

Abstract
Alfentanil is a validated probe for hepatic, first-pass, and intestinal cytochrome P450 (CYP) 3A activity, using plasma clearances, single-point concentrations and noninvasive pupil diameter change (miosis). Assessing intravenous and oral drug disposition typically requires separate dosing. This investigation evaluated concurrent administration of oral deuterated and intravenous unlabeled alfentanil, to assess both intestinal and hepatic CYP3A, and compare sequential and simultaneous dosing. Alfentanil disposition was evaluated after strong hepatic and/or intestinal CYP3A induction and inhibition by rifampin, ketoconazole, and grapefruit juice. Using plasma alfentanil concentrations and area under the curve, clearance, or single-point concentrations, both simultaneous and sequential dosing provided equivalent results and detected hepatic and intestinal CYP3A induction and inhibition. Miosis better detected CYP3A modulation with sequential vs simultaneous dosing. These results show that concurrent oral deuterated and intravenous alfentanil, administered either sequentially or simultaneously, is an efficient and effective approach to assessing hepatic and intestinal CYP3A activity.
Keywords: alfentanil, cytochrome P450 3A, CYP3A, in vivo probe, phenotyping
Cytochrome P450 (CYP) 3A subfamily enzymes are among the most clinically significant in human drug metabolism, and a major determinant of systemic and first-pass drug clearance in adults and children.1–3 CYP3A4 is the most abundant CYP in the liver and intestine, metabolizes >50% of all drugs, and CYP3A5 metabolizes many CYP3A4 substrates and is polymorphically expressed.4,5 CYP3A activity exhibits marked inter- and intra-individual differences as a result of variable expression, genetic polymorphism, and exquisite sensitivity to drug and dietary interactions, causes significant alternations in drug bioavailability, clearance and clinical effect, and challenges the development and safe use of CYP3A drugs.1
Considerable effort has been expended toward identifying and developing an in vivo probe for assessment of human hepatic and intestinal CYP3A activity (or phenotype), prediction of CYP3A-dependent drug metabolism, and individualized dosing of CYP3A drugs with narrow therapeutic indices.6–10 FDA regulations for new drug development require clinical assessment of potential drug interactions, and preferably, their consequences. Clinically, there is profound interest in using CYP3A activity assessment to predict optimal dosing, improve therapeutic efficacy, and minimize adverse drug effects.11,12
The CYP3A substrates alfentanil (ALF) and midazolam exhibit most of the characteristics desired of an ideal CYP3A probe.6 Midazolam is the most widely used CYP3A probe. Both are exclusive CYP3A (CYPs 3A4 and 3A5) substrates, neither are P-glycoprotein substrates, and they have similar intestinal extraction, although ALF hepatic extraction is approximately half that of midazolam.13,14 ALF may have some advantages compared with midazolam, because ALF half-life is shorter and hence so also are required sampling periods, and midazolam clearance may be influenced by hepatic blood flow in addition to CYP3A isoform activity.15 The most common CYP3A phenotyping approach uses plasma concentrations and intravenous and oral clearance (or area under the curve). ALF has thus been used to probe hepatic, intestinal and first-pass CYP3A isoform activities.13,14,16–26
Towards the goal of simplifying CYP3A phenotyping, other approaches have also been implemented. For example, limited sampling strategies have been used as a surrogate for midazolam clearance, such as reduced or single-point plasma concentration measurements or metabolite:parent ratios, with variably reported accuracy, success and enthusiasm for their clinical utility.22,27–33 Single-point plasma ALF concentrations have also been used successfully as a surrogate for ALF clearance and to assess hepatic and first-pass CYP3A isoform activity.22,23
Assessment of intravenous and oral drug clearances typically require their administration by different routes on separate days. Stable-label isotope analogues have been used in pharmacokinetic investigations,34 and enable, in conjunction with mass spectrometry, the simultaneous administration of drugs by different routes. This reduces the number of study days, blood sampling and analytical determinations (typically by half), eliminates interday variability and thus improves results, shortens overall study duration, and reduces study costs. Simultaneous administration of oral stable isotope 15N3-midazolam and intravenous unlabeled midazolam has been used to assess hepatic and first-pass CYP3A isoform activity.35–37 Other approaches to increasing the efficiency of CYP3A isoform phenotyping with unlabeled midazolam have been reported, such as semisimultaneous administration, in which oral dosing was followed 6 hr later by an intravenous infusion.38
This investigation was a pilot study to evaluate administration of oral deuterated alfentanil (d3-ALF) and intravenous unlabeled alfentanil (d0-ALF) in healthy volunteers on the same day, to concurrently assess both hepatic and first-pass (intestinal) CYP3A isoforms. A second purpose was to evaluate two different paradigms for administration (simultaneous intravenous and oral dosing, or semi-simultaneous, sequential dosing of intravenous then oral ALF), with the optimal paradigm to be used in subsequent studies of CYP3A isoforms. ALF disposition was evaluated after strong hepatic and intestinal CYP3A induction, strong hepatic and intestinal CYP3A inhibition, and selective intestinal CYP3A inhibition.
Results
Preliminary experiments showed that CYP3A4-catalyzed ALF metabolism, based on substrate disappearance, was not different for d0- and d3-ALF (not shown). Hence d3-ALF was used clinically knowing that deuterium substitution on the propionyl side chain did not affect in vitro ALF metabolism.
The subjects for this investigation were 28±6 yr (range 21–33) and 79±17 kg (range 63–106). Intravenous (d0) ALF plasma concentrations after sequential and simultaneous administration of IV d0-ALF and oral d3-ALF, and the influence of hepatic CYP3A induction and inhibition, are shown in Figure 1; pharmacokinetic parameters are in Table 1. Both measured and dose-normalized concentrations are provided, the latter to show the true magnitude of ketoconazole effects (since ALF doses were reduced in these subjects). IV ALF, whether administered alone (sequential) or together (simultaneous) with oral ALF, detected hepatic CYP3A isoforms induction and inhibition, with significant differences between controls and rifampin- or ketoconazole-treated subjects in AUC0-∞/D, CLIV, t1/2, Vss and EH. There was little or no influence of grapefruit juice on AU C0-∞/D, CLIV, t1/2, or EH. There were no differences in ALF kinetic parameters between sequential or simultaneous administration in rifampin-, grapefruit juice, or ketoconazole-treated subjects.
Figure 1.

Effect of CYP3A induction and inhibition on intravenous alfentanil (d0-ALF) plasma concentrations. Subjects received nothing (controls, ●), rifampin (▲), grapefruit juice (■), or ketoconazole (▼). Results are shown as mean ± SD (n=6); some SD are omitted for clarity. The d0-ALF dose was 1.0 mg, except after ketoconazole pretreatment (0.5 mg d0-ALF). Panels A–C show measured concentrations, and panels D–F show dose-adjusted concentrations. Panels A and D show results for sequential IV d0-ALF and oral d3-ALF (separated by 3 hr) administration, panels B and E show results for simultaneous IV d0-ALF and oral d3-ALF administration, and panels C and E show the results superimposed (solid lines and symbols, and open symbols and dotted lines represent sequential and simultaneous dosing, respectively).
Table 1.
Intravenous alfentanil pharmacokinetic parameters
| Control | Rifampin | Grapefruit | Ketoconazole | |
|---|---|---|---|---|
| Sequential dosing | ||||
| Cmax/D (ng •ml−1 •mg−1) | 80±40 | 54±10 | 59±31 | 73±19 |
| AUC0-∞/D (ng •hr •ml−1 •mg−1) | 59±27 | 21±8a | 57±30 | 291±159a |
| AUC0-∞/D ratio (geometric mean, 90% CI) | 0.4 (0.3,0.4) | 1.0 (0.9,1.0) | 4.8 (4.3,5.3) | |
| CLIV (ml•kg−1•min−1) | 4.3±1.8 | 11.3±3.4a | 4.5±2.0 | 0.9±0.4a |
| Elimination t1/2 (hr) | 1.4±0.4 | 0.9±0.1a | 1.5±0.4 | 5.0±1.6a |
| Vss (L/kg) | 0.39±0.13 | 0.48±0.16a | 0.46±0.14a | 0.34±0.10 |
| EH | 0.26±0.09 | 0.71±0.22a | 0.28±0.11 | 0.05±0.02a |
| C4 hr/D (ng • ml−1 •mg−1) | 3.1±2.3 | 0.3±0.1a | 2.9±1.8 | 21.6±9.6a |
| C4 hr/D ratio (geometric mean, 90% CI) | 0.1 (0.1,0.2) | 1.0 (0.8,1.1) | 7.8 (5.6,11.0) | |
| Simultaneous dosing | ||||
| Cmax/D (ng •ml−1 •mg−1) | 58±15b | 57±8 | 55±12 | 73±26 |
| AUC0-∞/D (ng •hr •ml−1 •mg−1) | 51±19 | 18±3a | 58±22 | 302±244a |
| AUC0-∞/D ratio (geometric mean, 90% CI) | 0.4 (0.2,0.7) | 1.2 (1.1,1.2) | 5.0 (2.9,8.6) | |
| CLIV (ml•kg−1•min−1) | 4.8±1.8 | 12.1±2.4a | 4.2±1.6 | 1.1±0.7a |
| Elimination t1/2 (hr) | 1.4±0.4 | 0.7±0.1a | 1.6±0.5 | 5.4±2.1a |
| Vss (L/kg) | 0.45±0.11 | 0.56±0.09a | 0.48±0.13 | 0.38±0.11a |
| EH | 0.30±0.10 | 0.76±0.16a | 0.25±0.08 | 0.06±0.03a |
| C4 hr/D (ng • ml−1 •mg−1) | 2.8±1.6 | 0.3±0.0a | 3.8±1.7b | 21.6±14.6a |
| C4 hr/D ratio (geometric mean, 90% CI) | 0.1 (0.1,0.2) | 1.4 (1.1,1.7) | 7.6 (5.2,11.0) | |
For sequential dosing subjects received 1 mg IV d0-ALF followed 3 hr later by 4 mg oral d3-ALF, except after ketoconazole pretreatment, where they received 0.5 mg IV d0-ALF and 1 mg oral d3-ALF. For simultaneous dosing subjects received IV d0-ALF and oral d3-ALF at the same time, in the same doses as for sequential administration. Results (n=6) are the arithmetic mean ± SD, except AUC ratios and single-point concentration ratios, which are also shown as the geometric mean and 90% confidence interval. Cmax/D, dose-normalized maximum concentration; AUC0-∞/D, dose-normalized area under plasma concentration–time curve extrapolated to infinity; CLIV, systemic clearance; t1/2, half-life; Vss, steady state volume of distribution; EH, hepatic extraction; C4 hr/D, dose-normalized plasma concentration 4 hr after dosing;
Significantly different from control (p<0.05)
Significantly different vs sequential dosing (p<0.05)
Single-point plasma concentrations, shown previously to be valid surrogates for both ALF AUC and clearance,22,23 were also evaluated. Dose-normalized IV ALF 4 hr concentrations (C4 hr/D), whether ALF was administered alone or with oral ALF, were significantly different from controls in rifampin- or ketoconazole-, but not grapefruit juice-treated subjects (Table 1). Dose-normalized ALF 2 hr concentrations gave similar results, detecting hepatic CYP3A induction and inhibition (not shown). With the exception of the 2 hr and 4 hr ALF/dose concentration in grapefruit juice-treated subjects, there were no differences in dose-normalized IV ALF single point concentrations between sequential or simultaneous ALF administration.
Oral (d3) ALF plasma concentrations after sequential and simultaneous administration of IV d0-ALF and oral d3-ALF, and the influence of intestinal CYP3A modulation are shown in Figure 2. Oral ALF, whether administered after (sequential) or together (simultaneous) with IV ALF, detected first-pass and intestinal CYP3A induction and inhibition, with significant differences between controls and rifampin-, grapefruit juice- and ketoconazole-treated subjects in ALF Cmax/D, AUC0-∞/D, CL/F, t1/2, bioavailability, and EG, except that ALF t1/2 was not affected by grapefruit juice (Table 2). There were small differences in some Cmax/D, AUC0-∞/D, and bioavailability parameters between sequential and simultaneous oral ALF administration.
Figure 2.

Effect of CYP3A induction and inhibition on oral alfentanil (d3-ALF) plasma concentrations. Subjects received nothing (controls, ●), rifampin (▲), grapefruit juice (■), or ketoconazole (▼). Results are shown as mean ± SD (n=6); some SD are omitted for clarity. The d3-ALF dose was 4.0 mg, except after ketoconazole pretreatment (1.0 mg d3-ALF). Panels A–C show measured concentrations, and panels D–F show dose-adjusted concentrations. Panels A and D show results for sequential IV d0-ALF and oral d3-ALF (separated by 3 hr) administration, panels B and E show results for simultaneous IV d0-ALF and oral d3-ALF administration, and panels C and E show the results superimposed (solid lines and symbols, and open symbols and dotted lines represent sequential and simultaneous dosing, respectively).
Table 2.
Oral alfentanil pharmacokinetic parameters
| Control | Rifampin | Grapefruit | Ketoconazole | |
|---|---|---|---|---|
| Sequential dosing | ||||
| Cmax/D (ng •ml−1 •mg−1) | 12±6 | 1.5±0.9a | 13±7 | 33±10a |
| Tmax (hr) | 1.0±0.8 | 0.4±0.2 | 1.4±1.0 | 1.3±1.0 |
| AUC0-∞/D (ng •hr •ml−1 •mg−1) | 27±17 | 1.6±0.8 | 45±27 | 253±178a |
| AUC0-∞/D ratio (geometric mean, 90% CI) | 0.06 (0.05,0.07) | 1.7 (1.5,1.9) | 9.2 (8.0,10.7) | |
| CL/F (ml•kg−1•min−1) | 11.1±6.8 | 168±74a | 6.4±3.8a | 1.2±0.6a |
| Elimination t1/2 (hr) | 1.2±0.3 | 0.6±0.1a | 1.4±0.5 | 4.7±2.1a |
| Foral | 0.43±0.09 | 0.08±0.03a | 0.76±0.13a | 0.83±0.11a |
| EG | 0.42±0.07 | 0.81±0.06a | 0.02±0.04a | 0.14±0.08a |
| C4 hr/D (ng • ml−1 •mg−1) | 2.2±2.0 | 0.02±0.03a | 4.7±2.9a | 20.7±8.7a |
| C4 hr/D ratio (geometric mean, 90% CI) | 0.01 (0.00,0.02) | 2.3 (1.9,2.8) | 11.6 (7.5, 17.9) | |
| Simultaneous dosing | ||||
| Cmax/D (ng •ml−1 •mg−1) | 7.1±3.0b | 0.9±0.5a,b | 13±9a | 36±20a |
| Tmax (hr) | 1.4±0.4 | 0.6±0.2 | 1.6±1.3 | 1.7±0.9 |
| AUC0-∞/D (ng •hr •ml−1 •mg−1) | 22±14 | 1.1±0.5 | 46±21 | 298±255a,b |
| AUC0-∞/D ratio (geometric mean, 90% CI) | 0.06 (0.01,0.22) | 2.2 (1.9,2.5) | 12.0 (6.2,23.3) | |
| CL/F (ml•kg−1•min−1) | 13.6±8.2 | 224±101a,b | 6.0±3.7a | 1.2±0.8a |
| Elimination t1/2 (hr) | 1.4±0.5 | 0.6±0.1a | 1.8±0.8 | 5.2±1.9a |
| Foral | 0.41±0.11 | 0.06±0.03a | 0.76±0.14a | 0.95±0.08a,b |
| EG | 0.42±0.11 | 0.74±0.12a | 0.06±0.10a | 0.02±0.04a |
| C4 hr/D (ng • ml−1 •mg−1) | 2.2±1.1 | 0.03±0.02a | 5.6±2.4a | 23.6±16.7a |
| C4 hr/D ratio (geometric mean, 90% CI) | 0.01 (0.01,0.02) | 2.8 (2.2, 3.7) | 11.3 (8.4,15.1) | |
For sequential dosing subjects received 1 mg IV d0-ALF followed 3 hr later by 4 mg oral d3-ALF, except after ketoconazole pretreatment, where they received 0.5 mg IV d0-ALF and 1 mg oral d3-ALF. For simultaneous dosing subjects received IV d0-ALF and oral d3-ALF at the same time, in the same doses as for sequential administration. Results (n=6) are the arithmetic mean ± SD, except AUC ratios and single-point concentration ratios, which are also shown as the geometric mean and 90% confidence interval. Cmax/D, dose-normalized maximum concentration; Tmax, time of maximum concentration; AUC0-∞/D, dose-normalized area under plasma concentration–time curve extrapolated to infinity; CL/F, apparent oral clearance; t1/2, half-life; Foral, oral bioavailability; EG, intestinal extraction; C4 hr/D, dose-normalized plasma concentration 4 hr after dosing.
Significantly different from control (p<0.05)
Significantly different vs sequential dosing (p<0.05)
Dose-normalized oral ALF 4 hr concentrations (C4 hr/D), whether ALF was administered alone or with IV ALF, were significantly different from controls in rifampin-, grapefruit juice- and ketoconazole-treated subjects (Table 2). Dose-normalized oral ALF 2 hr concentrations gave similar results, detecting first-pass and intestinal CYP3A induction and inhibition (not shown). There were no differences in dose-normalized oral ALF single point concentrations between sequential or simultaneous ALF administration.
Total (summed IV d0- and oral d3-ALF) plasma concentrations are shown in Figure 3. Washout of both IV and oral ALF was clearly visible after sequential, but not simultaneous dosing. Total plasma ALF concentrations were not appreciably greater after simultaneous compared with sequential dosing.
Figure 3.
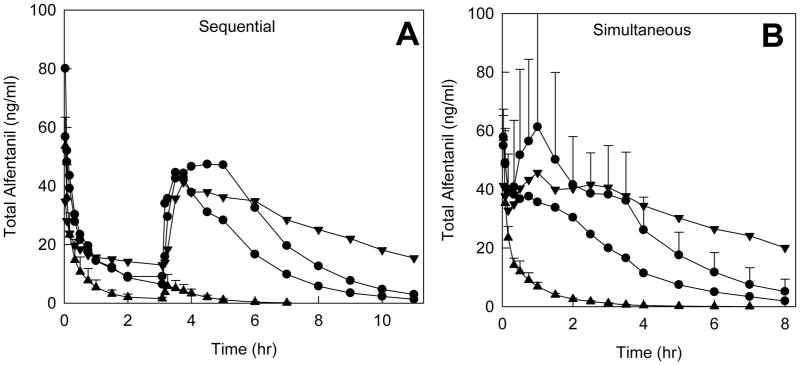
Effect of CYP3A induction and inhibition on total (d0 and d3-ALF) plasma concentrations. Drug doses and symbols are the same as described in the legends to Figs 1 and 2. Panels A and B show results for sequential and simultaneous IV and oral ALF administration, respectively. Results are the mean (n=6); SD are omitted for clarity.
Previous studies have shown that the decrease in dark-adapted pupil diameter (miosis) caused by ALF is a surrogate for plasma ALF concentrations, and that ALF miosis is a suitable noninvasive in vivo probe for both hepatic and first-pass CYP3A isoform activity.13,14,23 Using ALF pharmacodynamic parameters determined previously,14 total ALF concentrations were used to predict miosis after sequential and simultaneous IV and oral ALF dosing (Figure 4A and B). Actual pupil diameter changes are shown in Figure 4C and D. Washout of both IV and oral ALF was visible after sequential, but not simultaneous dosing.
Figure 4.

Effect of CYP3A induction and inhibition on ALF miosis (dark-adapted pupil diameter change from baseline). Panels A and C show results for sequential IV and oral ALF administration; panels B and D show results for simultaneous IV and oral ALF dosing. Panels A and B show predicted miosis, based on the total ALF concentrations shown in Figure 3. Panels C and D show actual results.
The experimental protocol was safely conducted without any serious adverse events. Respiratory depression, defined as an oxygen saturation <94% and need for supplemental oxygen, did not occur in any subject. Nausea or vomiting requiring treatment was experienced by only one subject, after grapefruit juice and sequential dosing, out of 48 total study sessions. Investigators’ subjective assessment of side effects was that some subjects experienced mild sedation, which was greater with simultaneous compared with sequential ALF administration, and more subjects noted feeling a drug effect with simultaneous dosing.
Discussion
A requisite predicate for using stable-label analogs in drug disposition is that biotransformation is unaffected by the substitution. Since the propionyl side chain is not a site of ALF metabolism,39 it was therefore selected for deuterium substitution. In addition, since propionylation is the final step in ALF synthesis, deuteration at this step is most efficient and least likely to result in unwanted deuterium exchange. Since deuteration did not affect CYP3A4-catalyzed ALF metabolism, d3-ALF was used clinically assuming that propionyl side chain deuteration did not affect clinical ALF metabolism or clearance. This assumption was verified, because the elimination rate constants for d0- and d3-ALF were not different, and the pharmacokinetic parameters for oral d3-ALF were similar to those reported previously for oral d0-ALF.13,14,20,21,23–26
The first major finding of this investigation was that oral (d3) ALF could be safely administered together with IV ALF, either sequentially or simultaneously, and in a fixed-dose paradigm. Both sequential and simultaneous dosing were well-tolerated, with no serious adverse events. This was no different than previous experience with ALF, given either orally or IV alone, on different days.8,13,14,21,23–26,40 More subjects reported experiencing a drug effect, and observers reported that subjects appeared mildly drowsy, after simultaneous compared with sequential administration. Nevertheless, objective measures (maximum miosis and peak ALF concentrations) were similar, regardless of the administration paradigm (Figures 3 and 4). This suggests that subjects’ and observers’ perceptions were related more to the time course than magnitude of ALF plasma concentrations.
In previous studies, ondansetron was administered for antiemetic prophylaxis because of theoretical concerns about opioid-related nausea/vomiting. In this investigation, prophylactic ondansetron was not administered, which was without consequence (there was only one instance of nausea/vomiting, and it is not known whether it was caused by the grapefruit juice or ALF). Prophylactic ondansetron therefore does not appear necessary with ALF for CYP3A isoform phenotyping.
ALF in this investigation was administered in a fixed dose, while previous investigations used weight-based dosing.13,14,16–26 The latter simply reflected the clinical practice of weight-based dosing for fentanyl-series (alfentanil, sufentanil, fentanyl, remifentanil) opioids. Present ALF dosing was 1 mg IV and 4 mg orally (0.5 and 1 mg with ketoconazole). IV ALF doses used previously were 15 μg/kg in controls and where CYP3A induction or weak CYP3A inhibition was anticipated, and 5–10 μg/kg with strong CYP3A inhibition. The fixed doses used herein were equivalent to 13 ± 2 μg/kg in control, rifampin, and grapefruit juice sessions, and 6 ± 1 μg/kg after ketoconazole. Oral ALF doses used previously were 40, 43, 60 or 75 μg/kg with controls, CYP3A induction, or weak CYP3A inhibition, and 4.3 or 23 μg/kg with strong or weak CYP3A inhibition. The fixed oral ALF doses used herein were equivalent to 52 ± 10 μg/kg control, rifampin, and grapefruit juice sessions, and 13 ± 3 μg/kg after ketoconazole. Total (IV and oral) ALF doses were 65 ± 13 μg/kg in control, rifampin, and grapefruit juice sessions, and 20 ± 4 μg/kg after ketoconazole. Thus the present fixed doses were within the range of doses used previously.
A potential concern with simultaneous IV and oral ALF is total plasma ALF concentrations and risk of side effects. Because oral ALF concentrations peak about 1 hr after dosing, and IV concentrations typically decrease to half by 1 hr, total ALF plasma concentrations were in the same range as in previous studies. Variability in the dose per kg, which occurs with fixed dosing, did not interfere with the ability of ALF to detect alterations in CYP3A isoform activity. Plasma concentrations and miosis were also within ranges observed previously. Thus the fixed doses selected, 1 mg IV and 4 mg orally, appear appropriate, and can be recommended for subsequent use. Higher fixed oral doses may also be used (with controls, CYP3A induction, moderate CYP3A inhibition), if greater miosis, comparable to that seen previously at higher ALF doses, is desired.13,20
The second major finding of this investigation was that concurrent administration of oral d3-ALF and IV d0-ALF, either sequentially or simultaneously, allowed concurrent assessment of hepatic, first-pass, and intestinal CYP3A isoform activity, and detected hepatic and intestinal CYP3A isoform modulation. Hepatic and intestinal CYP3A isoform induction and inhibition were detected using both ALF plasma AUCs and single point concentrations, using either sequential or simultaneous IV/oral dosing. Results were similar to those obtained with IV and oral ALF on separate days.8,13,14,21,23–26,40 The major advantage of concurrent IV and oral dosing is the ability to simultaneously assess hepatic, first-pass, and intestinal CYP3A isoform activities, and in one rather than two separate days, with consequent advantages of greater efficiency, reduced analytical and subject reimbursement costs, and the elimination of interday variability between IV and oral sessions. In addition to these cost-effectiveness considerations, there are other practical aspects of the proposed approach. Synthesis of d3-ALF is facile, and well-described, deuteration occurs at the final synthetic step, and gram quantities may be synthesized while clinical dosing requires only 1–4 mg.
One minor difference in this, compared with previous investigations, was the use of measured hematocrits compared with population estimates, for calculating hepatic plasma flow. Previous studies used population hematocrits of 40 and 36 in men and women, respectively.13,14,23 In the present investigation, hematocrits overall were 42±1 and 31±3 in males and females, respectively, and differed little across sessions (42±1, 42±1, 42±1, and 40±1 in males, and 33±1, 32±3, 31±2, and 30±4 in females, in control, rifampin, grapefruit juice, and ketoconazole sessions, respectively). Interday variations, and differences between measured and population hematocrits, had little influence on pharmacokinetic parameter estimates. For example, with sequential dosing, EH in control, rifampin, grapefruit juice, and ketoconazole sessions was 0.26±0.09, 0.71±0.22, 0.28±0.11, and 0.05±0.02 using measured hematocrits, and 0.27±0.10, 0.72±0.22, 0.29±0.12, and 0.06±0.03 using population hematocrits. Therefore, using actual vs population hematocrit values had little effect on the use of ALF as a CYP3A isoform probe.
The third major finding of this investigation was that both paradigms for ALF administration (simultaneous IV and oral dosing, or sequential IV ALF followed 3 hr later by oral ALF), gave similar although not identical results regarding ALF plasma concentrations and pharmacokinetic parameters, and the same conclusions regarding the influence of CYP3A isoform induction or inhibition. The conclusion, therefore, is that either simultaneous or sequential IV/oral dosing is applicable for CYP3A isoform phenotyping with ALF plasma concentrations. Previous investigations showed that simultaneous IV and oral administration of unlabeled and 15N3-midazolam, respectively, successfully probed hepatic, first-pass, and intestinal CYP3A isoform activity.35–37 Another investigation used sequential administration or unlabeled midazolam, in which oral dosing was followed 6 hr later by a 30 min IV infusion and 6 hr blood sampling.38 ALF may have advantages over these approaches with midazolam. The shorter half-life of ALF compared with midazolam (1.5 vs 4 hr) means that sampling duration (for example, to capture 3–4 half-lives) can be much shorter with ALF, particularly with simultaneous dosing. In addition, with semisimultaneous dosing of unlabeled midazolam, washout curves overlap and the curve fitting and parameter estimates require more complex modeling, and 6 hr sampling captures only 1.5 half-lives under control conditions (and even less with CYP3A isoform inhibition).
Although either simultaneous or sequential ALF dosing is applicable for CYP3A isoform phenotyping with ALF plasma concentrations, phenotyping with miosis suggests different conclusions. With simultaneous IV and oral ALF, miosis cannot distinguish hepatic from intestinal changes in CYP3A isoform activity caused by induction or inhibition, while this is possible with sequential dosing. Thus with simultaneous dosing, miosis can be used to provide an overall assessment of CYP3A isoform induction/inhibition, followed by plasma ALF quantification to specifically assess hepatic and intestinal CYP3A. Alternatively, with sequential dosing both miosis and ALF concentrations provide specific information on both hepatic and first-pass CYP3A isoforms.
There are some limitations to this investigation. The study session order (sequential followed the next day by simultaneous administration) was not randomized. Therefore with simultaneous dosing subjects had one additional day of rifampin (6 vs 5 doses), ketoconazole (4 vs 3 doses) and grapefruit juice (4 vs 2 doses). Although rifampin is typically administered for 5d in drug interaction studies, after which CYP3A induction is approximately 80% complete, induction does increase somewhat with continuing administration for up to 10–14 days.41,42 In this investigation, the extra rifampin dose had no effect on the magnitude of intestinal or hepatic CYP3A isoform induction. Ketoconazole effects were somewhat greater (AUC0-∞/D and Foral but not CL/F, EG, or C4 hr/D) after an additional dose. With grapefruit juice, intestinal and hepatic CYP3A isoforms can be differentially affected.43,44 For example, one glass of double-strength grapefruit juice inhibited intestinal but not hepatic CYP3A isoforms, while four glasses over 2 days (or longer) inhibited hepatic and intestinal CYP3A. Nonetheless, in this investigation, the extra day of grapefruit juice had no effect on the magnitude of intestinal CYP3A inhibition, and did not alter hepatic CYP3A isoform activity. Therefore, the ordered design had no overall effect on the conclusions. Another potential limitation was that only six subjects were studied, because the investigation was powered to assess CYP3A isoform modulation, rather than differences between sequential vs simultaneous dosing. CYP3A isoform induction and inhibition were both detected by the sequential and the simultaneous dosing designs, with little difference between these paradigms in results and no difference in conclusions regarding CYP3A isoforms modulation. It is possible that a larger sample size might have detected small differences between sequential and simultaneous dosing, but would not have affected the conclusion that either approach appears valid for concurrent hepatic/intestinal CYP3A phenotyping.
In conclusion, deuterated oral ALF administered either simultaneously or sequentially after IV ALF can be used effectively and safely to probe hepatic, first-pass, and intestinal CYP3A isoform activities. Using plasma ALF concentrations, and ALF AUCs and clearances as the measure of CYP3A activity, both simultaneous and sequential dosing provide equivalent results and can detect hepatic and intestinal CYP3A isoform induction and inhibition. Using single point (2 or 4 hr) plasma ALF concentrations, both simultaneous and sequential dosing provide equivalent results and can detect hepatic and intestinal CYP3A isoform modulation. Using ALF miosis alone, without plasma ALF determinations, sequential dosing provides better estimates than simultaneous dosing, of CYP3A isoform induction and inhibition. Concurrent use of oral deuterated and IV ALF is an efficient and effective approach to phenotyping hepatic, first-pass, and intestinal CYP3A isoform activities.
Methods
Study Population and Protocol
Six healthy volunteers (3 males and 3 females) participated in this investigation, which was approved by the Washington University Institutional Review Board, after written informed consent was obtained. Subjects (28±6 yr, range 21–33) were in good health with no remarkable medical problems, within 30% of ideal body weight (79±17 kg, range 63–106), had no history of hepatic or renal disease, and were taking no medications or natural products known to alter CYP3A activity. Subjects using oral or implanted contraceptives were excluded, due to potential influences of rifampin on their effectiveness. Subjects were enrolled without regard to tobacco use.
The study was a 4-way randomized balanced crossover protocol, to compare simultaneous vs sequential dosing of d0- and d3-ALF, in the setting of normal, induced (by rifampin), and inhibited (by ketoconazole or grapefruit juice) CYP3A activity. The design was based on previous protocols with oral and IV ALF, most notably a similar study of normal, induced (by rifampin), and inhibited (by troleandomycin or grapefruit juice) CYP3A activity, in which IV and oral ALF were administered on different, rather than the same days.13,16,19 The present protocol used ketoconazole to inhibit CYP3A because troleandomycin is no longer available.
Subjects were studied in the clinical research center. They were instructed to abstain from alcohol for 24 hr prior to and during study days, caffeine-containing beverages on the day of ALF administration, and from grapefruit or grapefruit juice for 5 days before and during each study session, except for study purposes. For each visit, an IV catheter was placed in each arm for drug administration and blood sampling. Subjects were monitored with an automated blood pressure cuff and pulse oximeter after ALF administration, and received supplemental oxygen for an oxygen saturation less than 94%.
All ALF doses are expressed as that of the free base. Control subjects received IV unlabeled d0-ALF (1 mg bolus), followed 3 hr later by 4 mg oral deuterium-labeled d3-ALF. Deuterated ALF solid was dissolved in 25 ml water with thorough mixing immediately prior to oral ingestion. The vial was rinsed with an additional 25 ml water, which subjects also swallowed, followed by 100 ml water. The following day they received both 1 mg IV d0-ALF and 4 mg oral d3-ALF administered simultaneously. For hepatic and intestinal CYP3A induction, rifampin (600 mg orally) was administered at bedtime for 5 days, and the next day subjects received 1 mg IV bolus d0-ALF, followed 3 hr later by 4 mg oral d3-ALF with 100 ml water. That night, subjects received an additional dose of rifampin, and the next day they received both 1 mg IV d0-ALF and 4 mg oral d3-ALF simultaneously. For hepatic and intestinal CYP3A inhibition, ketoconazole (400 mg orally) was administered at bedtime for 3 days, and the next day subjects received 0.5 mg IV bolus d0-ALF, followed 3 hr later by 1 mg oral d3-ALF. That night, subjects received an additional dose of ketoconazole, and the next day they received both 0.5 mg IV d0-ALF and 1 mg oral d3-ALF simultaneously. For selective intestinal CYP3A inhibition, subjects drank grapefruit juice (8 oz orally) at bedtime. The next morning they received 1 mg IV bolus d0-ALF. Two hr after IV ALF they received double-strength grapefruit juice (3 oz). One hr later (3 hr after IV ALF) they received 4 mg oral d3-ALF. That night they drank an additional 8 oz of grapefruit juice. The following morning they received double-strength grapefruit juice (3 oz) followed 1 hr later by both 1 mg IV d0-ALF and 4 mg oral d3-ALF administered simultaneously. Subjects were fed a standard meal 5 hr after IVALF (2 hr after oral ALF) on sequential dosing sessions, or 3 hr after simultaneous IV and oral ALF, and had free access to food and water thereafter. Venous blood samples were obtained 0,2,5,10,20,30,45,60,90,120,180,185,190,195,210,225,240,270,300,360,420,480,540,600,660 min after IV d0-ALF (corresponding to 5,10,15,30,45,60,90,120,180,240,300,360,420,480 min after oral d3-ALF) on sequential dosing study days, and 0,2,5,10,20,30,45,60,90,120,150,180,210,240,300,360,420,480 min after IV d0-ALF and oral d3-ALF on simultaneous dosing study days. Plasma was separated and stored at −20°C for later analysis. Coincident with blood sampling, and at intermediate times, dark-adapted pupil diameter was measured using an infrared pupilometer as described previously,13,14 except that a NeurOptics PLR-200™ Pupillometer (NeurOptics, Irvine, CA) was used. Each recorded value was the mean of triplicate measurements. Simultaneous dosing occurred the day after the sequential study day. Session pairs were typically separated by approximately 1–3 weeks.
Study Drug
2H3-alfentanil (N-[1-[2-(4-ethyl-4,5-dihydro-5-oxo-1H-tetrazol-1-yl)ethyl]-4-(methoxymethyl)-4-piperidinyl]-N-phenyl-2H3-propanamide monohydrochloride; alfentanil-d3; d3-ALF) was synthesized as a nonradioactive, stable-label, trideuterated, pharmacologically identical analog of alfentanil (N-{1-[2-(4-ethyl-5-oxo-4,5-dihydro-1H-1,2,3,4-tetrazol-1-yl)ethyl]-4-(methoxymethyl) piperidin-4-yl}-N-phenylpropanamide monohydrochloride). It was synthesized at Washington University from 2H3-propionyl chloride using slight modifications of published techniques (United States Patent 7074935B2) and in adherence to FDA Good Manufacturing Practice Guidelines for Phase 1 Investigational Drugs. Based on gas and liquid chromatography-mass spectrometry analysis, the synthetic d3-alfentanil HCl monohydrate had at least 99.4% purity. The relative isotope incorporation was >98% d3-alfentanil, approximately 2% d2-alfentanil, and <0.03% d0-alfentanil. Deuterated ALF was used under IND 106010, and the solid was dissolved in 25 ml water with thorough mixing immediately before use.
Alfentanil metabolism
Metabolic stability of d0-ALF and d3-ALF (5 μM) was assessed using expressed human CYP3A4 with coexpressed P450 reductase and cytochrome b5 (BD Biosciences, San Jose, CA) as described previously.45 ALF disappearance was determined by mass spectrometry.
Analytical Methods
Plasma ALF concentrations were measured by HPLC-tandem mass spectrometry (LC-MS/MS) with multiple reaction monitoring (MRM) using a modification of previously described methods.13 Briefly, the chromatographic separation was accomplished using a Agilent Zorbex Eclipse XDB C18 column, 50 mm × 2.1 mm × 5 μm (Wilmington, DE) attached to a Shimadzu Prominence HPLC system (Columbia, MD). The MS/MS analysis was achieved using an ABI Sciex API 4000 QTRAP mass spectrometer (Foster City, CA. USA) equipped with a Turbo Ion Spray ionization source. The MRM transitions were 417.0 to 197.2 for d0-ALF and 420.0 to 197.2 for d3-ALF. Interday coefficients of variation were 8, 6, and 5% at 0.3, 5 and 60 ng/ml d0-ALF, and 6, 7, and 6% at the same concentrations of d3-ALF.
Pharmacokinetic Analysis
Data were analyzed using noncompartmental methods, as described previously.13,14 Systemic clearance of IV ALF was (CLIV)=DoseIV,/AUCIV,, apparent oral clearance was (CL/F)=Doseoral/AUCoral,, bioavailability was (Foral)=(AUCoral/Doseoral) × (DoseIV/AUCIV), steady-state volume of distribution was (Vss)=mean residence time × CL. Hepatic extraction (EH) was determined as (CLIV/Qp), where hepatic plasma flow (Qp) was estimated as the product of hepatic blood flow (25.3 and 25.5 ml/kg in males and females)36 and hematocrit (determined on the morning of the simultaneous ALF study day), and negligible extrahepatic ALF metabolism was assumed. Hepatic availability (FH) was 1−EH. Gastrointestinal availability (FG) was calculated as Foral/(FH × Fabs), where the oral dose was assumed to be entirely absorbed and thus Fabs was considered to be unity. Gastrointestinal extraction was (EG) = 1−FG.13,14
Statistical Analysis
Results are expressed as the mean ± SD or median. Two-way analysis of variance followed by the Student-Newman-Keuls test was used to test for significant differences between groups (SigmaPlot 11.2, Systat Corp, San Jose, CA). Non-normal data were log transformed for analysis of variance. The primary outcome measures were ALF clearance and AUC. Sample size was based on ALF clearance. Based on prior data,13 the standard deviation of the changes in ALF clearance were approximately one-quarter the magnitude of the mean change (3-fold) induced by rifampin. Assuming a similar relationship, studying 6 subjects would provide 95% power, at the α=0.01 level, to detect the change induced by rifampin. Ketoconazole was expected to produce similarly large changes.
Acknowledgments
This work was supported by National Institutes of Health Grants R01-GM63674, R01-DA14211, and K24-DA00417 (to EDK), and NCRR grant UL1 RR024992.
Footnotes
Clinical trials.gov number NCT01008059
Conflict of Interest/Disclosure
No author has any conflict of interest.
References
- 1.Wilkinson GR. Drug metabolism and variability among patients in drug response. N Engl J Med. 2005;352:2211–21. doi: 10.1056/NEJMra032424. [DOI] [PubMed] [Google Scholar]
- 2.Stevens JC. New perspectives on the impact of cytochrome P450 3A expression for pediatric pharmacology. Drug Discov Today. 2006;11:440–5. doi: 10.1016/j.drudis.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Plant N. The human cytochrome P450 sub-family: transcriptional regulation, inter-individual variation and interaction networks. Biochim Biophys Acta. 2007;1770:478–88. doi: 10.1016/j.bbagen.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 4.Williams JA, et al. Comparative metabolic capabilities of CYP3A4, CYP3A5, and CYP3A7. Drug Metab Dispos. 2002;30:883–91. doi: 10.1124/dmd.30.8.883. [DOI] [PubMed] [Google Scholar]
- 5.Huang W, et al. Evidence of significant contribution from CYP3A5 to hepatic drug metabolism. Drug Metab Dispos. 2004;32:1434–45. doi: 10.1124/dmd.104.001313. [DOI] [PubMed] [Google Scholar]
- 6.Watkins PB. Noninvasive tests of CYP3A enzymes. Pharmacogenetics. 1994;4:171–84. doi: 10.1097/00008571-199408000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Benet LZ. There are no useful CYP3A probes that quantitatively predict the in vivo kinetics of other CYP3A substrates and no expectation that one will be found. Mol Intervent. 2005;5:79–83. doi: 10.1124/mi.5.2.5. [DOI] [PubMed] [Google Scholar]
- 8.Kharasch ED, Thummel KE, Watkins PB. CYP3A probes can quantitatively predict the in vivo kinetics of other CYP3A substrates and can accurately assess CYP3A induction and inhibition. Mol Interv. 2005;5:151–3. doi: 10.1124/mi.5.3.3. [DOI] [PubMed] [Google Scholar]
- 9.Liu YT, Hao HP, Liu CX, Wang GJ, Xie HG. Drugs as CYP3A probes, inducers, and inhibitors. Drug Metab Rev. 2007;39:699–721. doi: 10.1080/03602530701690374. [DOI] [PubMed] [Google Scholar]
- 10.Kirwan C, Macphee I, Philips B. Using drug probes to monitor hepatic drug metabolism in critically ill patients: midazolam, a flawed but useful tool for clinical investigation of CYP3A activity? Expert Opin Drug Metab Toxicol. 2010;6:1–11. doi: 10.1517/17425255.2010.482929. [DOI] [PubMed] [Google Scholar]
- 11.Zaigler M, Tantcheva-Poor I, Fuhr U. Problems and perspectives of phenotyping for drug-metabolizing enzymes in man. Int J Clin Pharmacol Ther. 2000;38:1–9. doi: 10.5414/cpp38001. [DOI] [PubMed] [Google Scholar]
- 12.Dahl ML. Cytochrome P450 phenotyping/genotyping in patients receiving antipsychotics: useful aid to prescribing? Clin Pharmacokinet. 2002;41:453–70. doi: 10.2165/00003088-200241070-00001. [DOI] [PubMed] [Google Scholar]
- 13.Kharasch ED, Walker A, Hoffer C, Sheffels P. Intravenous and oral alfentanil as in vivo probes for hepatic and first-pass CYP3A activity. Noninvasive assessment using pupillary miosis. Clin Pharmacol Ther. 2004;76:452–66. doi: 10.1016/j.clpt.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 14.Kharasch ED, et al. Influence of CYP3A5 genotype on the pharmacokinetics and pharmacodynamics of the cytochrome P4503A probes alfentanil and midazolam. Clin Pharmacol Ther. 2007;82:410–26. doi: 10.1038/sj.clpt.6100237. [DOI] [PubMed] [Google Scholar]
- 15.Rogers JF, Rocci ML, Jr, Haughey DB, Bertino JS., Jr An evaluation of the suitability of intravenous midazolam as an in vivo marker for hepatic cytochrome P4503A activity. Clin Pharmacol Ther. 2003;73:153–8. doi: 10.1067/mcp.2003.23. [DOI] [PubMed] [Google Scholar]
- 16.Kharasch ED, et al. The role of cytochrome P450 3A4 in alfentanil clearance. Implications for interindividual variability in disposition and perioperative drug interactions. Anesthesiology. 1997;87:36–50. doi: 10.1097/00000542-199707000-00006. [DOI] [PubMed] [Google Scholar]
- 17.Kharasch ED, Russell M, Garton K, Lentz G, Bowdle TA, Cox K. Assessment of cytochrome P450 3A4 activity during the menstrual cycle using alfentanil as a noninvasive probe. Anesthesiology. 1997;87:26–35. doi: 10.1097/00000542-199707000-00005. [DOI] [PubMed] [Google Scholar]
- 18.Kharasch ED, Jubert C, Senn T, Bowdle TA, Thummel KT. Intraindividual variability in male hepatic CYP3A4 activity assessed by alfentanil and midazolam clearance. J Clin Pharmacol. 1999;39:664–9. doi: 10.1177/00912709922008290. [DOI] [PubMed] [Google Scholar]
- 19.Phimmasone S, Kharasch ED. A pilot evaluation of alfentanil-induced miosis as a noninvasive probe for hepatic cytochrome P450 3A4 (CYP3A4) activity in humans. Clin Pharmacol Ther. 2001;70:505–17. doi: 10.1067/mcp.2001.119994. [DOI] [PubMed] [Google Scholar]
- 20.Kharasch ED, Hoffer C, Walker A, Sheffels P. Disposition and miotic effects of oral alfentanil: a potential noninvasive probe for first-pass cytochrome P4503A activity. Clin Pharmacol Ther. 2003;73:199–208. doi: 10.1067/mcp.2003.30. [DOI] [PubMed] [Google Scholar]
- 21.Kharasch ED, Walker A, Hoffer C, Sheffels P. Evaluation of first-pass cytochrome P4503A (CYP3A) and P-glycoprotein activities using alfentanil and fexofenadine in combination. J Clin Pharmacol. 2005;45:79–88. doi: 10.1177/0091270004269705. [DOI] [PubMed] [Google Scholar]
- 22.Chaobal HN, Kharasch ED. Single point sampling for assessment of constitutive, induced and inhibited CYP3A activity with alfentanil. Comparison with midazolam. Clin Pharmacol Ther. 2005;78:529–39. doi: 10.1016/j.clpt.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 23.Kharasch ED, Walker A, Hoffer C, Sheffels P. Sensitivity of intravenous and oral alfentanil and pupillary miosis as minimally invasive and noninvasive probes for hepatic and first-pass CYP3A activity. J Clin Pharmacol. 2005;45:1187–97. doi: 10.1177/0091270005280077. [DOI] [PubMed] [Google Scholar]
- 24.Kharasch ED, Bedynek PS, Walker A, Whittington D, Hoffer C. Mechanism of ritonavir changes in methadone pharmacokinetics and pharmacodynamics. II Ritonavir effects on CYP3A and P-glycoprotein activities. Clin Pharmacol Ther. 2008;84:506–12. doi: 10.1038/clpt.2008.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kharasch ED, Walker A, Whittington D, Hoffer C, Bedynek PS. Methadone metabolism and clearance are induced by nelfinavir despite inhibition of cytochrome P4503A (CYP3A) activity. Drug Alcohol Depend. 2009;101:158–68. doi: 10.1016/j.drugalcdep.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kharasch ED, Hoffer C, Whittington D, Walker A, Bedynek PS. Methadone pharmacokinetics are independent of cytochrome P4503A (CYP3A) activity and gastrointestinal drug transport. Insights from methadone interactions with ritonavir/indinavir. Anesthesiology. 2009;110:660–72. doi: 10.1097/ALN.0b013e3181986a9a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin YS, et al. In-vivo phenotyping for CYP3A by a single-point determination of midazolam plasma concentration. Pharmacogenetics. 2001;11:781–91. doi: 10.1097/00008571-200112000-00006. [DOI] [PubMed] [Google Scholar]
- 28.Zhu B, Ou-Yang DS, Cheng ZN, Huang SL, Zhou HH. Single plasma sampling to predict oral clearance of CYP3A probe midazolam. Acta Pharmacol Sin. 2001;22:634–8. [PubMed] [Google Scholar]
- 29.Kim JS, et al. Limited sampling strategy to predict AUC of the CYP3A phenotyping probe midazolam in adults: application to various assay techniques. J Clin Pharmacol. 2002;42:376–82. [PubMed] [Google Scholar]
- 30.Rogers JF, et al. Single plasma concentrations of 1′-hydroxymidazolam or the ratio of 1′-hydroxymidazolam:midazolam do not predict midazolam clearance in healthy subjects. J Clin Pharmacol. 2002;42:1079–82. doi: 10.1177/009127002401382614. [DOI] [PubMed] [Google Scholar]
- 31.Lee LS, Bertino JS, Jr, Nafziger AN. Limited sampling models for oral midazolam: midazolam plasma concentrations, not the ratio of 1-hydroxymidazolam to midazolam plasma concentrations, accurately predicts AUC as a biomarker of CYP3A activity. J Clin Pharmacol. 2006;46:229–34. doi: 10.1177/0091270005283466. [DOI] [PubMed] [Google Scholar]
- 32.Krupka E, et al. Probe of CYP3A by a single-point blood measurement after oral administration of midazolam in healthy elderly volunteers. Eur J Clin Pharmacol. 2006;62:653–9. doi: 10.1007/s00228-006-0159-2. [DOI] [PubMed] [Google Scholar]
- 33.Penzak SR, Busse KH, Robertson SM, Formentini E, Alfaro RM, Davey RT., Jr Limitations of using a single postdose midazolam concentration to predict CYP3A-mediated drug interactions. J Clin Pharmacol. 2008;48:671–80. doi: 10.1177/0091270008317305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Browne TR. Stable isotopes in clinical pharmacokinetic investigations. Advantages and disadvantages. Clin Pharmacokinet. 1990;18:423–33. doi: 10.2165/00003088-199018060-00001. [DOI] [PubMed] [Google Scholar]
- 35.Gorski JC, Jones DR, Haehner-Daniels BD, Hamman MA, O’Mara EM, Jr, Hall SD. The contribution of intestinal and hepatic CYP3A to the interaction between midazolam and clarithromycin. Clin Pharmacol Ther. 1998;64:133–43. doi: 10.1016/S0009-9236(98)90146-1. [DOI] [PubMed] [Google Scholar]
- 36.Gorski JC, et al. The effect of age, sex, and rifampin administration on intestinal and hepatic cytochrome P450 3A activity. Clin Pharmacol Ther. 2003;74:275–87. doi: 10.1016/S0009-9236(03)00187-5. [DOI] [PubMed] [Google Scholar]
- 37.Quinney SK, Haehner BD, Rhoades MB, Lin Z, Gorski JC, Hall SD. Interaction between midazolam and clarithromycin in the elderly. Br J Clin Pharmacol. 2008;65:98–109. doi: 10.1111/j.1365-2125.2007.02970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee JI, Chaves-Gnecco D, Amico JA, Kroboth PD, Wilson JW, Frye RF. Application of semisimultaneous midazolam administration for hepatic and intestinal cytochrome P450 3A phenotyping. Clin Pharmacol Ther. 2002;72:718–28. doi: 10.1067/mcp.2002.129068. [DOI] [PubMed] [Google Scholar]
- 39.Meuldermans W, et al. Alfentanil pharmacokinetics and metabolism in humans. Anesthesiology. 1988;69:527–34. doi: 10.1097/00000542-198810000-00012. [DOI] [PubMed] [Google Scholar]
- 40.Kharasch ED, Bedynek PS, Park S, Whittington D, Walker A, Hoffer C. Mechanism of ritonavir changes in methadone pharmacokinetics and pharmacodynamics. I Evidence against CYP3A mediation of methadone clearance. Clin Pharmacol Ther. 2008;84:497–505. doi: 10.1038/clpt.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohnhaus EE, Breckenridge AM, Park BK. Urinary excretion of 6 β-hydroxycortisol and the time course measurement of enzyme induction in man. Eur J Clin Pharmacol. 1989;36:39–46. doi: 10.1007/BF00561021. [DOI] [PubMed] [Google Scholar]
- 42.Venkatesan K. Pharmacokinetic drug interactions with rifampicin. Clin Pharmacokinet. 1992;22:47–65. doi: 10.2165/00003088-199222010-00005. [DOI] [PubMed] [Google Scholar]
- 43.Veronese ML, et al. Exposure-dependent inhibition of intestinal and hepatic CYP3A4 in vivo by grapefruit juice. J Clin Pharmacol. 2003;43:831–9. doi: 10.1177/0091270003256059. [DOI] [PubMed] [Google Scholar]
- 44.Culm-Merdek KE, et al. Effect of extended exposure to grapefruit juice on cytochrome P450 3A activity in humans: comparison with ritonavir. Clin Pharmacol Ther. 2006;79:243–54. doi: 10.1016/j.clpt.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 45.Klees TM, Sheffels P, Dale O, Kharasch ED. Metabolism of alfentanil by cytochrome P4503A (CYP3A) enzymes. Drug Metab Dispos. 2005;33:303–11. doi: 10.1124/dmd.104.002709. [DOI] [PubMed] [Google Scholar]