

Abstract
Partially tryptic peptides are often identified in shotgun proteomics using trypsin as the proteolytic enzyme; however, their sources have been controversial. Herein we investigate the impact of in-source fragmentation on shotgun proteomics profiling of three biological samples: a standard protein mixture, a mouse brain tissue homogenate, and mouse plasma. Since the in-source fragments of peptide ions have the same LC elution time as its parental peptide, partially tryptic peptide ions from in-source fragmentation can be distinguished from the other partially tryptic peptides based on their elution time differences from those computationally predicted data. The percentage of partially tryptic peptide identifications resulting from in-source fragmentation in a standard protein digest was observed to be ~60 %. In more complex mouse brain or plasma samples, in-source fragmentation contributed to a less degree of 1–3 % of all identified peptides due to the limit dynamic range of LC-MS/MS measurements. The other major source of partially tryptic peptides in complex biological samples is presumably proteolytic cleavage by endogenous proteases in the samples. Our work also provides a method to identify such proteolytic-derived partially tryptic peptides due to endogenous proteases in the samples by removing in-source fragmentation artifacts from the identified peptides.
Keywords: in-source fragmentation, partially tryptic, trypsin specificity, predicted elution time
INTRODUCTION
Trypsin has been commonly used in mass spectrometry (MS)-based bottom-up proteomics for several reasons.1–4 First, this protease has been reported to strictly cleave the C-terminal amide bond of arginine or lysine residues,1 resulting in peptides with a MS-friendly mass range of 10 amino acid residues (on average). Furthermore, the resulting basic residue located at the C-terminus in peptides enhances electrospray ionization efficiency and contributes to the generation of an abundant y-ion series in tandem mass spectra to facilitate confident identification. While the bottom-up proteomics strategy predominately identifies fully tryptic peptides in complex samples when trypsin is used, a significant number of partially tryptic peptides are often confidently identified.2, 3, 5–8 Such observations led to the controversy on whether trypsin provides strict specificity on its cleavage residues arginine and lysine. For example, Olsen et al. reported that trypsin cleaved exclusively C-terminal to arginine and lysine residues based on their data from mouse liver tissue. On the other hand, Picotti et al. reported that the percentage of partially tryptic peptides identified from a single standard protein beta-lactoglobulin was >70%, although most of them were of relatively low-abundance compared to fully tryptic peptides.3
There are several potential sources for generating partially tryptic peptides, including possible chymotrypsin contamination in trypsin, pseudotrypsin activity due to partial autolysis of trypsin, proteolytic products due to endogenous proteases, and in-source or “nozzle-skimmer” fragmentation of fully tryptic peptides.1–3, 8 Among these sources, the chymotrypsin contamination and autolysis issues have been largely alleviated by using the sequencing grade of modified trypsin. The modified trypsin is reductively dimethylated on lysine residues and subjected to TPCK treatment to eliminate chymotryptic activity,9 followed by affinity purification. The dimethylated trypsin also has a strong resistance against auto-proteolysis, which can generate pseudotrypsin activity.10 Therefore, there are only two main sources for generating partially tryptic peptides: proteolysis by endogenous proteases present in the sample and in-source fragmentation (ISF), which fragments fully tryptic peptides within the electrospray ionization source. We hypothesize that the observed high percentage of partially tryptic peptides in a single protein sample as reported by Picotti et al.3 was largely due to ISF since ISF products are more identifiable in a less complex peptide mixture. In this work, we aim to examine the impact of ISF on peptide identifications from both standard protein mixtures and complex biological samples.
In-source fragmentation is directly related with the desolvation and activation energy at the interface of electrospray ionization (ESI) source. The desolvation/activation energy is controlled by the cone voltage, which is also called nozzle-skimmer voltage, or declustering voltage.11–17 Higher desolvation/activation energy can enhance ion yield but simultaneously facilitate ISF due to elevated ion energy,14, 16 which compromises the final ion yield. In practice, researchers typically optimize the ESI-MS desolvation process by balancing these two inversely proportional factors to maximize ion gain. Therefore, some degree of ISF is an inherent phenomenon in ESI-MS, regardless of whether the resulting in-source fragments could be identified or not.
A substantial number of partially tryptic peptides were identified in different LC-MS/MS based shotgun proteomic studies, but they were often considered as less confident identifications than fully tryptic peptides. To clarify the sources of partially tryptic peptides and the specificity of trypsin, we investigated the impact of ISF on peptide identifications in a mixture of six standard proteins, mouse brain tissue, and mouse plasma. Since ISF-generated partially tryptic peptides will have the same LC elution times as their corresponding fully tryptic peptides, these peptides can be differentiated from other proteolytic-derived partially tryptic peptides based on examining their elution times in comparison with their parental tryptic peptides. By removing the ISF-generated artifacts from the identified partially tryptic peptides, it becomes possible to directly survey proteolytic cleavage products resulting from endogenous proteases in biological samples.
EXPERIMENTAL METHODS
Protein samples
Six standard proteins, including bovine carbonic anhydrase II, bovine beta-lactoglobulin, E. coli beta-galactosidase, equine skeletal muscle myoglobin, chicken ovalbumin, and bovine cytochrome c, were obtained from Sigma-Aldrich (St. Louis, MO). Mouse brain tissues dissected from adult C57BL/6J male mice (9 weeks, 21–27 g) were obtained from Jackson Laboratories (Bar Harbor, ME). All tissues were homogenized in 25 mM NH4HCO3 (pH 8.2), and protein concentration was determined by BCA assay (Pierce, Rockford, IL). Mouse plasma from Equitech-Bio (Kerrville, TX) was determined to have a concentration of ~40 mg/mL using the BCA protein assay.
Protein digestion
Protein samples were initially denatured and reduced with 8 M urea and 10 mM dithiothreitol in 50 mM NH4HCO3 buffer (pH 8.2) for 1 h at 37 °C and followed by alkylation of cysteine residues with 40 mM iodoacetamide for 1 h at 37 °C in the dark. Following a 10-fold dilution with 50 mM NH4HCO3, resulting samples were digested using sequencing grade modified porcine trypsin (Promega, Madison, WI) at a trypsin-to-protein ratio of 1:50 (w/w) for 5 h at 37 °C. The digested samples were individually loaded onto a 1-mL solid-phase extraction (SPE) C18 column (Supelco, Bellefonte, PA) and washed with 4 mL of 0.1% trifluoroacetic acid (TFA)/5% acetonitrile (ACN). Peptides were eluted from the SPE column with 1 mL of 0.1% TFA/80% ACN, and then lyophilized. After reconstituting the resulting peptide samples in 25 mM NH4HCO3, the final peptide concentration for each sample was measured with the BCA protein assay.
Reversed-phase capillary LC-MS/MS
Peptides were analyzed using a custom-built automated four-column high-pressure capillary LC system coupled on-line to a LTQ-Orbitrap mass spectrometer (Thermo Scientific) via a nanoelectrospray ionization interface manufactured in-house with 500 μL/min flow rate.18 The reversed-phase capillary column was prepared by slurry packing 3-μm Jupiter C18 bonded particles (Phenomenex, Torrence, CA) into a 65-cm-long, 75-μm-inner diameter fused silica capillary (Polymicro Technologies, Phoenix, AZ). After loading 2.5 μg of peptides onto the column, the mobile phase was held at 100% A (0.1% formic acid) for 20 min, followed by a linear gradient from 0 to 70% buffer B (0.1% formic acid in 90% acetonitrile) over 85 min. Each full MS scan (m/z 400–2000) was followed by collision-induced MS/MS spectra (normalized collision energy setting of 35%) for the 6 or 10 most abundant ions for standard protein mixture/mouse plasma or mouse brain sample, resulting in an overall cycle time of ~2.5 s or ~3.6 s, respectively. The automatic gain control setting for the orbitrap was 1,000,000 with a resolution of 60,000. The dynamic exclusion duration was set to 1 min. The heated capillary was maintained at 200 °C and. Parameters for ion optics were automatically tuned fby Xcalibur Tune Plus to maximize ion gain and the tuning parameters were: ESI voltage 2.2 kV, heated capillary temperature 200 °C, capillary voltage 30 V, and tube lens voltage 80 V.
Data analysis
LC-MS/MS raw data were converted into .dta files using Extract_MSn (version 3.0) in Bioworks Cluster 3.2 (Thermo Fisher Scientific, Cambridge, MA), and the SEQUEST algorithm with a parameter file consisting of no enzyme criteria and static cysteine modification (+57.022 Da) was used for all MS/MS spectra against the collected standard protein sequence list or the mouse Uniprot database (25 338 proteins, released May 5, 2010). A decoy database search was employed, and an extremely low false discovery rate (FDR) of 0.05% was achieved by filtering peptides using a MS generating function score threshold19, 20 in combination with a 10 ppm mass accuracy cut-off for the final identified peptides.
RESULTS AND DISCUSSION
Partially tryptic peptides from in-source fragmentation
To investigate how much the ISF-generated ‘artifacts’ can impact the peptide identifications, we initially examined all the identified partially tryptic peptides using a digest of six standard proteins to determine if the peptides were in-source fragments of potential parental peptides at the same elution time. Surprisingly, an almost complete series of y-ion type in-source fragments within detectable m/z range were observed with the majority having <10% intensity of their respective fully tryptic parental peptides in the standard protein digest. It should be noted that the N-terminal portion b-ion type ISF products are not readily identified in typically SEQUEST database searching because the loss of a water molecule at the C-terminus during ISF is often not accounted for in the searching parameters. Nevertheless, the b-ion type in-source fragments with significantly low abundances were also observed. Fig. 1A shows a complete series of extracted ion chromatograms of the in-source fragments from a fully tryptic parental peptide, AVVQDPALKPLALVYGEATSR, from bovine carbonic anhydrase II. All y-ion type in-source fragments, from y19 to y4, with detectable m/z range in orbitrap-MS (400–2000, m/z) and nine b-ions were observed with the same elution time (35.79 min). Among the sixteen y-ion type in-source fragments, ten ions, y19 - y10, were identified by SEQUEST search as partially tryptic peptides while any b-ion type in-source fragments were not identified, not surprising due to the loss of a water molecule in the C-terminus.
Figure 1.

Representative extracted ion chromatograms of AVVQDPALKPLALVYGEATSR from bovine carbonic anhydrase II in standard protein mixture (A) and VIISAPSADAPMFVMGVNHEK from glyceraldehyde-3-phosphate dehydrogenase in mouse brain lysate (B) along with their detectable in-source fragments.
When examining much more complex samples such as mouse brain, several in-source fragments were also frequently observed, but with much lower intensities. Fig. 1B shows the extracted ion chromatogram of VIISAPSADAPMFVMGVNHEK from glyceraldehyde-3-phosphate dehydrogenase in mouse brain and its co-eluted in-source fragments, five y-ion and two b-ion type in-source fragments with <2% and <0.1% intensities of the fully tryptic parental peptide, respectively. For these in-source fragments, only the y19 and y16 ions were identified by the SEQUEST search. It is notable that in-source fragments retain lower charge states than their corresponding parental peptides because the charge of the parental peptide is divided between daughter fragments. Similar patterns of ISF were observed in other mouse brain peptides (S. Fig. 1 and S. Table 2)
Proportions of ISF-generated peptides
Partially tryptic peptides can be classified into three groups as the [K/R].P-type, Y-ion type, and B-ion type (Table 1). The [K/R].P type of partially tryptic peptides retains -K.P- or -R.P- sequence on either N- or C-terminus, which are well-known as trypsin-inhibitory sequences. The Y-ion type has C-terminal lysine or arginine residue, while the B-ion type does not. Among the partially tryptic peptides, the [K/R].P type starting with the proline residue and Y-ion type peptides can be originated from in-source fragmentation, whereas the other types of in-source fragments cannot be identified by SEQUEST analysis using fully-tryptic search parameters without considering the water loss on the C-terminus. Considering that the partially tryptic peptides from ISF have the same elution time as their parental peptides, the elution times of partially tryptic peptides from ISF should often be much different from their predicted elution times. In other words, peptides whose actual elution times correlate well with their predicted elution times are more likely the result of proteolysis, not ISF. Based on this concept, we plotted the first observed scan number for all peptides identified against the predicted normalized elution times (NET) using an in-house NET prediction tool based on a previously published algorithm from our laboratory21, 22 (publicly available in http://omics.pnl.gov/software). Fig. 2 shows the plots from three different samples: a standard protein mixture, mouse brain, and mouse plasma. Almost all the fully tryptic peptides show a linear correlation (R2 > 0.97) between predicted NET values and the observed scan numbers, as expected. The identified B-ion type partially tryptic peptides also show similar correlation as the fully tryptic peptides for all three cases. As we noted, since the identified B-ion type partially tryptic peptides cannot be from ISF without considering water loss at the C-terminus, those well-aligned B-ion type partially tryptic peptides can be regarded as products of proteolytic cleavage. Unexpectedly, even the most [K/R].P type partially tryptic peptides show well-matched linear correlation (Fig. 2A–C), which suggests that some of the inhibitory sequences might still be cleavable by trypsin.4 Nevertheless, a significant number of misaligned points were observed as the Y-ion type partially tryptic peptides, which are potentially from ISF, especially in the standard protein mixture sample (Fig. 2A).
Table 1.
Types of partially tryptic peptides.
| Type of partially tryptic peptides | Description |
|---|---|
| [K/R].P type | [K/R].P---X[K/R].X′ or [K/R].X--P.[K/R] |
| Y-ion type | [not K/R].X---X′[K/R].X″ |
| B-ion type | [K/R].X---X′.[not K/R] |
X indicates any amino acid residue.
Figure 2.

Plots of first scan number of identified peptides versus predicted NET in standard protein mixture (A), mouse brain (B), and mouse plasma (C).
Manual inspection of the data confirmed our original speculation that the partially tryptic peptides observed at distant LC elution times from their expected elution times are most likely from ISF since these partially tryptic peptides were well aligned with their parental tryptic peptides in elution times. Furthermore, we did not observe any fully tryptic parental peptides for the [K/R].P-type or B-ion type partially tryptic peptides at the same elution time for all three datasets, which indicates that these types of partially tryptic peptides were generated by proteolytic cleavage.
Fig. 3 displays the percentages of each type of peptides in the three samples. At the unique peptide level, in-source fragments represent a large portion (~60 %) of the total peptide identifications from the standard protein mixture, although the in-source fragments are typically one to three orders of magnitude lower in their observed intensities compared to their fully tryptic parental peptides (Fig. 1). The portion of in-source fragments was smaller in the complex samples, where 1.1 % and 3.1 % of in-source fragments were observed from mouse brain and plasma, respectively. The observed significant portion of partially tryptic peptides (71% of 470 unique peptides) in standard proteins is highly consistent with the work of Picotti et al.,3 where it was reported that 78% (87/112) of the unique peptides in a bovine beta-lactoglobulin digest were partially tryptic; however, they concluded that trypsin might generate a number of non-specific cleavage products with much lower abundance by regarding most of the ISF-generated partially tryptic peptides as non-specific trypsin cleavage products. The lower charge state and significantly lower abundance of the ISF partially tryptic peptides as shown in Fig. 1 and S. Fig. 1 are also consistent with the data from Picotti et al.3 In principle, the level of in-source fragments should be dependent on ESI conditions and not dependent on sample complexity, the detectability of in-source fragments are largely dependent on the overall sample complexity due to the much lower abundance of in-source fragments compared to fully tryptic parental peptides. We attribute the large number of in-source artifacts identified in the standard protein mixture to the increased likelihood for low-abundance ions to be selected for MS/MS fragmentation for the low complex sample. However, in highly complex mouse brain lysate only a small portion (1.1% of 5270 unique peptides) of in-source fragments were observed (Fig. 2B and Fig. 3), while ~96% of identified peptides were fully tryptic. The result from mouse brain is consistent with the work of Olsen et al.,1 where they reported that trypsin cleaved exclusively C-terminal to arginine and lysine residues by demonstrating 97% (593/607) of peptides were fully tryptic in mouse liver lysate. It is notable that even if the numbers of identified in-source artifacts were significantly smaller in complex samples compared with standard protein digest, these in-source fragments still exist as shown in Fig. 1B, but they were not selected for MS/MS fragmentation in mouse brain sample due to their low abundance and so-called ‘under sampling’ in shotgun proteomics (S. Fig. 1A).
Figure 3.

Relative portions of each type of tryptic peptides at the unique peptide level in the three biological samples.
The identified in-source fragments in mouse plasma digest were mostly from highly abundance plasma proteins, e.g. serum albumin, transferrin, and apolipoprotein A-1 (S. Table 3). The increased portion (3.1%) of the ISF artifacts in plasma vs. mouse brain tissue can be attributed to the nature of plasma sample, which contains a number of highly abundant proteins. The in-source fragments from tryptic peptides of highly abundant proteins will have relatively competitive abundances compared to those tryptic peptides from mid- or low-abundant proteins, thus making these ISF-generated peptides to be more likely detectable. Also, a large portion (42%) of unique peptides in mouse plasma corresponds to the partially tryptic peptides unrelated to ISF (Fig. 3), in which the observed elution times of these peptides are well-aligned with the predicted ones similar to those fully tryptic peptides (Fig. 2C). This observation is reasonable for the plasma sample since plasma retains numerous secreted proteases and proteolytic processing events are more common in plasma. It is worthy to note that the removal of ISF-driving artifacts from all partially tryptic peptides by aligning predicted elution times with observed ones provide a means to identify in-vivo proteolytic cleavage or degradation products in biological samples. As an example, for serum albumin we identified a number of potential truncated products by aminopeptidases or carboxypeptidases (S. Table 4).
Sequence motifs of partially tryptic peptides
The cleavage motifs of partially tryptic peptides in mouse brain and plasma were investigated separately. Fig. 4 shows the sequence motifs of the ISF-generated partially tryptic peptides (Fig. 4A and B) and the other partially tryptic peptides, which are regarded as in-vivo or in-vitro proteolytic products (Fig. 4 C and D). The ISF artifacts show quite different sequence motifs from proteolysis-generated partially tryptic peptides. In both mouse brain (Fig. 4A) and plasma (Fig. 4B) samples, it appears that proline residue at P1’ position (right side of the cleaved peptide bond) and/or aliphatic residues such as valine, leucine, or isoleucine at P1 position (left side of the cleaved peptide bond) are more favorable for ISF. These residue-specific fragmentations are also well-known patterns for CID in MS/MS spectra,23, 24 providing further evidence that these partially tryptic peptides were from collisional dissociation in the electrospray source region.
Figure 4.
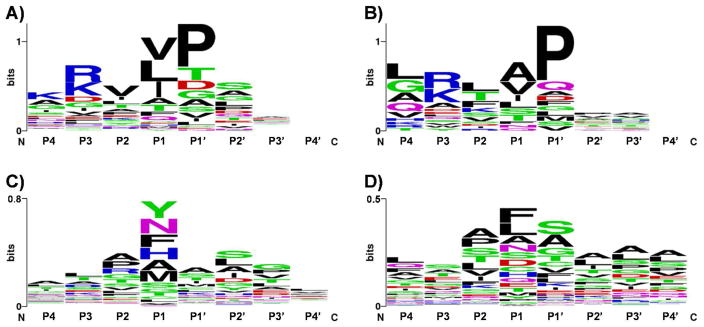
Sequence motifs around cleavage sites in in-source fragmentation artifacts from mouse brain (A) and mouse plasma (B). Sequence motifs of partially tryptic peptides after excluding in-source artifacts from mouse brain (C) and mouse plasma (D).
The sequence motifs from the proteolysis-generated partially tryptic peptides in mouse brain show a slightly higher frequency of chymotrypsin-susceptible residues, i.e. tyrosine and phenylalanine, at P1 position (Fig. 4C), which might indicate the presence of chymotrypsin-like proteases in the tissue sample, whereas no outstanding motif was observed in mouse plasma (Fig. 4D), which is actually expected because the plasma sample contains many different types of in-vivo proteolyzed proteins by various proteases. The sequence motif from partially tryptic peptides of trypsin only shows the preference of asparagine or alanine residue at P1 position (S. Fig. 2), which suggests that the pseudotrypsin25, 26 might have different specificity from the original trypsin due to partially altered three-dimensional structure. The alanine and asparagine residues with high frequency at P1 position in Fig. 4C might be partially counted to pseudotryptic activity of trypsin.
Interestingly, all of the [K/R].P type partially tryptic peptides in both brain lysate and plasma samples were observed with well-matched elution times as predicted for fully tryptic peptides, and no co-eluted potential fully tryptic parental peptide was observed for them. These observations strongly support that [K/R].P type partially tryptic peptides actually can be considered as fully tryptic peptides; i.e. some of [K/R]-P peptide bonds can be cleaved by trypsin as recently reported by Rodriguez et al.4 Furthermore, it appears that the [K/R].P type peptides contain small amino acids like glycine, alanine, and serine with relatively high frequency at both P2 and P2′ positions (S. Fig. 3A), whereas there is no preference of amino acid at P2 and P2′ positions in non-cleaved peptides. Of interest, however, is that carboxylic acid residues are occasionally present (S. Fig. 3B), and these residues are known to be unfavorable for trypsin digestion.27
Sources and impacts of partially tryptic peptides in shotgun proteomics
At first glance the stringency of trypsin specificity appears to be inconsistent between biological samples if the impact of in-source fragments is not considered, and which explains the discordance between several previous reports about trypsin specificity.1, 3, 4, 6 Biological samples with different levels of complexity were used in these reports, and the groups making shotgun data dependent measurements for complex proteome samples such as liver tissue and having significant under-sampling, report good trypsin specificity,1, 4 whereas the other groups dealing with much less complex samples, e.g. standard protein3 or in-gel digest6, observe significant levels of partially tryptic peptides. Our observation of ISF provides a solid explanation of the observed data and trypsin specificity. As shown in Fig. 3, the predominant source of partially tryptic peptides in standard proteins was ISF (81% of partially tryptic peptides). In mouse brain, since the [K/R].P-cleaved partially tryptic peptides could be regarded as fully tryptic peptides4 as we discussed, only 2% of peptides identified may arise from potentially unspecific activity of trypsin or in-vivo proteolysis. Therefore, it appears that the impact of trypsin-induced partially tryptic peptides is minimal (< 2% in number of unique peptides) in LC-MS/MS proteomics data for complex biological samples. For this reason, the identification of partially tryptic peptides is mostly likely from in-vivo or in-vitro proteolytic processing or degradation events after ISF-generated artifacts are removed. Table 2 and S. Table 4 provide some examples of the sources of identified partially tryptic peptides.
Table 2.
Example of sources of partially tryptic peptides in mouse plasma.
| Protein | Peptide | Origin |
|---|---|---|
| APOA1_MOUSE | S.PVAEEFR.D | in-source |
| Q.LAPHSEQMR.E | in-source | |
| A.PLGAELQESAR.Q | in-source | |
| L.SPVAEEFR.D | protease | |
| D.FWDNLEK.E | protease | |
| D.TLGSTVSQLQER.L | protease | |
| W.DTLGSTVSQLQER.L | protease | |
| N.LLENWDTLGSTVSQLQER.L | protease | |
| N.LNLLENWDTLGSTVSQLQER.L | protease | |
| S.SLGQQLNLNLLENWDTLGSTVSQLQER.L | protease | |
|
| ||
| HEMO_MOUSE | N.PVTGEVPPR.Y | in-source |
| R.WKNPITSVD.A | protease | |
| N.PITSVDAAFR.G | protease | |
| W.FWDFATR.T | protease | |
|
| ||
| IGG2B_MOUSE | L.PPPAEQLSR.K | in-source |
| S.VAHPASSTTVDK.K | protease | |
| C.SVAHPASSTTVDK.K | protease | |
| T.CSVAHPASSTTVDK.K | protease | |
| T.CSVAHPASSTTVDKK.L | protease | |
In summary, we report ISF is a major source of partially tryptic peptides in LC-MS/MS proteomics. The impact of ISF artifacts is much less significant for data-dependent measurements for more complex mixtures due to the larger extent of under sampling of low-abundance species. The in-source fragments were distinguished from proteolysis-generated partially tryptic peptides based on elution time criteria, in which the majority of the in-source fragments showed much later elution times compared to their predicted elution times. By excluding such ISF artifacts one can much more effectively identify in-vivo or in-vitro proteolytic products, as well as better characterize protease activities.
Supplementary Material
Acknowledgments
Portions of this work were supported by the NIH Director’s New Innovator Award Program DP2OD006668, and NIH grants 8P41 GM103493 and 5P41 RR018522. The experimental work described herein was performed in the Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by the DOE and located at Pacific Northwest National Laboratory, which is operated by Battelle Memorial Institute for the DOE under Contract DE-AC05-76RL0 1830.
Abbreviations
- ISF
in-source fragmentation
- NET
normalized elution times
Footnotes
Supporting Information Available: This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Olsen JV, Ong SE, Mann M. Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol Cell Proteomics. 2004;3 (6):608–614. doi: 10.1074/mcp.T400003-MCP200. [DOI] [PubMed] [Google Scholar]
- 2.Chalkley RJ, Baker PR, Hansen KC, Medzihradszky KF, Allen NP, Rexach M, Burlingame AL. Comprehensive analysis of a multidimensional liquid chromatography mass spectrometry dataset acquired on a quadrupole selecting quadrupole collision cell time-of-flight mass spectrometer - I. How much of the data is theoretically interpretable by search engines? Mol Cell Proteomics. 2005;4(8):1189–1193. doi: 10.1074/mcp.D500001-MCP200. [DOI] [PubMed] [Google Scholar]
- 3.Picotti P, Aebersold R, Domon B. The implications of proteolytic background for shotgun proteomics. Mol Cell Proteomics. 2007;6(9):1589–1598. doi: 10.1074/mcp.M700029-MCP200. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez J, Gupta N, Smith RD, Pevzner PA. Does trypsin cut before proline? J Proteome Res. 2008;7(1):300–305. doi: 10.1021/pr0705035. [DOI] [PubMed] [Google Scholar]
- 5.Strader MB, Tabb DL, Hervey WJ, Pan C, Hurst GB. Efficient and specific trypsin digestion of microgram to nanogram quantities of proteins in organic-aqueous solvent systems. Anal Chem. 2006;78(1):125–34. doi: 10.1021/ac051348l. [DOI] [PubMed] [Google Scholar]
- 6.Sergeant K, Pinheiro C, Hausman JF, Ricardo CP, Renaut J. Taking advantage of nonspecific trypsin cleavages for the identification of seed storage proteins in cereals. J Proteome Res. 2009;8(6):3182–90. doi: 10.1021/pr801093f. [DOI] [PubMed] [Google Scholar]
- 7.Gupta N, Hixson KK, Culley DE, Smith RD, Pevzner PA. Analyzing protease specificity and detecting in vivo proteolytic events using tandem mass spectrometry. Proteomics. 2010;10(15):2833–2844. doi: 10.1002/pmic.200900821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burkhart JM, Schumbrutzki C, Wortelkamp S, Sickmann A, Zahedi RP. Systematic and quantitative comparison of digest efficiency and specificity reveals the impact of trypsin quality on MS-based proteomics. J Proteomics. 2012;75(4):1454–1462. doi: 10.1016/j.jprot.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 9.Kostka V, Carpenter FH. Inhibition of Chymotrypsin Activity in Crystalline Trypsin Preparations. J Biol Chem. 1964;239:1799–803. [PubMed] [Google Scholar]
- 10.Rice RH, Means GE, Brown WD. Stabilization of bovine trypsin by reductive methylation. Biochim Biophys Acta. 1977;492(2):316–21. doi: 10.1016/0005-2795(77)90082-4. [DOI] [PubMed] [Google Scholar]
- 11.Loo JA, Edmonds CG, Smith RD. Primary sequence information from intact proteins by electrospray ionization tandem mass spectrometry. Science. 1990;248(4952):201–4. doi: 10.1126/science.2326633. [DOI] [PubMed] [Google Scholar]
- 12.Smith RD, Loo JA, Barinaga CJ, Edmonds CG, Udseth HR. Collisional Activation and Collision-Activated Dissociation of Large Multiply Charged Polypeptides and Proteins Produced by Electrospray Ionization. J Am Soc Mass Spectrom. 1990;1(1):53–65. doi: 10.1016/1044-0305(90)80006-9. [DOI] [PubMed] [Google Scholar]
- 13.Katta V, Chowdhury SK, Chait BT. Use of a single-quadrupole mass spectrometer for collision-induced dissociation studies of multiply charged peptide ions produced by electrospray ionization. Anal Chem. 1991;63(2):174–8. doi: 10.1021/ac00002a016. [DOI] [PubMed] [Google Scholar]
- 14.Purvine S, Eppel JT, Yi EC, Goodlett DR. Shotgun collision-induced dissociation of peptides using a time of flight mass analyzer. Proteomics. 2003;3(6):847–50. doi: 10.1002/pmic.200300362. [DOI] [PubMed] [Google Scholar]
- 15.Xia Y, Liang X, McLuckey SA. Sonic spray as a dual polarity ion source for ion/ion reactions. Anal Chem. 2005;77(11):3683–9. doi: 10.1021/ac0481811. [DOI] [PubMed] [Google Scholar]
- 16.Ramos AA, Yang H, Rosen LE, Yao X. Tandem parallel fragmentation of peptides for mass spectrometry. Anal Chem. 2006;78(18):6391–7. doi: 10.1021/ac060672t. [DOI] [PubMed] [Google Scholar]
- 17.Chakraborty AB, Berger SJ, Gebler JC. Use of an integrated MS-multiplexed MS/MS data acquisition strategy for high-coverage peptide mapping studies. Rapid Commun Mass Spectrom. 2007;21(5):730–744. doi: 10.1002/rcm.2888. [DOI] [PubMed] [Google Scholar]
- 18.Livesay EA, Tang K, Taylor BK, Buschbach MA, Hopkins DF, LaMarche BL, Zhao R, Shen Y, Orton DJ, Moore RJ, Kelly RT, Udseth HR, Smith RD. Fully automated four-column capillary LC-MS system for maximizing throughput in proteomic analyses. Anal Chem. 2008;80(1):294–302. doi: 10.1021/ac701727r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim S, Gupta N, Pevzner PA. Spectral probabilities and generating functions of tandem mass spectra: a strike against decoy databases. J Proteome Res. 2008;7(8):3354–63. doi: 10.1021/pr8001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim S, Mischerikow N, Bandeira N, Navarro JD, Wich L, Mohammed S, Heck AJR, Pevzner PA. The Generating Function of CID, ETD, and CID/ETD Pairs of Tandem Mass Spectra: Applications to Database Search. Mol Cell Proteomics. 2010;9(12):2840–2852. doi: 10.1074/mcp.M110.003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petritis K, Kangas LJ, Ferguson PL, Anderson GA, Pasa-Tolic L, Lipton MS, Auberry KJ, Strittmatter EF, Shen YF, Zhao R, Smith RD. Use of artificial neural networks for the accurate prediction of peptide liquid chromatography elution times in proteome analyses. Anal Chem. 2003;75(5):1039–1048. doi: 10.1021/ac0205154. [DOI] [PubMed] [Google Scholar]
- 22.Petritis K, Kangas LJ, Yan B, Monroe ME, Strittmatter EF, Qian WJ, Adkins JN, Moore RJ, Xu Y, Lipton MS, DGC, Smith RD. Improved peptide elution time prediction for reversed-phase liquid chromatography-MS by incorporating peptide sequence information. Anal Chem. 2006;78(14):5026–5039. doi: 10.1021/ac060143p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang Y, Triscari JM, Tseng GC, Pasa-Tolic L, Lipton MS, Smith RD, Wysocki VH. Statistical characterization of the charge state and residue dependence of low-energy CID peptide dissociation patterns. Anal Chem. 2005;77(18):5800–13. doi: 10.1021/ac0480949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Y, Tseng GC, Yuan S, Pasa-Tolic L, Lipton MS, Smith RD, Wysocki VH. A data-mining scheme for identifying peptide structural motifs responsible for different MS/MS fragmentation intensity patterns. J Proteome Res. 2008;7(1):70–9. doi: 10.1021/pr070106u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith RL, Shaw E. Pseudotrypsin. A modified bovine trypsin produced by limited autodigestion. J Biol Chem. 1969;244(17):4704–12. [PubMed] [Google Scholar]
- 26.Keil-Dlouha VV, Zylber N, Imhoff J, Tong N, Keil B. Proteolytic activity of seudotrypsin. FEBS Lett. 1971;16(4):291–295. doi: 10.1016/0014-5793(71)80373-3. [DOI] [PubMed] [Google Scholar]
- 27.Riviere LR, Tempst P. Enzymatic digestion of proteins in solution. Curr Protoc Protein Sci. 2001;Chapter 11(Unit 11.1) doi: 10.1002/0471140864.ps1101s00. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.