .001) |
| Iso et al17 (epalrestat; 2001) | Epalrestat, 35 | Epalrestat 150 mg/d (5 y) | Albuminuria remained unchanged in epalrestat group |
| Gaede et al66 (vitamins C and E; 2001) |
Vitamin C and E, 28 | Vitamin C 1,250 mg/d and vitamin E 680 IU/d (4 wk) |
Significant decreases in albuminuria (by 19%; P = .04) |
| Hopper et al25 (aspirin-dipyridamole; 1989) |
Aspirin-dipyridamole, 16 | Aspirin 990 mg/d or dipyridamole 225 mg/d (6 wk) versus placebo |
Significant decreases in proteinuria (1.9 to 1.4 g/24 h; P < .05) |
| Sinsakul et al27 (celecoxib; 2007) |
Celecoxib, 12; placebo, 12 | Celecoxib 200 mg/d (6 wk) versus placebo |
No significant difference in urinary proteinuria |
| Guerrero-Romero et al67 (pentoxifylline; 1995) |
Pentoxifylline, 65; captopril, 65 |
Pentoxifylline 1,200 mg/d or captopril 75 mg/d (6 mo) |
Significant decreases in UAE (pentoxifylline 101.1 to 23.1 μg/ min, captopril 102.0 to 23.9 μg/ min) |
| Mehdi et al37 (spironolactone; 2009) |
Spironolactone, 27; Losartan, 26; placebo, 27 |
Spironolactone 25 mg/d + lisinopril 80 mg/d (12 mo) or losartan 100 mg/ d (12 mo) + lisinopril 80 mg/d versus placebo |
Significant decreases in UAE (spironolactone + lisinopril by 34% from baseline, losartan + lisinopril by 16.8% from baseline; P = .007) |
| de Zeeuw et al38 (paricarcitol; 2010) |
Paricarcitol (1 μg), 93; paricarcitol (2 μg); placebo, 93 |
1 μg/d (20 mo) or 2 μg/d (20 mo) versus placebo |
Significant decreases in UAE (paricarcitol (2 μg/d) by 18% to 28% from baseline; P = .014) |
UAE, urinary albumin excretion; SCr, serum creatinine.
Pyridoxamine
Pyridoxamine inhibits the formation of AGE from preglycated (Amadori-modified) proteins12 and the formation of advanced lipoxidation end products on the protein moieties during lipid peroxidation reactions.13 Clinical trials have indicated the safety and tolerability of pyridoxamine in patients with type 1 and 2 diabetes with overt proteinuria (Table 1). Furthermore, decreases in urinary excretion of TGF-β also were noted.14 However, the most recent placebo-controlled clinical trial did not substantiate the efficacy of pyridoxamine to delay the profusion of DN in diabetic patients with overt proteinuria and a serum creatinine level greater than 2.2 mg/dL.15
Aldose Reductase Inhibitor
The polyol pathway is a glucose shunt that becomes activated at hyperglycemic conditions because aldose reductase (AR), the first and rate-limiting enzyme of the pathway, has a high Michaelis Constant (Km) for glucose to form sorbitol with its co-factor, nicotinamide adenine dinucleotide phosphate (NADPH). The second enzyme of the pathway, sorbitol dehydrogenase, then converts sorbitol to fructose with its co-factor NAD+. Increased sorbitol levels and the alteration of NADPH, NAD+/NADH levels via the metabolism of sorbitol have been postulated to damage vascular cells either by osmotic effect or lower antioxidant defense. Six months of treatment with the AR inhibitor, tolrestat, significantly reduced albuminuria in type 1 diabetes.16 Another AR inhibitor, epalrestat, was studied in type 2 diabetes and showed that it maintained renal function chronically.17 However, clear demonstration of the efficacy of ARI in delaying progression of DN has not been reported.
Antioxidants as Therapeutics
Increases in oxidant production clearly have been shown to occur when vascular or glomerular cells are exposed to hyperglycemia. Glucose’s metabolism via mitochondria pathways and the activation of NADPH oxidases via PKC activation has been shown to contribute significantly to oxidant production.18 Multiple trials using antioxidants such as vitamins C and E have been reported to have beneficial effects in rodent models of nephropathy.19 In addition, several reports involving small numbers of diabetic patients have shown improvements in oxidative stress markers in the plasma, urine, and circulatory cells, as well as endothelial dysfunction and microalbuminuria.20 However, no positive efficacy in definitive renal functions such as improvement in glomerular filtration rate has been reported. Long-term studies using these antioxidants have not shown any beneficial effects in cardiovascular or retinal end points. Recently, there has been intriguing evidence using activators of NF-E2-related factor 2 (Nrf-2), a transcription factor regulating multiple genes for antioxidant enzymes such as superoxide dismutase, glutathione synthase, and others.21 Bardoxolone methyl interacts with cysteine residues on Keap1, allowing Nrf2 translocation to the nucleus and subsequent up-regulation of a multitude of cytoprotective genes. The structure and activity profile of bardoxolone methyl resemble those of the cyclopentenone prostaglandins, endogenous Nrf2 activators that promote the resolution of inflammation.22 In clinical trials reported recently, bardoxolone methyl had beneficial effects in diabetic patients with chronic renal disease stage III. The bardoxolone-treated group showed an improvement in eGFR in type 2 diabetes mellitus patients with chronic kidney disease compared with placebo after 52 weeks of treatments.21
Anti-Inflammatory Drugs as Therapeutics
Inflammatory cells are observed in the renal glomeruli before the establishment of glomerular and interstitial pathologies in several models of diabetic nephropathy.23 Thus, anti-inflammatory drugs in addition to antioxidants could be helpful for DN.
Aspirin and Cyclooxygenase-2 Inhibitors
The prevalence of cataracts is significantly lower in diabetic patients treated with high doses of aspirin for rheumatoid arthritis compared with a matched population on no aspirin.24 Aspirin has been reported to decrease albuminuria in DN patients without affecting blood pressure and blood glucose control.25 Selective inhibitors of cyclooxygenase 2 have been reported to provide potential benefit in patients with renal disease on renal hemodynamics and decreases profibrotic cytokine levels.26 However, a study with celecoxib 200 mg/d, a cyclooxygenase 2 inhibitor, for 6 weeks did not alter proteinuria in DN patients.27
Pentoxifylline
Pentoxifylline is a methylxanthine derivate with significant anti-inflammatory, antiproliferative, and antifibrotic properties.28 It has been reported to inhibit the expression of tumor necrosis factor-α (TNF-α) messenger RNA levels and reduce the accumulation of TNF-α messenger RNA.29 Pentoxifylline has been reported to significantly reduce albuminuria in those treated with ACEI or ARB30,31 (Table 1).
Chemokine C-C Motif ligand 2 Inhibitor
It has been reported that deletion or inhibition of chemokine C-C motif ligand 2 (CCL2) signaling can prevent glomerulosclerosis by blocking macrophage recruitment to the glomeruli of diabetic mice.32 However, the complete deletion of CCL2 only reduced albuminuria and renal fibrosis in diabetic mice. Thus, it is likely that clinical inhibitors of CCL2 that will only partially reduce CCL2 expression will have only mild effects on diabetic nephropathy.33
Anti-Extracellular Matrix Production
CTGF is a critical mediator of extra cellular matrix accumulation and coordinates a final common pathway of fibrosis. FG-3019, a monoclonal antibody to CTGF, significantly decreased albuminuria in DN patients.34 It also has been reported that antifibrotic actions of pirfenidone and tranilast have been shown in the kidney of diabetic rats.35 Another control of accumulation of ECM is thought to be determined by the balance between the synthesis of matrix and its degradation by metalloproteases. XL 784, a bioavailable metalloprotease inhibitor, reduced proteinuria and glomerulosclerosis in diabetic rats.36 Because the description of inhibitors for TGF-β is covered in another article in this issue of Seminars in Nephrology, we will not describe it here.
Mineralocorticoid Antagonist
Aldosterone can cause renal injury and subsequent fibrosis by promoting tissue inflammation. It has been reported that the addition of spironolactone to a regimen including maximal ACEI affords greater renoprotection in DN despite a similar effect on blood pressure37 (Table 1). The additional effects that spironolactone will have in reducing renal fibrosis in diabetes are unclear. Side effects of spironolactone in diabetic patients with renal impairment also will need to be considered.
Vitamin D
In a multivariable analysis of patients with chronic kidney disease, lower calcitriol concentrations significantly correlated with high risk of albuminuria and eGFR. A randomized trial showed that paricarcitol can effectively reduce residual albuminuria in patients with type 2 diabetic nephropathy who were receiving stable treatment with an ACEI or ARB.38 The effects of vitamin D are likely to be mediated by its anti-oxidative stress or anti-inflammatory actions. However, because well-known anti-oxidants and anti-inflammatory agents have not been effective in human trials for DN, it is unclear whether vitamin D will provide significant effects to reduce renal damage in hyperglycemic conditions.
Roles of Endogenous Protective Factors
The presence of endogenous protective factors has been shown clearly in clinical studies. For diabetic nephropathy, Perkins et al39 reported in the New England Journal of Medicine that of 368 type 1 diabetic patients who initially had microalbuminuria and were followed up for 12 years, one third of the patients showed no progression and another third of the patients actually had regression with decreased amounts of microalbuminuria. This study and other subsequent studies clearly have shown that diabetic patients who had mild nephropathy as manifested by microalbuminuria potentially can stop the progression of the disease and possibly regress as well. Recently, the Joslin Medalists Study comprised a group of type 1 diabetic patients who have had insulin dependency for 50 years or longer.4 Extensive analysis of more than 351 Medalist patients has shown that more than 40% of these patients have no or mild diabetic retinopathy and only 13% manifested significant microalbuminuria or proteinuria.5 Longitudinal follow-up evaluation in a subset of these Medalists patients with regard to their retinopathy clearly showed that more than 30% to 40% of these patients may develop only mild retinopathy as quantitated by fundus photography. However, in this subset of patients, mild diabetic retinopathy did not progress or even may regress. These clinical studies clearly have shown that endogenous protective factors exist to neutralize the toxic effects of hyperglycemia and protect against the progression of DN and diabetic retinopathy (DR). In this article, we discuss some of the potential endogenous protective factors or mechanisms that may exist in the vascular tissues to counter the adverse actions of hyperglycemia.
Protein Kinase C Activation
A great deal of evidence has been accumulated to support the activation of PKC, especially the β and δ isoforms, to participate in the development of DN.40 Ruboxistaurin (RBX), a PKCβ isoform selective inhibitor, has been shown to prevent DN in rodent models of diabetes.41 In addition, compared with diabetic wild-type mice, diabetic PKCβ null mice showed attenuated renal abnormalities including reduced albuminuria and mesangial expansion.42 The effects of PKCβ inhibitor has been evaluated in diabetic patients. In a phase II clinical trial, RBX (32 mg/d) was used to determine whether PKCβ inhibition can be effective in type 2 diabetic patients with overt albuminuria and treated with ACEI or ARB (Table 1). Patients who were treated with RBX showed significant reductions in albuminuria and did not show increases of urinary (TGF-β. RBX-treated patients maintained a stable eGFR over 1 year.43
Recently, we showed that hyperglycemia persistently can activate PKCδ and p38 mitogen-activated protein kinases (MAPK) to increase Src homology-2 domain containing phosphatase-1 (SHP-1), and leads to PDGF receptor β dephosphorylation and reduction of its downstream actions to induce pericyte apoptosis and acellular capillaries in diabetic retina6 (Fig. 3). In normoglycemia, PDGF-β increased DNA synthesis, inhibited cellular apoptosis, and induced phosphorylation of Akt and extracellular signal-regulated kinases (Erk) activation in retinal pericytes.44
Figure 3.

Dual pathways of the actions of hyperglycemia to induce accelerated apoptosis of renal glomerular podocytes and retinal capillary pericytes. ROS, reactive oxygen species; ERK, extracellular signal-regulated kinases.
Similar to retinal pericytes, our recent preliminary findings have suggested that hyperglycemia also can activate PKCδ/SHP-1 in the renal podocytes to induce its apoptosis. In addition, hyperglycemia also will activate NADPH oxidases, which also can cause nuclear factor-κB activation and podocyte apoptosis. These findings support the idea that effective therapies for DN need to neutralize the toxic effects of glucose’s toxic metabolites and enhance the actions of protective factors such as VEGF caused by SHP-1 activation (Fig. 3).
Insulin
In insulin-resistant and diabetic states, insulin’s actions in the endothelial cells are diminished, leading to endothelial dysfunction and acceleration of atherosclerosis.45 Normally, insulin can stimulate the production of NO by the activation of endothelial NO synthase (eNOS), which results in vasodilatation and antithrombosis in the short term, and can inhibit smooth muscle cell growth and migration chronically. Impaired action of insulin has been observed in the vasculature in both type 1 and type 2 diabetes, possibly by the activation of the PKCβ2 isoform.41,45
In diabetes and insulin-resistant states, insulin’s activation of the insulin receptor substrate (IRS1)/Akt/eNOS pathway is inhibited selectively, but another major pathway of insulin signaling, MAPK, is not inhibited.41,46 This selective insulin resistance has been shown in skeletal muscle from obesity and patients with type 2 diabetes,47 and in the vasculature, myocardium,48 and glomeruli41 of Zucker fatty and diabetic rats, which are the animal models of insulin resistance. We reported that insulin-stimulated phosphoinositide kinase-3 (PI3K)/ eNOS activity is inhibited by PKC activation in endothelial cells and vascular tissues of Zucker fatty and diabetic rats.46 Moreover, we found that PKCβ-specific inhibitor, RBX, improved insulin signaling on NO production in the vasculature, myocardium, and glomeruli of streptozotocin-induced diabetic rats or Zucker fatty rats.41,45
Insulin stimulates not only NO production from endothelial cells but also the expression of eNOS.45 The vascular endothelial cell–specific insulin receptor knockout mice showed significant decreases in eNOS expression in the aorta. Thus, insulin’s regulation of NO might be an important factor for vascular homeostasis or improving insulin sensitivity in the endothelium could decrease the risk for atherosclerosis in insulin resistance and diabetes.49 Also, phosphodiesterase 5, an exogenous NO donor, decreases blood pressure.50 However, clear demonstration of the efficacy of phosphodiesterase 5 in delaying progression of DN has not been reported.
Welsh et al51 recently showed that mice in which the gene encoding the insulin receptor was deleted specifically from podocytes caused excessive excretion of albumin in the urine, shortening of the podocyte foot processes, increased deposition of components of the basal membrane, and a higher frequency of programmed podocyte apoptosis.
We have reported that the beneficial effect of insulin may be attributed to its capacity to increase heme oxygenase-1, which is a potent antioxidant enzyme. Therefore, improving insulin sensitivity in vascular and glomerular tissue may decrease the risk for diabetic nephropathy and other vascular complications.
VEGF-A
VEGF promotes angiogenesis, inducing confluent microvascular endothelial cells to invade collagen gel.52 VEGF also is known to prevent endothelial apoptosis mediated by the activation of the PI3K/Akt pathway.53 VEGF also has actions in nonendothelial cells. During fetal development, one of the cell types producing the largest amounts of VEGF-A is the podocyte. Unlike many other tissues or cells, podocytes continue to express VEGF-A when they are fully differentiated, although the absolute levels of VEGF expression do decrease during differentiation.54
In early stages of DN, many reports have shown that the expression of VEGF-A is increased in the glomeruli.55 Some investigators have suggested that VEGF acts in a novel autocrine signaling mode to induce the podocytopathy of diabetes, especially the genesis of albuminuria. Thus, treatment with anti-VEGF antibodies may attenuate glomerular basement membrane thickening and progression of DN.
However, recently, Eremina et al56 reported proteinuria, hypertension, and renal failure in several patients treated with an anti-VEGF agent, suggesting that VEGF-A plays an important role in maintaining endothelial cell function and a glomerular filtration barrier. Supporting this, detailed reports by Eremina et al57 clearly showed that VEGF-A is necessary for forming and maintaining the glomerular filtration barrier. Furthermore, in several other glomerular diseases, a beneficial role of VEGF has been shown through the prevention of progressive capillary rarefaction, promotion of capillary repair, and improvement of renal injury.44
Activated Protein C
APC has profibrinolytic function by modifying clot formation and inhibiting plasminogen activator inhibitor-1.58 It has been reported that plasma thrombomodulin from the endothelium caused by reduction of APC are increased in diabetic patients, and the impairment of the thrombomodulin/protein C system is associated with DN.59 Isermann et al60 recently reported that APC may delay the onset of nephropathy in long-term experimental diabetes by modulating apoptosis of endothelial cells and podocytes.
Glucagon like Peptide-1
Glucagon like peptide-1 (GLP-1) is a gut incretin hormone that stimulates glucose-dependent insulin responses in the β cells.61 GLP-1 acts through the GLP-1 receptor, which is present abundantly in the gastrointestinal tract but it has been reported in many other tissues including endothelial cells and the kidney.62 In endothelial cells, GLP-1 has been reported to inhibit the expression of TNF-α and vascular cell adhesion molecule-1.63 Mechanistically, GLP-1 has been reported to stimulate NO production, a potential mechanism for improving endothelial function.64 Moreover, GLP-1 has been reported to improve renal pathologies in diabetic rodents.63 However, no mechanism is known to reflect the GLP-1 protective action on endothelial cells.
SUMMARY
During the past 3 decades, considerable progress has been made in delaying the progression of DN. Although good glycemic control clearly is the best method to prevent diabetic complications, it often is difficult to maintain, and vascular complications develop despite good gylcemic treatment. Inhibitors of AGE, receptor for AGE, oxidative stress, diacylglycerol (DAG)-PKC, vascular inflammation, and the renin-angiotensin system, for example, should provide useful targets for therapy; however, many clinical trials using agents directly against these targets have not shown dramatic efficacy to prevent or delay the progression of DN. The presence and the importance of endogenous protective factors against the development of DN and DR clearly have been shown clinically by patients who have had diabetes of extreme duration yet do not have manifestations of DN and DR. Thus, we propose that it is equally important to enhance endogenous protective factors, such as insulin, VEGF, APC, GLP-1, and others to neutralize the adverse effects of hyperglycemia and prevent DN (Fig. 1).
ACKNOWLEDGMENT
The authors wish to thank Dr. Glorian Qi and Ms. Patti Muehter for their reading of the manuscript.
Financial support Supported by grants from the National Institutes of Health/National Eye Institute (EY016150), and the National Institutes of Health (RO1DK053105-10, R24DK090961, and DERC P30DK036836 to G.L.K.). A.M. is the recipient of a Research Fellowship (Manpei Suzuki Diabetes Foundation, Kanzawa Medical Research Foundation, NOVARTIS Foundation, Japan).
Footnotes
Conflict of interest statement: none.
REFERENCES
- 1.The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329:977–86. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 2.Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:854–65. [PubMed] [Google Scholar]
- 3.Nobakht N, Kamgar M, Rastogi A, Schrier RW. Limitations of angiotensin inhibition. Nat Rev Nephrol. 2011;7:356–9. doi: 10.1038/nrneph.2011.29. [DOI] [PubMed] [Google Scholar]
- 4.Keenan HA, Costacou T, Sun JK, et al. Clinical factors associated with resistance to microvascular complications in diabetic patients of extreme disease duration: the 50-year medalist study. Diabetes Care. 2007;30:1995–7. doi: 10.2337/dc06-2222. [DOI] [PubMed] [Google Scholar]
- 5.Sun JK, Keenan HA, Cavallerano JD, et al. Protection from retinopathy and other complications in patients with type 1 diabetes of extreme duration: the Joslin 50-year medalist study. Diabetes Care. 2011;34:968–74. doi: 10.2337/dc10-1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geraldes P, Hiraoka-Yamamoto J, Matsumoto M, et al. Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat Med. 2009;15:1298–306. doi: 10.1038/nm.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rask-Madsen C, King GL. Kidney complications: factors that protect the diabetic vasculature. Nat Med. 2010;16:40–1. doi: 10.1038/nm0110-40. [DOI] [PubMed] [Google Scholar]
- 8.Wendt TM, Tanji N, Guo J, et al. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol. 2003;162:1123–37. doi: 10.1016/S0002-9440(10)63909-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bolton WK, Cattran DC, Williams ME, et al. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am J Nephrol. 2004;24:32–40. doi: 10.1159/000075627. [DOI] [PubMed] [Google Scholar]
- 10.Tilton RG, Chang K, Hasan KS, et al. Prevention of diabetic vascular dysfunction by guanidines. Inhibition of nitric oxide synthase versus advanced glycation end-product formation. Diabetes. 1993;42:221–32. doi: 10.2337/diab.42.2.221. [DOI] [PubMed] [Google Scholar]
- 11.Suji G, Sivakami S. DNA damage by free radical production by aminoguanidine. Ann N Y Acad Sci. 2006;1067:191–9. doi: 10.1196/annals.1354.023. [DOI] [PubMed] [Google Scholar]
- 12.Chetyrkin SV, Mathis ME, Ham AJ, et al. Propagation of protein glycation damage involves modification of tryptophan residues via reactive oxygen species: inhibition by pyridoxamine. Free Radic Biol Med. 2008;44:1276–85. doi: 10.1016/j.freeradbiomed.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 13.Onorato JM, Jenkins AJ, Thorpe SR, Baynes JW. Pyridoxamine, an inhibitor of advanced glycation reactions, also inhibits advanced lipoxidation reactions. Mechanism of action of pyridoxamine. J Biol Chem. 2000;275:21177–84. doi: 10.1074/jbc.M003263200. [DOI] [PubMed] [Google Scholar]
- 14.Williams ME, Bolton WK, Khalifah RG, et al. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am J Nephrol. 2007;27:605–14. doi: 10.1159/000108104. [DOI] [PubMed] [Google Scholar]
- 15.Lewis EJ, Greene T, Spitalewiz S, et al. Pyridorin in type 2 diabetic nephropathy. J Am Soc Nephrol. 2011;23:131–6. doi: 10.1681/ASN.2011030272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Passariello N, Sepe J, Marrazzo G, et al. Effect of aldose reductase inhibitor (tolrestat) on urinary albumin excretion rate and glomerular filtration rate in IDDM subjects with nephropathy. Diabetes Care. 1993;16:789–95. doi: 10.2337/diacare.16.5.789. [DOI] [PubMed] [Google Scholar]
- 17.Iso K, Tada H, Kuboki K, Inokuchi T. Long-term effect of epalrestat, an aldose reductase inhibitor, on the development of incipient diabetic nephropathy in type 2 diabetic patients. J Diabetes Complications. 2001;15:241–4. doi: 10.1016/s1056-8727(01)00160-x. [DOI] [PubMed] [Google Scholar]
- 18.Inoguchi T, Sonta T, Tsubouchi H, et al. Protein kinase C-dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: role of vascular NAD(P)H oxidase. J Am Soc Nephrol. 2003;14(Suppl 3):S227–32. doi: 10.1097/01.asn.0000077407.90309.65. [DOI] [PubMed] [Google Scholar]
- 19.Koya D, Lee IK, Ishii H, Kanoh H, King GL. Prevention of glomerular dysfunction in diabetic rats by treatment with d-alphatocopherol. J Am Soc Nephrol. 1997;8:426–35. doi: 10.1681/ASN.V83426. [DOI] [PubMed] [Google Scholar]
- 20.Beckman JA, Goldfine AB, Gordon MB, Creager MA. Ascorbate restores endothelium-dependent vasodilation impaired by acute hyperglycemia in humans. Circulation. 2001;103:1618–23. doi: 10.1161/01.cir.103.12.1618. [DOI] [PubMed] [Google Scholar]
- 21.Pergola PE, Raskin P, Toto RD, et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med. 2011;365:327–36. doi: 10.1056/NEJMoa1105351. [DOI] [PubMed] [Google Scholar]
- 22.Yates MS, Tauchi M, Katsuoka F, et al. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol Cancer Ther. 2007;6:154–62. doi: 10.1158/1535-7163.MCT-06-0516. [DOI] [PubMed] [Google Scholar]
- 23.Rodriguez-Iturbe B, Pons H, Herrera-Acosta J, Johnson RJ. Role of immunocompetent cells in nonimmune renal diseases. Kidney Int. 2001;59:1626–40. doi: 10.1046/j.1523-1755.2001.0590051626.x. [DOI] [PubMed] [Google Scholar]
- 24.Blakytny R, Harding JJ. Prevention of cataract in diabetic rats by aspirin, paracetamol (acetaminophen) and ibuprofen. Exp Eye Res. 1992;54:509–18. doi: 10.1016/0014-4835(92)90129-g. [DOI] [PubMed] [Google Scholar]
- 25.Hopper AH, Tindall H, Davies JA. Administration of aspirindipyridamole reduces proteinuria in diabetic nephropathy. Nephrol Dial Transplant. 1989;4:140–3. [PubMed] [Google Scholar]
- 26.Cheng HF, Wang CJ, Moeckel GW, et al. Cyclooxygenase-2 inhibitor blocks expression of mediators of renal injury in a model of diabetes and hypertension. Kidney Int. 2002;62:929–39. doi: 10.1046/j.1523-1755.2002.00520.x. [DOI] [PubMed] [Google Scholar]
- 27.Sinsakul M, Sika M, Rodby R, et al. A randomized trial of a 6-week course of celecoxib on proteinuria in diabetic kidney disease. Am J Kidney Dis. 2007;50:946–51. doi: 10.1053/j.ajkd.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 28.Turgut F, Bolton WK. Potential new therapeutic agents for diabetic kidney disease. Am J Kidney Dis. 2010;55:928–40. doi: 10.1053/j.ajkd.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 29.Han J, Thompson P, Beutler B. Dexamethasone and pentoxifylline inhibit endotoxin-induced cachectin/tumor necrosis factor synthesis at separate points in the signaling pathway. J Exp Med. 1990;172:391–4. doi: 10.1084/jem.172.1.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harmankaya O, Seber S, Yilmaz M. Combination of pentoxifylline with angiotensin converting enzyme inhibitors produces an additional reduction in microalbuminuria in hypertensive type 2 diabetic patients. Ren Fail. 2003;25:465–70. doi: 10.1081/jdi-120021159. [DOI] [PubMed] [Google Scholar]
- 31.Navarro JF, Mora C, Muros M, Garcia J. Additive antiproteinuric effect of pentoxifylline in patients with type 2 diabetes under angiotensin II receptor blockade: a short-term, randomized, controlled trial. J Am Soc Nephrol. 2005;16:2119–26. doi: 10.1681/ASN.2005010001. [DOI] [PubMed] [Google Scholar]
- 32.Chow FY, Nikolic-Paterson DJ, Ozols E, et al. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 2006;69:73–80. doi: 10.1038/sj.ki.5000014. [DOI] [PubMed] [Google Scholar]
- 33.Chow FY, Nikolic-Paterson DJ, Ma FY, et al. Monocyte chemoattractant protein-1-induced tissue inflammation is critical for the development of renal injury but not type 2 diabetes in obese db/db mice. Diabetologia. 2007;50:471–80. doi: 10.1007/s00125-006-0497-8. [DOI] [PubMed] [Google Scholar]
- 34.Adler SG, Schwartz S, Williams ME, et al. Phase 1 study of anti-CTGF monoclonal antibody in patients with diabetes and microalbuminuria. Clin J Am Soc Nephrol. 2010;5:1420–8. doi: 10.2215/CJN.09321209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan SM, Zhang Y, Cox AJ, Kelly DJ, Qi W. Tranilast attenuates the up-regulation of thioredoxin-interacting protein and oxidative stress in an experimental model of diabetic nephropathy. Nephrol Dial Transplant. 2011;26:100–10. doi: 10.1093/ndt/gfq355. [DOI] [PubMed] [Google Scholar]
- 36.Williams JM, Zhang J, North P, et al. Evaluation of metalloprotease inhibitors on hypertension and diabetic nephropathy. Am J Physiol Renal Physiol. 2011;300:F983–98. doi: 10.1152/ajprenal.00262.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mehdi UF, Adams-Huet B, Raskin P, Vega GL, Toto RD. Addition of angiotensin receptor blockade or mineralocorticoid antagonism to maximal angiotensin-converting enzyme inhibition in diabetic nephropathy. J Am Soc Nephrol. 2009;20:2641–50. doi: 10.1681/ASN.2009070737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Zeeuw D, Agarwal R, Amdahl M, et al. Selective vitamin D receptor activation with paricarcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL study): a randomised controlled trial. Lancet. 2010;376:1543–51. doi: 10.1016/S0140-6736(10)61032-X. [DOI] [PubMed] [Google Scholar]
- 39.Perkins BA, Ficociello LH, Silva KH, et al. Regression of microalbuminuria in type 1 diabetes. N Engl J Med. 2003;348:2285–93. doi: 10.1056/NEJMoa021835. [DOI] [PubMed] [Google Scholar]
- 40.Koya D, Jirousek MR, Lin YW, et al. Characterization of protein kinase C beta isoform activation on the gene expression of transforming growth factor-beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. J Clin Invest. 1997;100:115–26. doi: 10.1172/JCI119503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mima A, Ohshiro Y, Kitada M, et al. Glomerular-specific protein kinase C-beta-induced insulin receptor substrate-1 dysfunction and insulin resistance in rat models of diabetes and obesity. Kidney Int. 2011;79:883–96. doi: 10.1038/ki.2010.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohshiro Y, Ma RC, Yasuda Y, et al. Reduction of diabetes-induced oxidative stress, fibrotic cytokine expression, and renal dysfunction in protein kinase Cbeta-null mice. Diabetes. 2006;55:3112–20. doi: 10.2337/db06-0895. [DOI] [PubMed] [Google Scholar]
- 43.Gilbert RE, Kim SA, Tuttle KR, et al. Effect of ruboxistaurin on urinary transforming growth factor-beta in patients with diabetic nephropathy and type 2 diabetes. Diabetes Care. 2007;30:995–6. doi: 10.2337/dc06-2079. [DOI] [PubMed] [Google Scholar]
- 44.Kang DH, Joly AH, Oh SW, et al. Impaired angiogenesis in the remnant kidney model: I. Potential role of vascular endothelial growth factor and thrombospondin-1. J Am Soc Nephrol. 2001;12:1434–47. doi: 10.1681/ASN.V1271434. [DOI] [PubMed] [Google Scholar]
- 45.Naruse K, Rask-Madsen C, Takahara N, et al. Activation of vascular protein kinase C-beta inhibits Akt-dependent endothelial nitric oxide synthase function in obesity-associated insulin resistance. Diabetes. 2006;55:691–8. doi: 10.2337/diabetes.55.03.06.db05-0771. [DOI] [PubMed] [Google Scholar]
- 46.Jiang ZY, Lin YW, Clemont A, et al. Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats. J Clin Invest. 1999;104:447–57. doi: 10.1172/JCI5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cusi K, Maezono K, Osman A, et al. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J Clin Invest. 2000;105:311–20. doi: 10.1172/JCI7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He Z, Opland DM, Way KJ, et al. Regulation of vascular endothelial growth factor expression and vascularization in the myocardium by insulin receptor and PI3K/Akt pathways in insulin resistance and ischemia. Arterioscler Thromb Vasc Biol. 2006;26:787–93. doi: 10.1161/01.ATV.0000209500.15801.4e. [DOI] [PubMed] [Google Scholar]
- 49.Vicent D, Ilany J, Kondo T, et al. The role of endothelial insulin signaling in the regulation of vascular tone and insulin resistance. J Clin Invest. 2003;111:1373–80. doi: 10.1172/JCI15211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolk R, Smith WB, Neutel JM, et al. Blood pressure lowering effects of a new long-acting inhibitor of phosphodiesterase 5 in patients with mild to moderate hypertension. Hypertension. 2009;53:1091–7. doi: 10.1161/HYPERTENSIONAHA.109.132225. [DOI] [PubMed] [Google Scholar]
- 51.Welsh GI, Hale LJ, Eremina V, et al. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 2010;12:329–40. doi: 10.1016/j.cmet.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nicosia RF, Nicosia SV, Smith M. Vascular endothelial growth factor, platelet-derived growth factor, and insulin-like growth factor-1 promote rat aortic angiogenesis in vitro. Am J Pathol. 1994;145:1023–9. [PMC free article] [PubMed] [Google Scholar]
- 53.Gerber HP, McMurtrey A, Kowalski J, et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem. 1998;273:30336–43. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 54.Sison K, Eremina V, Baelde H, et al. Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J Am Soc Nephrol. 2011;21:1691–701. doi: 10.1681/ASN.2010030295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ziyadeh FN. Different roles for TGF-beta and VEGF in the pathogenesis of the cardinal features of diabetic nephropathy. Diabetes Res Clin Pract. 2008;82(Suppl 1):S38–41. doi: 10.1016/j.diabres.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 56.Eremina V, Sood M, Haigh J, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–16. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eremina V, Jefferson JA, Kowalewska J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358:1129–36. doi: 10.1056/NEJMoa0707330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fulcher CA, Gardiner JE, Griffin JH, Zimmerman TS. Proteolytic inactivation of human factor VIII procoagulant protein by activated human protein C and its analogy with factor V. Blood. 1984;63:486–9. [PubMed] [Google Scholar]
- 59.Hafer-Macko CE, Ivey FM, Gyure KA, Sorkin JD, Macko RF. Thrombomodulin deficiency in human diabetic nerve microvasculature. Diabetes. 2002;51:1957–63. doi: 10.2337/diabetes.51.6.1957. [DOI] [PubMed] [Google Scholar]
- 60.Isermann B, Vinnikov IA, Madhusudhan T, et al. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med. 2007;13:1349–58. doi: 10.1038/nm1667. [DOI] [PubMed] [Google Scholar]
- 61.Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696–705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- 62.Park CW, Kim HW, Ko SH, et al. Long-term treatment of glucagon-like peptide-1 analog exendin-4 ameliorates diabetic nephropathy through improving metabolic anomalies in db/db mice. J Am Soc Nephrol. 2007;18:1227–38. doi: 10.1681/ASN.2006070778. [DOI] [PubMed] [Google Scholar]
- 63.Kodera R, Shikata K, Kataoka HU, et al. Glucagon-like peptide-1 receptor agonist ameliorates renal injury through its anti-inflammatory action without lowering blood glucose level in a rat model of type 1 diabetes. Diabetologia. 2011;54:965–78. doi: 10.1007/s00125-010-2028-x. [DOI] [PubMed] [Google Scholar]
- 64.Erdogdu O, Nathanson D, Sjoholm A, Nystrom T, Zhang Q. Exendin-4 stimulates proliferation of human coronary artery endothelial cells through eNOS-, PKA- and PI3K/Akt-dependent pathways and requires GLP-1 receptor. Mol Cell Endocrinol. 2010;325:26–35. doi: 10.1016/j.mce.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 65.Tuttle KR, Bakris GL, Toto RD, et al. The effect of ruboxistaurin on nephropathy in type 2 diabetes. Diabetes Care. 2005;28:2686–90. doi: 10.2337/diacare.28.11.2686. [DOI] [PubMed] [Google Scholar]
- 66.Gaede P, Poulsen HE, Parving HH, Pedersen O. Double-blind, randomised study of the effect of combined treatment with vitamin C and E on albuminuria in type 2 diabetic patients. Diabet Med. 2001;18:756–60. doi: 10.1046/j.0742-3071.2001.00574.x. [DOI] [PubMed] [Google Scholar]
- 67.Guerrero-Romero F, Rodriguez-Moran M, Paniagua-Sierra JR, et al. Pentoxifylline reduces proteinuria in insulin-dependent and non insulin-dependent diabetic patients. Clin Nephrol. 1995;43:116–21. [PubMed] [Google Scholar]