

Abstract
Bacillus cereus strains harboring a pXO1-like virulence plasmid cause respiratory anthrax-like disease in humans, particularly in welders. We developed mouse models for intraperitoneal as well as aerosol challenge with spores of B. cereus G9241, harboring pBCXO1 and pBC218 virulence plasmids. Compared to wild-type B. cereus G9241, spores with a deletion of the pBCXO1-carried protective antigen gene (pagA1) were severely attenuated, whereas spores with a deletion of the pBC218-carried protective antigen homologue (pagA2) were not. Anthrax vaccine adsorbed (AVA) immunization raised antibodies that bound and neutralized the pagA1-encoded protective antigen (PA1) but not the PA2 orthologue encoded by pagA2. AVA immunization protected mice against a lethal challenge with spores from B. cereus G9241 or B. cereus Elc4, a strain that had been isolated from a fatal case of anthrax-like disease. As the pathogenesis of B. cereus anthrax-like disease in mice is dependent on pagA1 and PA-neutralizing antibodies provide protection, AVA immunization may also protect humans from respiratory anthrax-like death.
INTRODUCTION
Several cases of severe or fatal respiratory disease have been reported in the United States where sputum, bronchoalveolar lavage, or postmortem samples revealed the presence of Bacillus cereus strains with pXO1-like virulence plasmids (1–5). For most of these cases, occupational records included welding, which may predispose humans to respiratory infections with B. cereus (1). In comparison with respiratory anthrax (6), the clinical course of anthrax-like disease is similarly fulminant, and most cases are revealed by postmortem diagnosis (3–5). Geographical restriction of clinical cases to Louisiana and Texas suggests that welders in the southern United States may be at risk of acquiring respiratory B. cereus infections (3). We therefore tested the hypothesis that a preventive strategy for B. cereus-induced respiratory anthrax-like disease can be developed.
B. cereus and Bacillus anthracis are close relatives and belong to the Bacillus cereus sensu lato group (7). B. anthracis is unique among the B. cereus group due to its ability to cause anthrax in animals and humans (8). Anthrax is transmitted by spores, which germinate and replicate as vegetative forms throughout many organ systems (8, 9). All clinical isolates of B. anthracis harbor two large plasmids, pXO1 and pXO2 (10, 11). pXO1 harbors genes for protective antigen (pag) (12), lethal factor (lef) (13), and edema factor (cya) (14). The secreted gene products function together as anthrax toxins to kill immune cells or promote tissue edema (15). Following association with its host cell receptors (16, 17), protective antigen (PA) is cleaved, which triggers assembly of a heptameric pore for transport of lethal factor and edema factor into host cells (18–20). pXO2 provides for the expression of the poly-d-γ-glutamic acid (PDGA) capsule (10, 21), which endows bacilli with resistance to opsonophagocytic clearance during infection (22, 23).
Most B. cereus isolates lack anthrax toxin (pXO1) and PDGA capsule (pXO2) plasmids and cause human disease in both immunocompromised as well as healthy individuals (24–26). The emergence of anthrax-like disease has focused research efforts on B. cereus G9241, a strain that was isolated from a severe case of respiratory disease (2). Genome sequencing of B. cereus G9241 revealed the presence of two plasmids, pBCXO1 and pBC218, as well as the prophage lin29 (2). pBCXO1 exhibits a high level of synteny with B. anthracis pXO1 and includes the toxin genes pagA1, lef1, and cya1. In contrast, pBC218 is unique and does not display synteny to other known B. cereus group plasmids (2).
Following intraperitoneal injection, B. cereus G9241 spores cause anthrax-like disease in C57BL/6 mice, and the disease is dependent on each of the two virulence plasmids (27). The dependence on pBCXO1 can in part be explained as the requirement for protective antigen (PA1): pagA1 mutant spores are severely attenuated in the intraperitoneal challenge model (27). Further, the hasACB cluster on pBCXO1 provides for the synthesis of the B. cereus hyaluronic acid capsule (27). pBC218 harbors the bpsX-H genes, whose products synthesize a second capsule that, together with hasACB, is essential for B. cereus G9241 escape from phagocytic clearance (27). pBC218 also carries pagA2 and lef2 (2). PagA2, an orthologue of PagA1 (PA), has been hypothesized to translocate Lef2 (CerADPr), an ADP-ribosyltransferase, into host cells (2, 28). However, the contributions of pagA1 and pagA2 to B. cereus G9241-mediated respiratory anthrax-like disease have not been studied. These questions are addressed here in an effort to test whether a protective antigen-derived vaccine can protect against B. cereus G9241 respiratory disease.
MATERIALS AND METHODS
Bacillus cereus growth and spore preparation.
B. cereus G9241 was obtained from the Biodefense and Emerging Infections Research Program (BEI). B. cereus Elc4 was isolated from a fatal case of respiratory anthrax-like disease in Texas (4). B. cereus G9241, its mutants, and B. cereus Elc4 were grown in tryptic soy broth (TSB) or propagated on tryptic soy agar (TSA). Kanamycin (Kan) was added at a concentration of 50 μg ml−1 for Escherichia coli and B. cereus to retain plasmids or select for mutant alleles. For spore preparations, B. cereus strains were inoculated into TSB and grown overnight at 30°C. Bacilli were diluted into modG medium (29) or 2× SG medium (30) and grown at 200 rpm and 30°C for at least 4 days. The cultures were heated at 68°C for 150 min to kill the remaining vegetative cells. Spore preparations were then washed and suspended in water. Ten-fold serial dilutions were spread on Luria-Bertani (LB) agar followed by incubation and enumeration of CFU. The purity and extent of sporulation were assessed by phase-contrast microscopy. Experiments with B. cereus G9241 and Elc4 were conducted according to protocols that had been reviewed and approved by the Institutional Biosafety Committee at the University of Chicago. The experimental work was carried out in biological safety level 3 containment laboratories at the Howard Taylor Ricketts Laboratory.
Bacillus cereus G9241 mutants.
Construction of the B. cereus G9241 pagA1 mutant has been previously described (27). The pagA2 deletion mutant was generated with the temperature-sensitive replication plasmid pLM4 (31). PCR products derived from the primer pairs P205 (TTTGAATTCAGATGCAGCATTGCAAAAGTC)/P206 (TTTGCTAGCCAGCTAATAATGGGATGAATAC) and P207 (AAAGCTAGCGTTTTAGTCCTTTCGCTAAAGAAATAA)/P208 (TTTGGTACCCAAATACAATAAACTACCCTC) were inserted into pLM4 via its EcoRI, NheI, and KpnI restriction sites (31). This generated pSY109, which carries the mutant pagA2 allele. Plasmids were electroporated into B. cereus G9241 via a previously established protocol (32). Transformants were screened and verified for allelic replacement as described in an earlier report (27).
Purification of the D1 and D4 domains of PA2.
The coding sequences for the PA2 D1 and D4 domains were amplified with the primers p288 (AAAGGATCCACAACGCAAGAGGACAG)/p289 (AAAGAATTCTTATTACGAAGCAGTGCTCC) and p286 (AAAGGATCCTTCCGTTATGTGGATGGG)/p287 (AAAGAATTCTTATTTCTTTAGCGAAAGG) by using B. cereus G9241 genomic DNA as a template. PCR products were cleaved with BamHI and EcoRI, inserted into pGEX-2T to generate pSY140 (GST-D1PA2) and pSY141 (GST-D4PA2). GST-D1PA2 and GST-D4PA2 were purified by affinity chromatography using a protocol developed for the D4 domain of B. anthracis PA (GST-D4PA) (33).
Immunoblotting.
B. cereus cultures were grown in TSB supplemented with 0.8% sodium bicarbonate. Cultures were centrifuged, and the supernatant was precipitated with 9% trichloroacetic acid (TCA). TCA precipitates were washed in acetone and dried, and proteins were solubilized in 50 μl sample buffer. Proteins were separated on SDS-PAGE, transferred to polyvinylidene difluoride membrane and probed with guinea pig antibodies specific for B. anthracis PA (33).
Toxin neutralization assay.
J774A.1 murine macrophages were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and 1% Glutamax (Gibco). Cells were seeded in 96-well tissue culture dishes (Corning) at a concentration of 1 × 104 cells/well and incubated at 37°C with 5% CO2. Approximately 2 × 105 cells/well were washed with prewarmed DMEM prior to use. Purified recombinant lethal toxin (LT), i.e., PA and lethal factor (LF), were purchased from List Biologicals (Campbell, CA). LT was reconstituted from equimolar amounts of PA and LF. Pooled serum from either mock-immunized (phosphate-buffered saline [PBS]) or anthrax vaccine adsorbed (AVA)-immunized mice was incubated with 0.1 μg of LT for 20 min at room temperature. LT or LT-serum mix was transferred to J774A.1 cells. After 3 h of incubation at 37°C with 5% CO2, cell viability was determined via the lactate dehydrogenase (LDH)-based cytotoxicity detection kit (Roche) according to the manufacturer's protocol.
Animal models of B. cereus infection.
Animal experiments were conducted according to protocols that had been reviewed and approved by the Institutional Animal Care and Use Committee at the University of Chicago. All infections were carried out in animal biological safety level 3 containment laboratories at the Howard Taylor Ricketts Laboratory. C57BL/6 female mice were housed in cages with HEPA filters and infected at 8 to 9 weeks of age. Infected animals were monitored at 8- to 12-h intervals for survival or a moribund state (inability to remain upright, weight loss, nonresponsive to touch) for 14 days. Moribund animals were euthanized by inhalation of compressed CO2, subjected to necropsy, and their spleen, liver, kidneys, and lungs were removed. Sections of organ tissue were immediately fixed by submersion in 10% neutral-buffered formalin and embedded in paraffin. Samples were submitted to the University of Chicago Animal Pathology Core for serial 4-μm sections and staining with hematoxylin-eosin. Tissue samples were viewed by light microscopy, and images were acquired with a cooled charge-coupled-device camera. Organ samples isolated during necropsy were also homogenized in PBS with or without heat treatment (68°C for 30 min), serially diluted, and plated on TSA to enumerate the load of vegetative bacilli and spores as CFU. Alternatively, samples were fixed with neutral buffered formalin and stained with India ink to visualize the capsule of bacilli. All animal infection experiments were repeated at least once, and representative data are shown. Experimental protocols were reviewed, approved, and performed under regulatory supervision of The University of Chicago's Institutional Biosafety Committee (IBC). Animals were managed by the University of Chicago Animal Resource Center. Animals that were judged to be moribund were euthanized with CO2. For intraperitoneal (i.p.) challenge, mice were given a spore 1 × 105 B. cereus spore suspension in 100 μl PBS. The spore inoculum was serially diluted and spread on agar plates to enumerate the challenge dose.
Aerosol challenge.
For aerosol challenge, mice were exposed to aerosolized spores of B. cereus G9241 in distilled water with 0.01% Tween 80 by using a nose-only inhalation exposure system (CH Technologies, NJ) operated inside a class III biosafety cabinet. Spores were aerosolized with a six-jet collision nebulizer holding 8 ml of inoculum suspension in its liquid reservoir. Up to 22 animals were held in restraint tubes attached to their respective inhalation ports of the exposure chamber, which was operated under continuous negative pressure relative to the biosafety cabinet. In addition, two ports were utilized for real-time measurement of aerosol particle size and particle collection via a Teflon impinger. Prior to challenge, mice were supplied with fresh air for 10 min to allow their respiratory rates to normalize. Exposure was initiated by directing the airflow to the nebulizer for 10 or 20 min. Particle size distribution of the aerosol was registered using an aerodynamic particle sizer spectrometer (Promo 2070; Pallas GmbH, Germany). Viable spore exposure was determined by collecting aerosol with a Teflon impinger and plating the impinger reservoir liquid on LB agar for CFU counts. Immediately following exposure, 2 mice were euthanized with CO2 and necropsied; their lungs were homogenized and plated for colony formation to determine the number of organisms that had been deposited into lung tissues. Typically, cohorts of 20 infected mice per experiment were monitored for survival. To measure bacterial dissemination, mice were euthanized at variable time points following exposure. Lungs and spleens were removed during necropsy, homogenized, and plated on LB agar for CFU enumeration. In order to distinguish between spores and bacilli, a fraction of each tissue homogenate was heat treated at 68°C for 30 min to kill vegetative bacilli. Heat-treated and untreated samples were serially diluted and plated for CFU determination. The bacterial detection limit in animal tissues was determined to be 100 CFU.
AVA immunization.
Groups of female C57BL/6 mice (n = 10) were immunized on days 1 and 15 by intramuscular injection into the hind leg with 0.1 ml of AVA (Biothrax), the aluminum hydroxide-adsorbed precipitate of B. anthracis V63340 77/-NP1-R (pXO1+ pXO2−) culture supernatants (34). Animals were bled on day 25 to measure serum antibody titers, prior to challenge on day 29. Levels of mouse serum immunoglobulin G (IgG) reactive with PA were determined by enzyme-linked immunosorbent assay (ELISA) as described previously (33).
Statistical analysis.
Data were processed using GraphPad Prism 5.0 software to generate graphs for statistical analyses. Bacterial load data were analyzed for statistical significance with the two-tailed Student t test. Comparisons of survival between two groups were evaluated with the log rank test. Data from cytotoxicity experiments were analyzed with an unpaired t test with Welch's correction.
RESULTS
B. cereus G9241 harbors two orthologues of the protective antigen gene.
In response to a bicarbonate signal during host infection, B. anthracis synthesizes and secretes PA, LF, and edema factor (EF) (9, 35). PA is a major virulence factor of B. anthracis and is also the principal immunogen of AVA (BioThrax) (36). Similar to B. anthracis pXO1, B. cereus G9241 pBCXO1 harbors genes for protective antigen (pagA1), lethal factor (lef1), and edema factor (cya) (Fig. 1A). Furthermore, B. cereus G9241 pBC218 harbors an orthologue of pagA, here designated pagA2, as well as a paralogue of lef, here designated lef2. Comparison of amino acid sequences via BLAST (37) revealed that B. cereus PA1 is a close relative of B. anthracis PA, with 99.6%, 99.7%, 99%, and 100% amino acid identities for protective antigen domains 1 to 4, respectively (Fig. 1B). PA2, on the other hand, is more distantly related to either PA1 or PA and displayed 61%, 70%, 59%, and 45% amino acid identities with protective antigen domains 1 to 4 (Fig. 1B).
Fig 1.
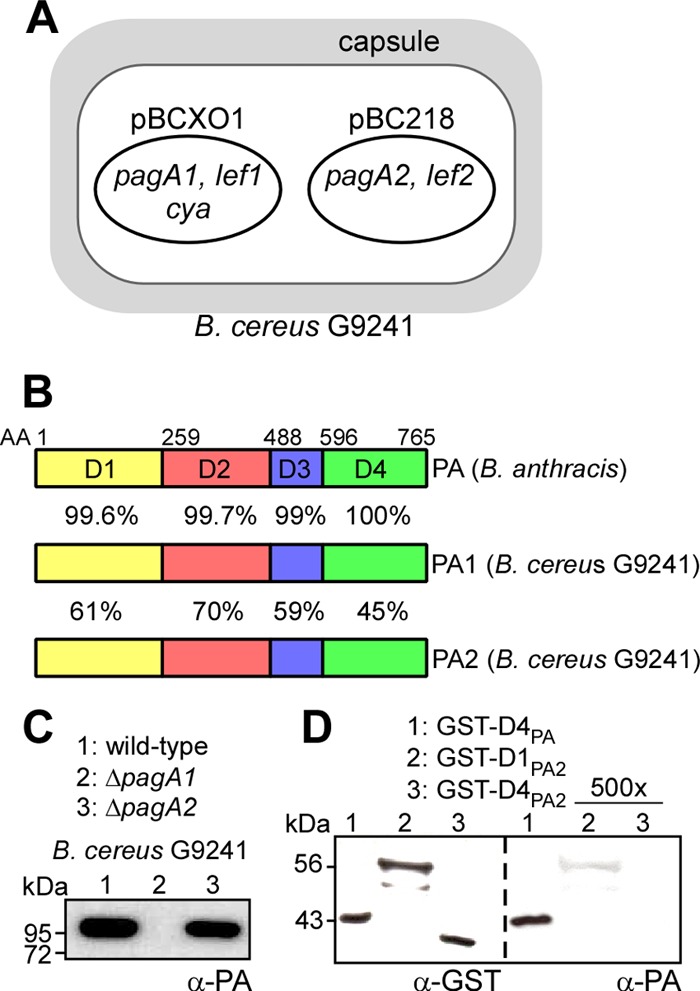
B. cereus G9421 virulence plasmids harbor two protective antigen orthologues. (A) B. cereus G9241 virulence plasmid pBCXO1 harbors anthrax toxin genes pagA1 (protective antigen, PA1), lef1 (lethal factor), and cya (edema factor). The B. cereus G9241 virulence plasmid pBC218 harbors genes for expression of BPS capsule as well as pagA2 (protective antigen, PA2) and lef2 (encoding the ADP-ribosylase CerADPr) (28). (B) The amino acid identities between Bacillus anthracis PA and B. cereus PA1 and PA2 were revealed with BLAST searches (37) and are listed for each of its four domains (D1 to D4) (60). (C) The extracellular medium of B. cereus G9241 wild type or its ΔpagA1 and ΔpagA2 mutants was precipitated with TCA and subjected to immunoblotting with PA-specific antibodies. (D) Purified GST hybrids with the N-terminal end of the D1 or D4 domain of PA2 or the D4 domain of PA were subjected to immunoblotting with PA- or GST-specific antibodies. In the right panel, the concentrations of GST-D1PA2 and GST-D4PA2 were 500-fold higher than that of GST-D4PA.
By using allelic replacement, the pagA2 open reading frame was deleted from pBC218. Under conditions of vegetative growth, the B. cereus ΔpagA2 mutant replicated at the same rate as the wild-type parent or the ΔpagA1 mutant strain. Following starvation, ΔpagA2 bacilli formed spores at a similar rate as the wild type and ΔpagA1 mutant B. cereus. To measure protective antigen synthesis and secretion, B. cereus G9241 cultures were induced for toxin and capsule expression by the addition of bicarbonate. Deletion of the pagA1 or pagA2 gene did not interfere with B. cereus G9241 capsule formation (data not shown). Following centrifugation of B. cereus cultures, proteins in the extracellular medium were removed with the supernatant, precipitated with TCA, and analyzed by immunoblotting (Fig. 1C). PA-specific antibodies detected PA1 secreted into the extracellular medium of B. cereus G9241 cultures. Deletion of the pagA2 gene did not affect PA1 secretion (Fig. 1C). As expected, deletion of the pagA1 gene abrogated the expression of protective antigen, as PA1 was not detected in the extracellular medium of ΔpagA1 mutant cultures (Fig. 1C). As a test of whether PA-specific antibodies recognize PA2, we expressed and purified two recombinant hybrids, GST-D1PA2 and GST-D4PA2, with a fusion of the D1 or D4 domains of PA2 (pagA2) to glutathione S-transferase (GST). Antibodies directed against GST bound to purified GST-D1PA2, GST-D4PA2, and to the GST-D4PA control. In contrast, antibodies raised against purified PA bound GST-D4PA (which is identical to D4PA1), but not GST-D4PA2, and associated only weakly with GST-D1PA2 (Fig. 1D). Thus, PA-specific antibodies recognize B. cereus PA1 but do not significantly cross-react with PA2.
B. cereus G9241 pagA1, not pagA2, contributes to virulence in mice.
Spores were derived from B. cereus G9241 as well as the ΔpagA1 and ΔpagA2 mutant strains. When 1 × 105 spores were inoculated into the peritoneal cavity of C57BL/6 mice (n = 10), all animals challenged with B. cereus G9241 succumbed to infection within 4 days (Fig. 2A). In contrast, only two animals that were challenged with the ΔpagA1 mutant strain succumbed to infection, one on day 4 and another on day 11 (wild type versus ΔpagA1, P < 0.0001) (Fig. 2A). Animals challenged with the ΔpagA2 mutant succumbed at a similar rate as the cohort infected with wild-type B. cereus G9241 (wild type versus ΔpagA2, P = 0.5078) (Fig. 2A). Moribund animals were euthanized and necropsied. Tissue samples from the spleen, kidney, liver, and lung were homogenized, and bacterial load was determined as the CFU (Fig. 2B). Mice infected with B. cereus G9241 harbored high numbers of vegetative forms in the spleen (>108 CFU) as well as 106 to 108 CFU in kidney, liver, and lung tissues (Fig. 2B). Pathogen load in host tissues was similar for animals that had been challenged with the ΔpagA2 mutant strain (Fig. 2B). The bacterial load in moribund animals harboring ΔpagA1 bacilli was reduced by 2 to 4 log10 CFU (wild type versus ΔpagA1, P < 0.0001) (Fig. 2B).
Fig 2.
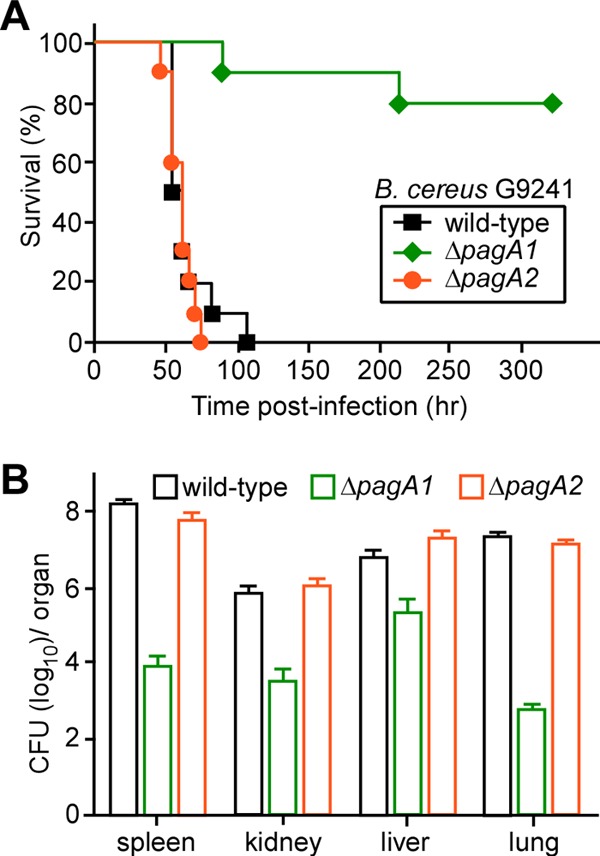
PA1 contributes to the pathogenesis of B. cereus G9241 anthrax-like disease in mice. (A) Survival of C57BL/6 mice (cohorts of 10 animals) following intraperitoneal injection of 1× 105 spores derived from B. cereus G9241 or its ΔpagA1 and ΔpagA2 mutant strains. (B) The bacterial loads in various organ tissues of dead or moribund C57BL/6 mice infected with either wild-type or ΔpagA1 or ΔpagA2 mutant B. cereus G9241 were quantified. Animals were necropsied, organs (spleen, kidney, liver, and lung) were removed, and tissue homogenates were spread on agar plates to enumerate colony formation. Data are representative of three independent experiments.
Histopathology of anthrax-like disease caused by B. cereus G9241 wild-type, ΔpagA1, or ΔpagA2 mutant strains.
Tissue samples of infected mice were fixed with formalin, embedded, thin sectioned, and stained with hematoxylin-eosin (Fig. 3). B. cereus G9241 infection resulted in massive replication of encapsulated bacilli in the spleen, predominantly in splenic sinuses (red pulp), but also infiltrating the lymphoid tissue (white pulp) (Fig. 3A). Histopathological disease features included large numbers of encapsulated bacilli with sparse infiltrations of phagocytes. Histopathological analysis of lung tissues revealed the interstitial replication of encapsulated bacilli in addition to a moderate phagocytic infiltration (Fig. 3D). In contrast, ΔpagA1 mutant bacilli displayed fewer signs of bacterial invasion and stronger immune responses. The histopathology of the spleen was dominated by massive invasion of polymorphonuclear leukocytes with few encapsulated bacilli (Fig. 3B). Similarly, the histopathology of lung tissues presented massive phagocytic infiltrates with few ΔpagA1 mutant bacilli (Fig. 3E). Animals infected with ΔpagA2 mutant bacilli displayed histopathological lesions in the spleen and lung tissues that were indistinguishable from those caused by wild-type B. cereus G9241 (Fig. 3CF).
Fig 3.

Histopathology of B. cereus G9241 wild-type and ΔpagA1 or ΔpagA2 mutant disease in mice. C57BL/6 mice were infected with 1 × 105 spores of B. cereus G9241 (A and D) or its ΔpagA1 (B and E) or ΔpagA2 mutant (C and F). Sections from infected spleen and lung tissues were fixed, thin sectioned, and stained with hematoxylin-eosin. Tissue sections were viewed by light microscopy, and images of the spleen (A, B, and C) and lung (D, E, and F) were captured. Low-magnification views are presented on the left, and higher magnifications of the same image are shown on the right of each panel.
AVA-immunized mice are protected against B. cereus G9241 challenge.
We hypothesized that AVA immunization, which raises PA-specific serum IgG protective for anthrax disease (34, 36), would protect animals against B. cereus-mediated anthrax-like disease. To test this, cohorts of C57BL/6 mice (n = 10) were immunized by intramuscular injection of 100 μl AVA by using a prime-booster regimen with a 14-day interval between injections. Ten days following the booster, immune sera were analyzed for the presence of PA-specific antibodies (IgG) via enzyme-linked immunosorbent assay. An average titer of 11,169 (±349 [standard error]) PA-specific IgG was determined in AVA-immune serum. PA-specific antibodies were not detected in mock (PBS)-immunized animals. To examine whether or not these antibodies neutralize anthrax toxin, purified PA and LF were mixed. Reconstituted LT was incubated for 20 min in the presence or absence of immune sera and was then added for 3 h to J774A.1 macrophages (Fig. 4A). LT (0.1 μg) lysed 77.5% (±5%) of 2 × 105 J774A.1 cells, as measured with the LDH release assay (Fig. 4A). When mixed with pooled serum from mock (PBS)-immunized animals, no significant reduction in LT toxicity for J774A.1 cells was observed (no serum treatment versus PBS immune serum, P = 0.1807) (Fig. 4A). However, incubation of LT with AVA-immune serum neutralized most of its toxin activity, as only 10.05% (±3.891%) of J774A.1 cells were lysed following the 3-h incubation period (no serum treatment versus AVA immune serum, P = 0.0006) (Fig. 4A).
Fig 4.
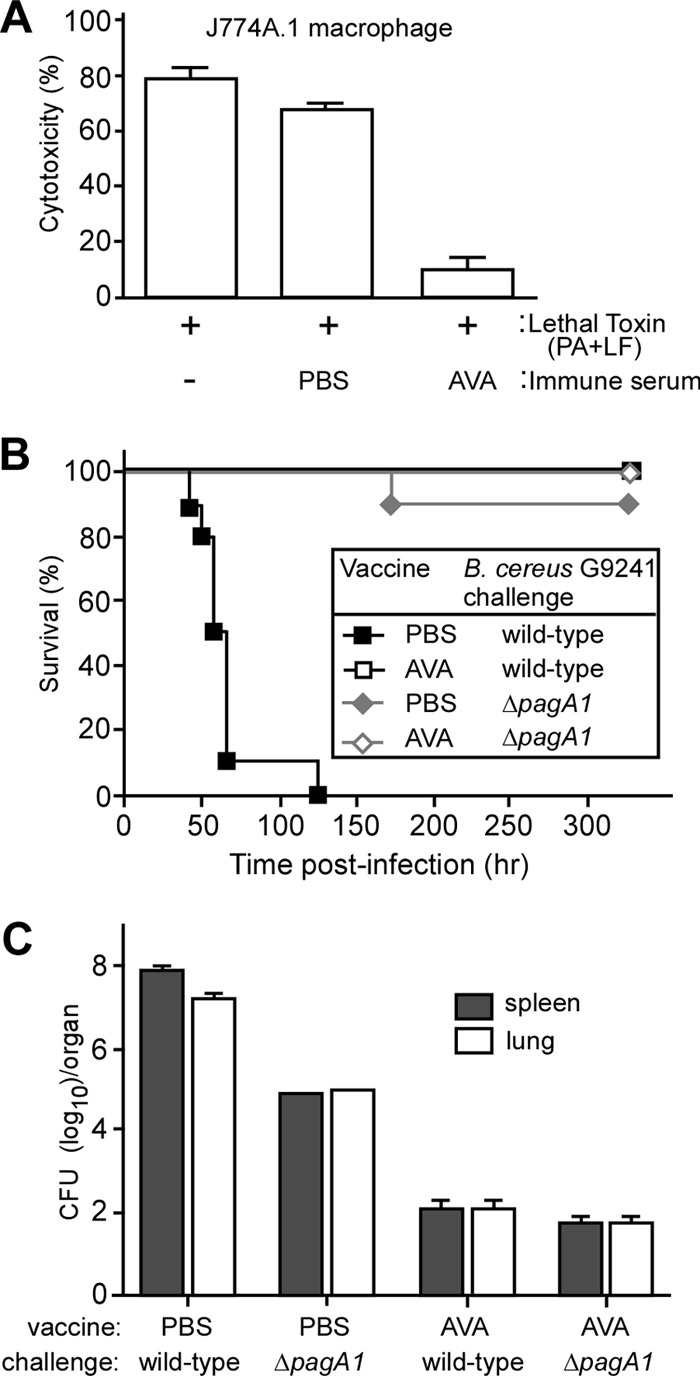
AVA-immunized mice are protected against B. cereus G9241 challenge. (A) Lethal toxin neutralization assay. Serum from mock (PBS)-immunized or AVA-immunized C57BL/6 mice was incubated with 0.1 μg anthrax lethal toxin (PA + LF), and the toxin-serum mix as well as a no-serum control reaction mixture was transferred to 105 J774A.1 cells. Cell viability was monitored with an LDH-based cytotoxicity assay after 3 h of incubation. (B) C57BL/6 mice (n = 10) were immunized with a prime-booster regimen of AVA or mock (PBS) in 14-day intervals. Animals were challenged by intraperitoneal inoculation with 1 × 105 spores derived from the B. cereus G9241 wild type or its ΔpagA1 mutant, and survival was monitored. Data are representative of three independent experiments. (C) Bacterial loads in spleen and lung tissues of immunized mice that had been infected with either B. cereus G9241 wild type or its ΔpagA1 mutant. The organs of mock-immunized mice were removed during necropsy when animals were either dead or moribund. Tissue homogenates were spread on agar plates to enumerate colony formation. Data are representative of three independent experiments.
Immunized mice were challenged via intraperitoneal injection with 1 × 105 spores derived from B. cereus G9241 wild-type or ΔpagA1 mutant strains (Fig. 4B). Compared to mock (PBS)-immunized animals, which died on days 2 to 5 following spore injection, AVA immunization afforded complete protection against B. cereus G9241 challenge (PBS versus AVA, P < 0.0001) (Fig. 4B). AVA immunization did not affect the outcome of the B. cereus ΔpagA1 spore challenge, as mock-immunized mice displayed a similar survival rate as cohorts receiving AVA vaccine (PBS versus AVA, P = 0.3173) (Fig. 4B).
Moribund animals as well as survivors at the end of their observation period were euthanized and necropsied. Tissue homogenates from the spleens and lungs of infected animals revealed average loads of 9.9 × 107 and 1.5 × 107 CFU B. cereus G9241 in PBS (mock)-immunized mice, respectively (Fig. 4C). As expected, the bacterial load of mock-immunized animals receiving the ΔpagA1 spore challenge was reduced, i.e., 9.1 × 104 CFU in the spleen and 1.0 × 105 CFU in lung tissues (Fig. 4C). Of note, AVA-immunized animals harbored 1.3.×102 CFU B. cereus G9241 and 66.7 CFU ΔpagA1 bacilli in their organ tissues (Fig. 4C). These data suggest that AVA-induced PA-specific antibodies provide cross-protection against B. cereus G9241 by neutralizing secreted PA1. As reported before, PA-specific antibodies cannot confer sterilizing immunity, because the antibodies cannot trigger opsonophagocytic killing of bacilli (33). Nevertheless, neutralization of PA1 via PA-specific antibodies ameliorates the pathogenesis of B. cereus G9241 in the intraperitoneal spore challenge model and allows injected animals to survive an otherwise lethal challenge.
AVA immunization protects against B. cereus Elc4 spore challenge.
We wondered whether AVA immunization provides universal protection against respiratory anthrax-like disease caused by virulent B. cereus strains. B. cereus Elc4 was isolated from a fatal case of respiratory anthrax-like disease (4). When tested in the mouse model of intraperitoneal challenge, 1 × 105 B. cereus Elc4 spores caused lethal disease in 70% of mock (PBS)-immunized animals (Fig. 5A). For comparison, all mice challenged with a similar dose of B. cereus G9241 spores developed anthrax-like disease and died (Fig. 5A). AVA-immunized mice were fully protected from a lethal challenge with 1 × 105 spores of either B. cereus G9241 or B. cereus Elc4 (B. cereus Elc4 PBS versus AVA, P = 0.0012; B. cereus G9241 PBS versus AVA, P < 0.0001) (Fig. 5A). In mock-immunized mice, the spores of B. cereus Elc4 germinated and disseminated from the site of infection to the peripheral organ tissues, including the spleen, kidney, liver, and lung. The tissue dissemination and organ load of vegetative forms were similar for B. cereus Elc4 and B. cereus G2941 (Fig. 5B).
Fig 5.
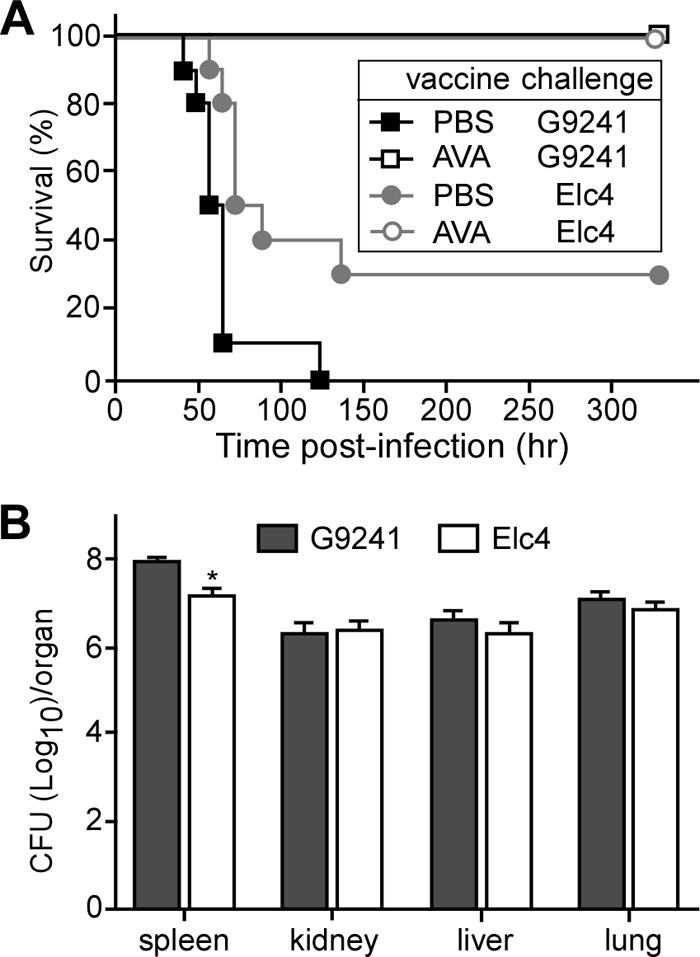
AVA immunization protects mice against B. cereus Elc4 spore challenge. (A) C57BL/6 mice (n = 10) were immunized with a prime-booster regimen of AVA or mock immunization (PBS) in 14-day intervals. Animals were challenged by intraperitoneal inoculation with 1 × 105 spores derived from B. cereus Elc4 or B. cereus G9241, and survival was monitored. Data are representative of three independent experiments. (B) Bacterial loads in organ tissues of mice infected with B. cereus Elc4 or B. cereus G9241. The organs of mock-immunized mice were removed during necropsy when animals were either dead or moribund. Tissue homogenates were spread on agar plates to enumerate colony formation. Data are representative of three independent experiments.
Histopathology of infected spleen tissues revealed large numbers of encapsulated B. cereus Elc4 bacilli in sinuses of the red pulp, together with an infiltrate of granulocytes and macrophages (Fig. 6A). In lung tissues, encapsulated B. cereus Elc4 vegetative forms were found as microcolonies in the interstitial space, presumably within lung capillaries, together with a modest infiltrate of phagocytes (Fig. 6D). In AVA-immunized animals that had been challenged with B. cereus Elc4 spores, we observed infiltrates of granulocytes and macrophages in splenic sinuses but failed to detect encapsulated bacilli (Fig. 6B). Similarly, we failed to detect B. cereus Elc4 vegetative forms in lung tissues, although a moderate infiltrate of granulocytes and macrophages could be detected in the interstitial space (Fig. 6E). AVA immunization abolished the appearance of encapsulated B. cereus G9241 in either spleen or lung tissues. Histopathology revealed infiltrates of phagocytes in splenic sinuses and the lung interstitium, similar to that observed with B. cereus Elc4-challenged animals (Fig. 6C and F).
Fig 6.
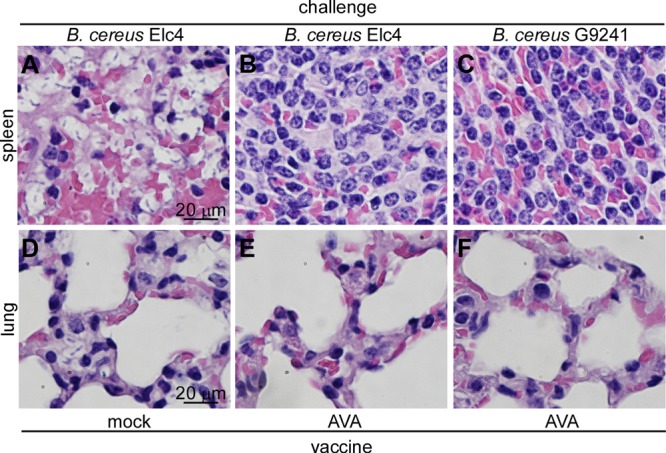
Histopathology of AVA-immunized mice following challenge with spores of B. cereus Elc4 or B. cereus G9241. C57BL/6 mice were infected with 1 × 105 spores of B. cereus Elc4 (A, B, D, and E) or B. cereus G9241 (C and F). Sections from infected spleen and lung tissues were fixed, thin sectioned, and stained with hematoxylin-eosin. Tissue sections were viewed by light microscopy, and images of the spleens (A, B, and C) and lung (D, E, and F) were captured.
B. cereus G9241 causes respiratory anthrax-like disease in mice.
The pathogenesis of B. cereus respiratory anthrax-like disease in humans is initiated via the inhalation of spores (4). To examine whether B. cereus G9241 causes respiratory anthrax-like disease in mice, a suspension of spores was aerosolized in a Collison 6-jet nebulizer (38) to infect the lung tissues of C57BL/6 mice, individually immobilized in a nose-only exposure chamber. Different aerosol challenge doses of B. cereus G9241 spores were obtained by varying the inoculum concentration between 1.0 × 108 and 2.0 × 1010 CFU/ml. Each challenge dose was experimentally verified in euthanized animals by homogenizing lung tissues and quantifying the spore load via enumeration of bacterial colonies (CFU). At a challenge dose of 2.3 × 105 B. cereus G9241 spores, all infected animals succumbed to respiratory anthrax-like disease within 5 days. Most mice remained clinically asymptomatic until 12 to 18 h prior to death; at this time, moribund animals displayed a gradual decrease in activity, labored gait, and were eventually immobilized and unable to remain upright. Spore challenge doses of 2.3 × 104, 1.3 × 104, and 4.3 × 103 precipitated lethal respiratory anthrax-like disease in 70%, 50%, and 20% of infected animals, respectively (Fig. 7A). From these data we derived a 50% lethal dose (LD50) of 1.1 × 104 spores for B. cereus G9241 (39), which is 20-fold higher than the LD50 for respiratory anthrax caused by B. anthracis Ames spores (40) and 4-fold higher than the LD50 for intraperitoneal challenge with B. cereus G9241 (27).
Fig 7.
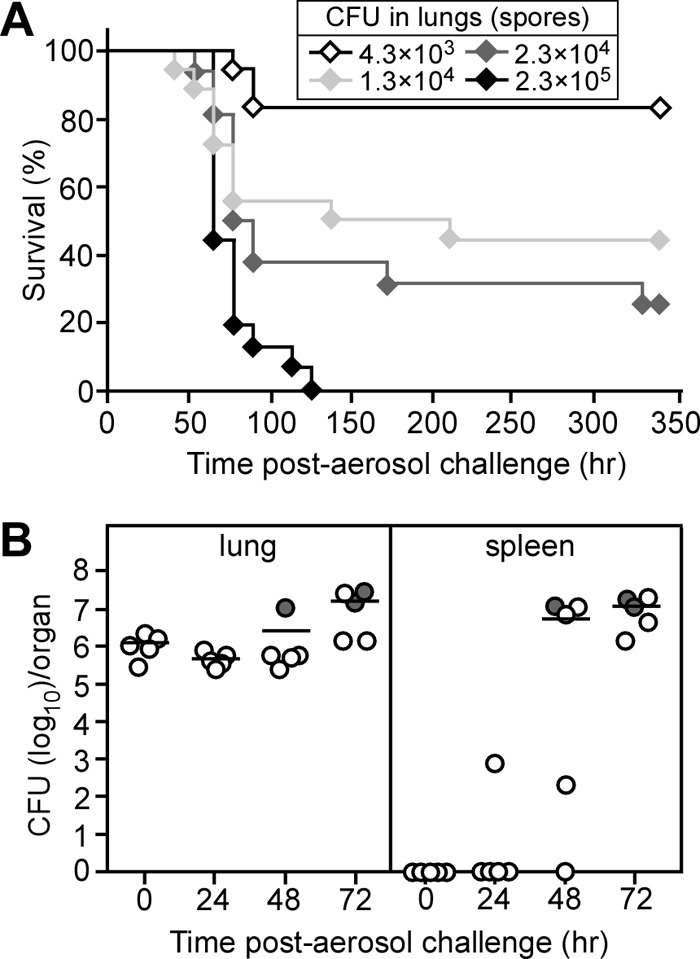
Virulence of B. cereus G9241 spores in an aerosol challenge model of respiratory anthrax-like disease in mice. (A) Survival of C57BL/6 mice following aerosol infection. Cohorts of 10 mice were exposed to aerosols of B. cereus G9241 spores and monitored over 14 days. The actual doses retained in the lungs of animals following exposure was determined for a subset of mice from each challenge group and are denoted in the legend. (B) Dissemination of vegetative forms following aerosol infection of B. cereus G9241 spores. Cohorts of 20 mice were infected with 1 × 106 spores derived from B. cereus G9241. Five mice were euthanized in 24-h intervals and necropsied. The spleen and lung homogenates were diluted and spread on agar plates to enumerate bacterial loads. Each circle represents data from one mouse. The closed circle denotes a mouse that succumbed to infection. The bars represent the arithmetic means of CFU values. Data shown are representative of two independent experiments.
To monitor the progression of respiratory anthrax-like disease, cohorts of C57BL/6 mice (n = 5) were euthanized and necropsied 0, 24, 48, and 72 h following infection. The load of B. cereus vegetative forms was determined in lung and spleen tissues. To evaluate germination of B. cereus G9241 spores in lung tissues of infected mice, the numbers of heat-resistant spores and heat-sensitive vegetative forms were determined. At the time of infection (0 h), mice harbored <0.1% vegetative forms in their lung tissues. Thus, we did not observe high numbers of vegetative forms in the lungs of animals that had been euthanized via CO2 inhalation, as has been reported for mice receiving an intratracheal instillation of B. anthracis Ames spores (40); CO2 inhalation has been reported to trigger germination of B. anthracis Ames spores in lung tissues (40). B. cereus vegetative forms in lung tissues increased to 60% and 93% of the bacterial load at 24 and 72 h post-aerosol challenge, respectively (Fig. 7B). At the time of death, >99.9% of bacilli in lung tissue were comprised of heat-sensitive vegetative forms with a concomitant increase in the bacterial load by 1 to 2 log10 CFU (Fig. 7B). The dissemination of vegetative forms from the lung to the spleen commenced 24 h postinfection (Fig. 7B). By 48 h, several animals harbored large numbers of bacilli in their splenic sinuses. The pathogenesis of splenic anthrax-like disease caused by disseminating B. cereus G9241 was completed by 72 h; at this time, most of the infected mice were moribund (Fig. 7B). Organ tissues of infected animals were also subjected to histopathological analysis. Shortly following aerosol challenge, neither lung nor spleen tissues revealed pathological changes (Fig. 8A and B). We could not detect B. cereus G9241 spores in lung tissues, likely because the spores were too small and sparse for detection. We also did not detect pathological changes in lung or spleen tissues at 24 h following challenge (Fig. 8C and D). Tissues obtained from animals that were euthanized 48 h following challenge revealed intra-alveolar and interstitial encapsulated bacilli and phagocytic infiltrates in lung tissues (Fig. 8E). Large numbers of encapsulated bacilli were detected in splenic sinusoids (Fig. 8F). At 72 h, lung tissues harbored small areas of hemorrhage in addition to encapsulated bacilli and phagocytic infiltrates (Fig. 8G). At this time point, B. cereus G9241 was found disseminated throughout splenic sinusoids (Fig. 8H). Thus, similar to B. anthracis (23), B. cereus G9241 causes respiratory infections in mice and disseminates rapidly from lung tissues to other organ systems, thereby triggering fatal anthrax-like disease.
Fig 8.
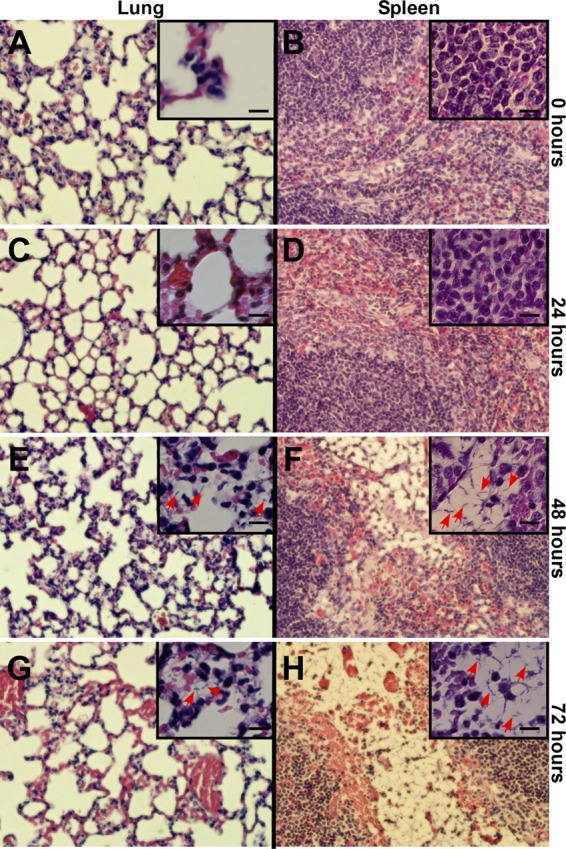
Histopathology of lung and spleen tissues in mice infected with aerosolized spores of B. cereus G9241. C57BL/6 mice (n = 20) were infected with 1 × 106 spores of B. cereus G9241 (see Fig. 7B). Immediately following challenge (A and B, 0 h) or after 24 h (C and D), 48 h (E and F), or 72 h (G and H), animals in each cohort were euthanized and necropsied, and lung or spleen tissues were prepared for histopathology. Thin sections were stained with hematoxylin-eosin and viewed by light microscopy using 400× or 1,000× (insets) magnification. Bar, 10 μm. Red arrows identify vegetative forms of B. cereus G9241. See the text for details.
AVA immunization protects mice against B. cereus G9241 respiratory anthrax-like disease.
Cohorts of C57BL/6 mice (n = 10) were immunized by intramuscular injection with 0.1 ml of AVA (BioThrax) or mock (PBS) control using a prime-booster schedule as described above. Fourteen days following the booster, mice were challenged via aerosol with either 3 × 105 B. cereus G9241 wild-type spores (30× LD50) or 1 × 106 ΔpagA1 mutant spores. All mock-immunized animals challenged with B. cereus G9241 spores succumbed to respiratory anthrax-like disease within 2 to 5 days (Fig. 9A). Half of the mock-immunized animals challenged with ΔpagA1 mutant spores succumbed to infection; these data suggest that the LD50 of the ΔpagA1 mutant is >80-fold higher than that of wild-type B. cereus G9241 spores. AVA immunization protected 90% of animals challenged with B. cereus G9241 spores and 80% of mice that were infected with ΔpagA1 mutant spores (for wild-type challenge, AVA versus mock, P < 0.0001; for ΔpagA1 challenge, AVA versus mock, P = 0.1384) (Fig. 9A).
Fig 9.
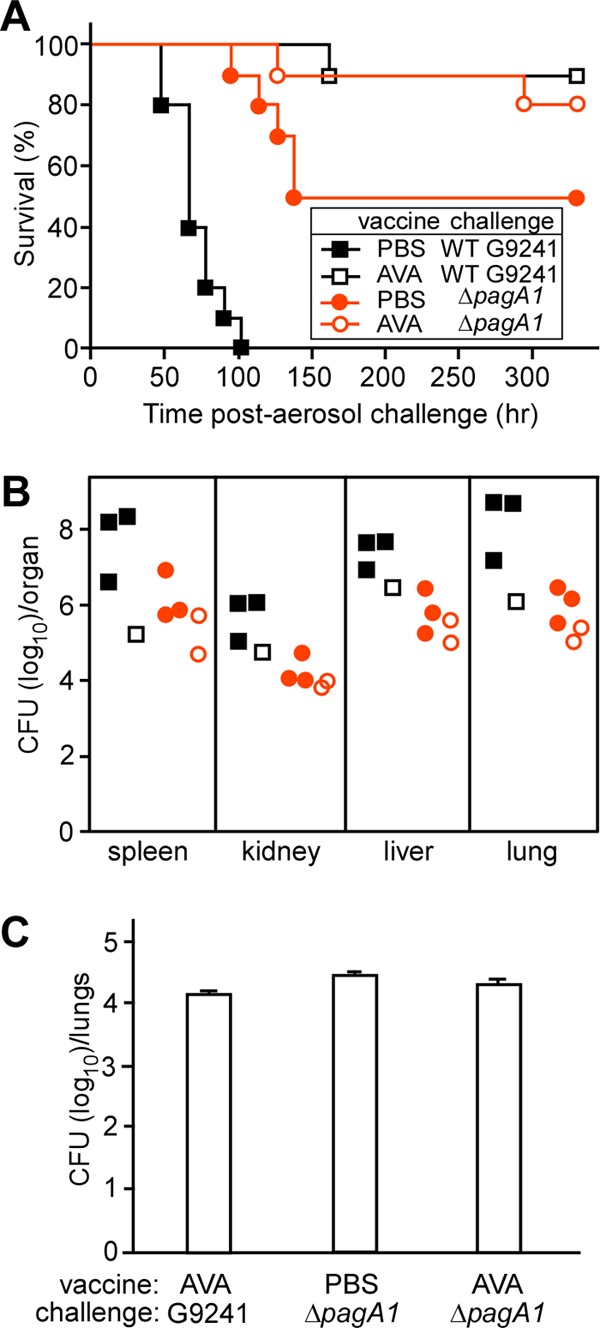
AVA-immunized mice are protected against aerosol challenge of B. cereus G9241 spores. (A) Survival of mock (PBS)- or AVA-immunized C57BL/6 mice (n = 10) following aerosol challenge with B. cereus G9241 wild-type or ΔpagA1 mutant spores. Doses of 3 × 105 wild-type G9241 spores or 1 × 106 ΔpagA1 mutant spores were delivered into the lungs of C57BL/6 mice. (B) Bacterial loads in various organ tissues of mice that succumbed to disease. (C) Numbers of spores retained in the lungs of AVA- or mock (PBS)-immunized animals that survived aerosol challenge with B. cereus G9241 wild-type or ΔpagA1 mutant spores.
When examined for its effect on the bacterial load in moribund animals, we observed that AVA immunization reduced the load of B. cereus G9241 vegetative forms in lung and spleen tissues by 2 to 3 log10 CFU and in kidney or liver tissues by 1 to 2 log10 CFU (Fig. 9B). As expected, AVA immunization did not affect pathogen load in host tissues infected with the ΔpagA1 mutant. Of note, in mock-immunized animals, the load of the ΔpagA1 mutant was reduced by 2 to 3 log10 CFU compared to wild-type B. cereus G9241 (Fig. 9B).
Earlier work reported that PA-specific antibodies promote opsonophagocytic clearance of B. anthracis spores by macrophages (41, 42). These antibodies are thought to bind PA that is produced during sporulation and remains bound to the surface of spores (42). We therefore wondered whether AVA immunization reduced the number of spores in lung tissues of animals that survived the respiratory challenge experiments. Survivors were euthanized on day 14, and homogenized lung tissues were heat treated to kill vegetative forms. AVA-immunized mice that had been challenged with either wild-type or ΔpagA1 mutant B. cereus G9241 still harbored 104 spores in their lung tissues. Thus, AVA immunization did not eliminate the load of B. cereus G9241 spores in lung tissues (Fig. 9C).
DISCUSSION
B. anthracis is a highly virulent pathogen whose spores cause fatal anthrax disease when inhaled (wool sorter's disease) (43). B. cereus strains harboring pXO1-like virulence plasmids for the expression of anthrax toxins have been isolated from welders with severe or fatal respiratory pneumonia (3). Unlike B. anthracis, these B. cereus strains are not designated select agents, and their virulence and pathogenic strategies have only recently been investigated. Wilson and coworkers studied the virulence of B. cereus G9241 spores in a rabbit model of inhalational anthrax-like disease (44). In contrast to B. anthracis strain Ames, which causes lethal respiratory disease in rabbits at a dose of ≥1 × 105 spores, only one out of seven rabbits infected with B. cereus G9241 spores succumbed to aerosol challenge; the moribund rabbit had received a dose of approximately 107 spores (44). By using intranasal spore challenge of anesthetized C57BL/6 mice, those authors calculated an LD50 of 6.3 × 105 B. cereus G9241 spores. This dose is approximately 20- to 50-fold higher (less virulent) than the LD50 of B. anthracis Ames spores administered intranasally into anesthetized BALB/c or C57BL/6 mice (40). However, when spores are inoculated directly into the trachea of anesthetized mice, the LD50 of B. anthracis Ames drops to 5 × 102 to 1 × 103 spores (40). Of note, the structural genes for anthrax toxins (cya, lef, and pagA) are not required for the pathogenesis of respiratory anthrax in mice, as the LD50 for the corresponding mutants does not vary significantly from that of the wild-type parent (45, 46). Thus, it seems likely that AVA immunization and PA-specific antibodies may not protect mice against B. anthracis Ames challenge, as pagA expression is not required for anthrax disease in mice challenged via subcutaneous, intraperitoneal, or intratracheal spore inoculation (33, 45, 46). These results were a surprise, as earlier work had defined protective antigen as the key immunogen for vaccine protection against toxin-mediated disease (47). Indeed, intravenous injection of purified LT into mice causes lethal disease at a dose of 100 μg, and PA-specific antibodies, when administered via passive immunization, provide protection against such challenge (48).
Here we characterized the contribution of protective antigen to B. cereus-mediated anthrax-like disease in C57BL/6 mice. Following intraperitoneal challenge, spores of B. cereus G9241 cause lethal anthrax-like disease, with an LD50 of 2,710 CFU; the disease is associated with massive replication of vegetative forms in all organ tissues (27). The intraperitoneal LD50 of B. cereus G9241 is only 5- to 10-fold higher than that of B. anthracis Ames spores (33). Unlike B. anthracis, secretion of protective antigen is required for B. cereus G924-mediated lethal disease, because the LD50 of the pagA1 mutant strains is increased >80-fold (27). The pBC218 (pagA2)-carried orthologue, PA2, presumably translocates Lef2 (CerADPr) into host cells (2). When purified as a recombinant polypeptide, the product of lef2, CerADPr, leads to ADP-ribosylation of a 120-kDa protein in HeLa tissue culture cells (28). Expression of lef2 as a hybrid with a fluorescent reporter (EGFP-CerADPr) caused HeLa cell rounding (28); however, the role of this activity in B. cereus G9241-mediated anthrax-like disease was hitherto not appreciated. We have shown here that the pagA2 gene and the presumed translocation activity of PA2 are not required for anthrax-like disease in mice. Encouraged by the phenotype of the pagA1 mutant strain, we developed a mouse model for respiratory anthrax-like disease by challenging mice with aerosolized spores. An LD50 of 1.1 × 104 spores was determined for B. cereus G9241, which is 20-fold higher (less virulent) than that of B. anthracis Ames (40). Unlike B. anthracis Ames (46, 49), B. cereus G9241 secretion of PA1 was required for the pathogenesis of respiratory anthrax-like disease.
We therefore reasoned that AVA immunization may be able to protect mice against B. cereus-mediated anthrax-like disease. AVA immunization indeed protected mice against lethal intraperitoneal spore challenge with B. cereus G9241 as well as B. cereus Elc4. Vaccinated animals displayed a significant reduction in bacterial load, but not sterilizing immunity. This is explained via the toxin-neutralizing activity of PA-specific antibodies, which cannot promote the opsonophagocytic clearance of either B. cereus spores or vegetative forms. Vaccine protection was also observed for AVA-immunized mice that were aerosol challenged with 30× the LD50 of B. cereus G9241 spores.
From these data, we conclude that B. cereus G9241 is a fully virulent pathogen that causes respiratory anthrax-like disease not only in humans but also in mice. Its LD50 is 20-fold higher than that of B. anthracis Ames, which is one of the most virulent B. anthracis strains (50). Unlike B. anthracis Ames, B. cereus G9241 requires PA1 (pagA1) for the establishment of respiratory anthrax-like disease. PA1-neutralizing antibodies can be derived via AVA immunization, which protects mice against B. cereus G9241 respiratory anthrax-like disease. We therefore propose that AVA immunization may also protect humans from B. cereus-mediated respiratory anthrax-like disease and provide a preventive strategy for this newly emerging disease.
The amino acid sequences of protective antigen in B. anthracis (PA) and B. cereus G9241 (PA1) are almost identical (2). How then can the same molecule be required for disease pathogenesis in one but not in the other species? B. anthracis Ames A35 (pXO1+ pXO2−), a variant that is attenuated similar to the B. anthracis Sterne vaccine strain (51, 52), causes lethal disease in mice at a subcutaneous challenge dose of 1 × 106 spores (53). The pathogenesis of B. anthracis A35-mediated disease in mice is dependent on the expression of CMG2 (capillary morphogenesis protein 2), the receptor for PA-mediated translocation of LF and EF into host cells (53). Recently, Liu and colleagues demonstrated that lethal disease in mice (following subcutaneous injection of B. anthracis A35 spores) was dependent on CMG2 expression in myeloid cells, such as macrophages, neutrophils, and dendritic cells (54). Of note, CMG2 expression in myeloid cells was not a requirement for toxin-mediated death, as the LD50 for intravenous LT injection was not altered in mice with or without CMG2 expression (54). These results suggest that secretion of PA and LT/ET-mediated intoxication of neutrophils and macrophages enable the disease pathogenesis of B. anthracis (54). However, to detect these phenotypes in mice, B. anthracis must be defective for expression of its PDGA capsule (55, 56). PDGA is a key virulence factor that not only interferes with opsonophagocytic killing of vegetative forms (23, 57), but also, when released from bacilli, displays immune-suppressive attributes (58). B. cereus G9241 does not express PDGA; its capsular material is comprised of hyaluronic acid and the B. cereus exopolysaccharide (BPS) (27). Therefore, the pathogenesis of B. cereus G9241 respiratory anthrax-like infection in mice requires the secretion of PA1 by vegetative bacilli, likely because LT-mediated destruction of neutrophils and macrophages enables pathogen escape from innate immune defenses (59). Support for this hypothesis can be found in the histopathology of B. cereus G9241 anthrax-like disease. As shown in Fig. 2 and 3, vegetative forms of B. cereus G9241 replicated in splenic sinuses to reach >108 CFU/g of tissue; however, bacterial colonies were associated with only a sparse infiltrate of neutrophils and macrophages. In contrast, the tissue load of the ΔpagA1 mutant was reduced by >4 log10 CFU, and isolated bacilli in splenic sinuses were accompanied by a massive infiltrate of macrophages and neutrophils (Fig. 2 and 3).
Finally, although AVA vaccine and PA-specific antibodies can neutralize anthrax toxin, the associated adaptive immune response does not establish sterilizing immunity. This is explained by the absence of antibodies against either capsule or another surface molecule that would promote opsonophagocytosis. It is our conjecture that an ideal anthrax vaccine must accomplish both toxin neutralization and opsonophagocytic clearance of the invading pathogen, i.e., of either spores and/or vegetative forms. For this reason, additional work is needed to develop anthrax vaccines for licensure and use in humans.
ACKNOWLEDGMENTS
We acknowledge membership within and support from the Region V Great Lakes Regional Center of Excellence (GLRCE) in Biodefense and Emerging Infectious Diseases Consortium (NIH award 1-U54-AI-057153). We thank the Animal Research and Immunology Core of the GLRCE for help with animal experiments. This work was supported by the NIH/NIAID, award R01-AI069227.
We thank members of our laboratory for discussions and acknowledge the contributions of the University of Chicago Dual Use Research of Concern (DURC) Task Force for comments on the manuscript. The following reagent was obtained from the NIH Biodefense and Emerging Infections Research Resources Repository, NIAID, NIH: Bacillus cereus G9241.
Footnotes
Published ahead of print 14 January 2013
REFERENCES
- 1. Miller JM, Hair JG, Hebert M, Hebert L, Roberts FJ, Jr, Weyant RS. 1997. Fulminant bacteremia and pneumonia due to Bacillus cereus. J. Clin. Microbiol. 35:504–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hoffmaster AR, Ravel J, Rasko DA, Chapman GD, Chute MD, Marston CK, De BK, Sacchi CT, Fitzgerald C, Mayer LW, Maiden MC, Priest FG, Barker M, Jiang L, Cer RZ, Rilstone J, Peterson SN, Weyant RS, Galloway DR, Read TD, Popovic T, Fraser CM. 2004. Identification of anthrax toxin genes in a Bacillus cereus associated with an illness resembling inhalation anthrax. Proc. Natl. Acad. Sci. U. S. A. 101:8449–8454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hoffmaster AR, Hill KK, Gee JE, Marston CK, De BK, Popovic T, Sue D, Wilkins PP, Avashia SB, Drumgoole R, Helma CH, Ticknor LO, Okinaka RT, Jackson PJ. 2006. Characterization of Bacillus cereus isolates associated with fatal pneumonias: strains are closely related to Bacillus anthracis and harbor B. anthracis virulence genes. J. Clin. Microbiol. 44:3352–3360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wright AM, Beres SB, Consamus EN, Long SW, Flores AR, Barrios R, Richter GS, Oh SY, Garufi G, Maier H, Drews AL, Stockbauer KE, Cernoch P, Schneewind O, Olsen RJ, Musser JM. 2011. Rapidly progressive, fatal, inhalation anthraxlike infection in a human: case report, pathogen genome sequencing, pathology, and coordinated response. Arch. Pathol. Lab. Med. 135:1447–1459 [DOI] [PubMed] [Google Scholar]
- 5. Avashia SB, Riggins WS, Lindley C, Hoffmaster A, Drumgoole R, Nekomoto T, Jackson PJ, Hill K, Williams K, Lehman L, Libal MC, Wilkins PP, Alexander J, Tvaryanas A, Betz T. 2007. Fatal pneumonia among metalworkers due to inhalation exposure to Bacillus cereus containing Bacillus anthracis toxin genes. Clin. Infect. Dis. 44:414–416 [DOI] [PubMed] [Google Scholar]
- 6. Guarner J, Jernigan JA, Shieh WJ, Tatti K, Flannagan LM, Stephens DS, Popovic T, Ashford DA, Perkins BA, Zaki SR; Inhalational Anthrax Pathogology Working Group 2003. Pathology and pathogenesis of bioterrorism-related inhalational anthrax. Am. J. Pathol. 163:701–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jensen GB, Hensen BM, Eilenberg J, Mahillon J. 2003. The hidden lifestyles of Bacillus cereus and relatives. Environ. Microbiol. 5:631–640 [DOI] [PubMed] [Google Scholar]
- 8. Koch R. 1876. Die Ätiologie der Milzbrand-Krankheit, begründet auf die Entwicklungsgeschichte des Bacillus anthracis. Beiträge Biol. Pflanz. 2:277–310 [Google Scholar]
- 9. Mock M, Fouet A. 2001. Anthrax. Annu. Rev. Microbiol. 55:647–671 [DOI] [PubMed] [Google Scholar]
- 10. Green BD, Battisti L, Koehler TM, Thorne CB, Ivins BE. 1985. Demonstration of a capsule plasmid in Bacillus anthracis. Infect. Immun. 49:291–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Uchida I, Hashimoto K, Terakado N. 1986. Virulence and immunogenicity in experimental animals of Bacillus anthracis strains harbouring or lacking 110 MDa and 60 MDa plasmids. J. Gen. Microbiol. 132:557–559 [DOI] [PubMed] [Google Scholar]
- 12. Vodkin MH, Leppla SH. 1983. Cloning of the protective antigen gene of Bacillus anthracis. Cell 34:693–697 [DOI] [PubMed] [Google Scholar]
- 13. Robertson DL, Leppla SH. 1986. Molecular cloning and expression in Escherichia coli of the lethal factor gene of Bacillus anthracis. Gene 44:71–78 [DOI] [PubMed] [Google Scholar]
- 14. Leppla SH. 1982. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclin AMP concentrations in eukaryotic cells. Proc. Natl. Acad. Sci. U. S. A. 79:3162–3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leppla SH. 2000. Anthrax toxin, p 445–472 In Aktories K, Just I. (ed), Bacterial protein toxins. Springer, Berlin, Germany [Google Scholar]
- 16. Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. 2001. Identification of the cellular receptor for anthrax toxin. Nature 414:225–229 [DOI] [PubMed] [Google Scholar]
- 17. Scobie HM, Rainey GJ, Bradley KA, Young JA. 2003. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. U. S. A. 100:5170–5174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Milne J, Furlong D, Hanna PC, Wall JS, Collier RJ. 1994. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J. Biol. Chem. 269:20607–20612 [PubMed] [Google Scholar]
- 19. Milne JC, Blanke SR, Hanna PC, Collier RJ. 1995. Protective antigen-binding domain of anthrax lethal factor mediates translocation of a heterologous protein fused to its amino- or carboxy-terminus. Mol. Microbiol. 15:661–666 [DOI] [PubMed] [Google Scholar]
- 20. Young JA, Collier JR. 2007. Anthrax toxin: receptor binding, internalization, pore formation and translocation. Annu. Rev. Biochem. 76:243–265 [DOI] [PubMed] [Google Scholar]
- 21. Preisz H. 1909. Experimentelle Studien über Virulenz, Empfänglichkeit und Immunität beim Milzbrand. Z. Immun. Forschung 5:341–452 [Google Scholar]
- 22. Tomcsik J, Szongott H. 1933. Über ein spezifisches Protein der Kapsel des Milzbrandbazillus. Z. Immun. Forschung 78:86–99 [Google Scholar]
- 23. Drysdale M, Heninger S, Hutt J, Chen Y, Lyons CR, Koehler TM. 2005. Capsule synthesis by Bacillus anthracis is required for dissemination in murine inhalation anthrax. EMBO J. 24:221–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Drobniewski FA. 1993. Bacillus cereus and related species. Clin. Microbiol. Rev. 6:324–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bottone EJ. 2010. Bacillus cereus, a volatile pathogen. Clin. Microbiol. Rev. 23:382–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stenfors Arnesen LP, Fagerlund A, Granum PE. 2008. From soil to gut: Bacillus cereus and its food poisoning toxins. FEMS Microbiol. Rev. 32:579–606 [DOI] [PubMed] [Google Scholar]
- 27. Oh SY, Budzik JM, Garufi G, Schneewind O. 2011. Two capsular polysaccharides enable Bacillus cereus G9241 to cause anthrax-like disease. Mol. Microbiol. 79:455–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Simon NC, Vergis JM, Ebrahimi AV, Ventura CL, O'Brien AD, Barbieri JT. 30 August 2012. Host cell cytotoxicity and cytoskeleton disruption by CerADPr, an ADP-ribosyltransferase of Bacillus cereus G9241. Biochemistry doi:10.1021/bi300692g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim HU, Goepfert JM. 1974. A sporulation medium for Bacillus anthracis. J. Appl. Bacteriol. 37:265–267 [DOI] [PubMed] [Google Scholar]
- 30. Leighton TJ, Doi RH. 1971. The stability of messenger ribonucleic acid during sporulation in Bacillus subtilis. J. Biol. Chem. 246:3189–3195 [PubMed] [Google Scholar]
- 31. Marraffini LA, Schneewind O. 2006. Targeting proteins to the cell wall of sporulating Bacillus anthracis. Mol. Microbiol. 62:1402–1417 [DOI] [PubMed] [Google Scholar]
- 32. Silo-Suh LA, Lethbridge BJ, Raffel SJ, He H, Clardy J, Handelsman J. 1994. Biological activities of two fungistatic antibiotics produced by Bacillus cereus UW85. Appl. Environ. Microbiol. 60:2023–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Garufi G, Wang Y-T, Oh S-Y, Maier H, Missiakas DM, Schneewind O. 2012. Sortase-conjugation generates a capsule vaccine that protects guinea pigs against Bacillus anthracis. Vaccine 30:3435–3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grabenstein JD. 2008. Vaccines: countering anthrax: vaccines and immunoglobulins. Clin. Infect. Dis. 46:129–136 [DOI] [PubMed] [Google Scholar]
- 35. Collier RJ, Young JA. 2003. Anthrax toxin. Annu. Rev. Cell Dev. Biol. 19:45–70 [DOI] [PubMed] [Google Scholar]
- 36. Singer DE, Schneerson R, Bautista CT, Rubertone MV, Robbins JB, Taylor DN. 2008. Serum IgG antibody response to the protective antigen (PA) of Bacillus anthracis induced by anthrax vaccine adsorbed (AVA) among U.S. military personnel. Vaccine 26:869–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 38. May KR. 1973. The Collison nebulizer: description, performance and applications. J. Aerosol Sci. 4:235–243 [Google Scholar]
- 39. Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am. J. Hyg. (Lond.) 27:493–497 [Google Scholar]
- 40. Lyons CR, Lovchik J, Hutt J, Lipscomb MF, Wang E, Heninger S, Berliba L, Garrison K. 2004. Murine model of pulmonary anthrax: kinetics of dissemination, histopathology, and mouse strain susceptibility. Infect. Immun. 72:4801–4809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cote CK, Van Rooijen N, Welkos SL. 2006. Roles of macrophages and neutrophils in the early host response to Bacillus anthracis spores in a mouse model of infection. Infect. Immun. 74:469–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cote CK, Rossi CA, Kang AS, Morrow PR, Lee JS, Welkos SL. 2005. The detection of protective antigen (PA) associated with spores of Bacillus anthracis and the effects of anti-PA antibodies on spore germination and macrophage interactions. Microb. Pathog. 38:209–225 [DOI] [PubMed] [Google Scholar]
- 43. Dixon TC, Meselson M, Guillemin J, Hanna PC. 1999. Anthrax. N. Engl. J. Med. 341:815–826 [DOI] [PubMed] [Google Scholar]
- 44. Wilson MK, Vergis JM, Alem F, Palmer JR, Keane-Myers AM, Brahmbhatt TN, Ventura CL, O'Brien AD. 2011. Bacillus cereus G9241 makes anthrax toxin and capsule like highly virulent B. anthracis Ames but behaves like attenuated toxigenic nonencapsulated B. anthracis Sterne in rabbits and mice. Infect. Immun. 79:3012–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chand HS, Drysdale M, Lovchik J, Koehler TM, Lipscomb MF, Lyons CR. 2009. Discriminating virulence mechanisms among Bacillus anthracis strains by using a murine subcutaneous infection model. Infect. Immun. 77:429–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Heninger S, Drysdale M, Lovchik J, Hutt J, Lipscomb MF, Koehler TM, Lyons CR. 2006. Toxin-deficient mutants of Bacillus anthracis are lethal in a murine model for pulmonary anthrax. Infect. Immun. 74:6067–6074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smith H, Keppie J, Stanley JL. 1955. The chemical basis of the virulence of Bacillus anthracis. V. The specific toxin produced by Bacillus anthracis in vivo. Br. J. Exp. Pathol. 36:460–472 [PMC free article] [PubMed] [Google Scholar]
- 48. Crowe SR, Ash LL, Engler RJ, Ballard JD, Harley JB, Farris AD, James JA. 2010. Select human anthrax protective antigen epitope-specific antibodies provide protection from lethal toxin challenge. J. Infect. Dis. 202:251–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Welkos SL, Vietri NJ, Gibbs PH. 1993. Non-toxigenic derivatives of the Ames strain of Bacillus anthracis are fully virulent for mice: role of plasmid pXO2 and chromosome in strain-dependent virulence. Microb. Pathog. 14:381–388 [DOI] [PubMed] [Google Scholar]
- 50. Ivins BE, Ezzell JWJ, Jemski J, Hedlund KW, Ristroph JD, Leppla SH. 1986. Immunization studies with attenuated strains of Bacillus anthracis. Infect. Immun. 52:454–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pezard C, Berche P, Mock M. 1991. Contribution of individual toxin components to virulence of Bacillus anthracis. Infect. Immun. 59:3472–3477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sterne M. 1937. Avirulent anthrax vaccine. Onderstepoort J. Vet. Sci. Anim. Ind. 21:41–43 [PubMed] [Google Scholar]
- 53. Liu S, Crown D, Miller-Randolph S, Moayeri M, Wang H, Hu H, Morley T, Leppla SH. 2009. Capillary morphogenesis protein-2 is the major receptor mediating lethality of anthrax toxin in vivo. Proc. Natl. Acad. Sci. U. S. A. 106:12424–12429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu S, Miller-Randolph S, Crown D, Moayeri M, Sastalla I, Okugawa S, Leppla SH. 2010. Anthrax toxin targeting of myeloid cells through the CMG2 receptor is essential for establishment of Bacillus anthracis infections in mice. Cell Host Microbe 8:455–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ivanovics G, Erdös L. 1937. Ein Beitrag zum Wesen der Kaspelsubstanz des Milzbrandbazillus. Z. Immun. Forschung 90:304 [Google Scholar]
- 56. Bruckner V, Kovacs J, Denes G. 1953. Structure of poly-d-glutamic acid isolated from capsulated strains of B. anthracis. Nature 172:508. [DOI] [PubMed] [Google Scholar]
- 57. Richter GS, Anderson VJ, Garufi G, Lu L, Joachimiak A, He C, Schneewind O, Missiakas D. 2009. Capsule anchoring in Bacillus anthracis occurs by a transpeptidation mechanism that is inhibited by capsidin. Mol. Microbiol. 71:404–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zwartouw HT, Smith H. 1956. Polyglutamic acid from Bacillus anthracis grown in vivo: structure and aggressin activity. Biochem. J. 63:437–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Friedlander A. 1986. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J. Biol. Chem. 261:7123–7126 [PubMed] [Google Scholar]
- 60. Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC. 1997. Crystal structure of the anthrax toxin protective antigen. Nature 385:833–838 [DOI] [PubMed] [Google Scholar]