

Abstract
Recurrent urinary tract infections (UTIs), primarily caused by uropathogenic Escherichia coli (UPEC), annually affect over 13 million patients in the United States. Menopausal women are disproportionally susceptible, suggesting estrogen deficiency is a significant risk factor for chronic and recurrent UTI. How estrogen status governs susceptibility to UTIs remains unknown, and whether hormone therapy protects against UTIs remains controversial. Here, we used a mouse model of surgical menopause by ovariectomy and demonstrate a protective role for estrogen in UTI pathogenesis. We found that ovariectomized mice had significantly higher bacteriuria, a more robust inflammatory response, and increased production of the proinflammatory cytokine interleukin-6 (IL-6) upon UPEC infection compared to sham-operated controls. We further show that response of the urothelial stem cell niche to infection, normally activated to restore homeostasis after infection, was aberrant in ovariectomized mice with defective superficial urothelial cell differentiation. Finally, UPEC-infected ovariectomized mice showed a significant increase in quiescent intracellular bacterial reservoirs, which reside in the urothelium and can seed recurrent infections. Importantly, this and other ovariectomy-induced outcomes of UTI were reversible upon estrogen supplementation. Together, our findings establish ovariectomized mice as a model for UTIs in menopausal women and pinpoint specific events during course of infection that are most susceptible to estrogen deficiency. These findings have profound implications for the understanding of the role of estrogen and estrogen therapy in bladder health and pathogen defense mechanisms and open the door for prophylaxis for menopausal women with recurrent UTIs.
INTRODUCTION
Urinary tract infections (UTIs), primarily caused by uropathogenic Escherichia coli (UPEC), are among the most common frequently recurring infectious diseases in humans (1). Menopausal women, who have greatly reduced levels of the sex hormone estrogen, are more likely to have recurrent/chronic UTIs than any other group (2–5); 53% of menopausal women with a UTI will have at least one recurrence (6). Consistent with evidence from animal and human studies, demonstrating that sex hormones have an important effect on the female lower urinary tract during adult life, estrogen receptors have been identified in the bladder, urethra, and pelvic floor. In addition, fluctuations in the circulating levels of estrogen during the menstrual cycle and in pregnancy influence the prevalence of urinary symptoms. Furthermore, decreased estrogen during menopause is a significant risk factor for UTIs (3, 7–9) and bladder barrier dysfunction (10), but little is known about the mechanisms underlying this increased susceptibility.
A murine model of UTI shows that UPEC infection of the urinary bladder follows a multistep pathogenic cycle: UPEC invades superficial urothelial cells by binding cell surface uroplakin receptors (11). During the acute stage of infection (0 to 72 h), intracellular UPEC replicates rapidly and establishes cytoplasmic biofilms termed intracellular bacterial communities (IBCs), which are also observed in humans (12, 13). The host response includes induction of proinflammatory cytokines, including interleukin 6 (IL-6), exfoliation of the superficial urothelial cells containing IBCs into the urine, and influx of innate immune cells, particularly neutrophils. This rapid proinflammatory response aids in defending against pathogens (14, 15). The damaged epithelial barriers are restored by urothelial stem cell niche activation and terminal differentiation of superficial cells (16). Despite the host defense response, however, a subset of UPEC can survive and establish long-term reservoirs, likely within autophagosomal compartments, in urothelial cells (17, 18). These reservoirs, termed quiescent intracellular reservoirs (QIRs), can serve as seeds for recurrent infection (19).
Estrogen has been used widely to treat urinary symptoms in postmenopausal women, but the evidence from randomized studies does not consistently show that estrogen therapy is effective in reducing recurrence of UTIs (20–28). These inconsistencies may be because the trials used variable dosages and durations of treatment. Given that the causes of UTIs are complex and multifactorial, gaining a clear understanding of the role of estrogen in UPEC pathogenesis requires development of an animal model with a defined genetic background in which estrogen levels can be manipulated in a controlled manner. However, there have been limited studies using animal models to examine the estrogenic modulation of UTI progression. One study has suggested that supplementation of 17β-estradiol increased the susceptibility of ascending UTI in the kidneys, but not in the bladder (22). Thus, the dynamic interaction between estrogen signaling and UPEC pathogenesis in the urothelium still remains to be elucidated. In this report, we employ a murine model of surgical menopause, ovariectomy, to directly test the hypothesis that changes in hormone levels play a role in regulating the course of UTIs in the bladder and present evidence that estrogenic deficiency adversely affects the course of UPEC pathogenesis, in particular UPEC persistence in the bladder wall and the urothelial regenerative response upon infection.
MATERIALS AND METHODS
Mice.
All protocols were approved by the animal studies committee of the Washington University School of Medicine (Animal Welfare Assurance number A-3381-01). Mice were maintained under pathogen-free conditions in a barrier facility under a strict 12-h light/dark cycle.
Ovariectomy.
Seven- to 8-week-old C57BL/6 female mice (NCI Mouse Repository, Frederick, MD) were anesthetized and the ovaries excised as described previously (29). For sham surgery, the same procedures were performed without removal of the ovaries. The animals were allowed at least 2 weeks to recover.
17β-estradiol supplementation.
A 90-day time-release pellet containing 0.01 mg of 17β-estradiol (Innovative Research of America, Sarasota, FL) was implanted under the side of the neck of each animal. The mice were maintained on the pellets for 60 to 90 days until sacrifice.
Inoculations of mice.
UTI89 (19), a pathogenic UPEC strain, was grown statically in Luria-Bertani (LB) broth for 17 h at 37°C. Mice were anesthetized and inoculated, via transurethral catheterization, with 50 μl of a bacterial suspension (107 CFU) of UTI89 in phosphate-buffered saline (PBS) as described previously (11, 13).
Tissue histopathology and inflammation scoring.
Bladders were processed as described previously (17). Briefly, bladders were aseptically removed immediately after sacrifice, fixed in methacarn (60% methanol, 30% chloroform, and 10% acetic acid), and embedded in paraffin. Five-micrometer-thick tissue sections were stained with hematoxylin and eosin. Inflammation scores of infected bladders were determined as described previously (30).
Histochemical and immunofluorescence analysis.
Bladders were processed as described above. The following primary antibodies were used on bladder tissue sections: rabbit polyclonal antibody (pAb) to E. coli (1:500; United States Biological, Swampscott, MA), mouse monoclonal antibody (MAb) to Uroplakin III (1:100; Fitzgerald, Acton, MA), goat pAb to BrdU (31), rat MAb to Lamp1 (1:50, clone ID4B; Developmental Studies Hybridoma Bank, Iowa City, IA), rabbit pAb to p27kip1 (1:500; Sigma, St. Louis, MO), rabbit pAb to cytokeratin 5 (CK5) (1:500; Abcam, Cambridge, MA), and mouse MAb to E-cadherin (1:500, BD Transduction Labs, San Jose, CA). After three 5-min PBS washes at room temperature, antigen-antibody complexes were detected with species-specific Alexa Fluor 488-, Alexa Fluor 594-, or Alexa Fluor 647-conjugated secondary antibodies (1:500; Invitrogen, Carlsbad, CA). Images were obtained using a Zeiss Apotome microscope.
QIR quantification.
Six separate 5-μm serial sections over a thickness of 300 μm were immunostained with antibodies against E. coli, Lamp1, and E-cadherin (listed above) and imaged at 63×. The total number of Lamp1-positive UPEC reservoirs in the six sections was counted and reported as the number of QIRs per bladder (n = 18 to 23 mice per group).
BrdU labeling.
BrdU labeling was performed as described previously (16).
Urinalysis and bacterial titers.
Urine specimens from infected mice were collected at 0 to 14 days postinfection (dpi) and serially diluted in PBS; 5 μl of each dilution was spotted onto LB plates six times as described previously (11). Bacterial titers were calculated as CFU/ml of urine. Urine sediments were obtained by cyto-centrifuging 50 μl of a 1:5 dilution of the urine onto poly-l-lysine-coated glass slides, which were then stained for inflammatory scoring as described previously (30).
Bioplex cytokine bead array assay.
Sera were obtained at 6 h postinfection (hpi), and cytokine levels were measured using the Bioplex kit from Bio-Rad (Bio-Rad, Hercules, CA) as described previously (17, 32).
Quantitative real-time PCR (qRT-PCR) analysis.
Bladders from sham-operated controls (SHAM) or ovariectomized (OVX) mice (3 mice per group) were removed at 6 hpi. RNA was isolated from the bladders using TRIzol (Invitrogen, Carlsbad, CA) and treated with DNase I (Ambion, Austin, TX) to remove contaminating DNA. cDNAs were synthesized from 2 μg of total RNA using Superscript II RNase H reverse transcriptase (Invitrogen, Carlsbad, CA). Expression of Bmp4 and p27kip1 was detected by real-time PCR using an ABI Prism 7700 sequence detection system and SYBR green PCR master mix (Applied Biosystems, Foster City, CA). Expression of each target was measured in triplicate. Relative quantification was determined by using the comparative threshold cycle (CT) method with 18S expression as a control, as described in the ABI Prism 7700 sequence detection system user bulletin. The following primers were used for real-time PCR: 18S (5′-CGGCTACCACATCCAAGGAA-3′ and 5′-GCTGGAATTACCGCGGCT-3′), Bmp4 (5′-CAACACCATGATTCCTGGTAACC-3′ and 5′-TCCCGGTCTCAGGTATCAAACT-3′), and p27kip1 (5′-CGGCGGCAAGGTTTGGAGAGG-3′ and 5′GGAGGAGGCAGGAGGAGGTGG-3′).
Statistical analysis.
Two-sample unpaired t tests, nonparametric Mann-Whitney U tests, and one-way analyses of variance (ANOVA) followed by a Tukey's multiple-comparison posttest were performed using GraphPad Prism software. In cases where n was >5, a Shapiro-Francia test was performed for normality. A t test was performed if the normality test was not significant. A Mann-Whitney U test was used if the normality test was significant or n was <5. For time course studies, the standard error (SE) used in the t test was estimated by ANOVA, and two-sample tests were performed at individual time points. To control for false positives, Bonferroni-adjusted P values at individual time points are reported. A P value of <0.05 was used as the cutoff for statistical significance.
RESULTS
Ovariectomized mice exhibit delayed bacterial clearance from the bladder.
To determine whether removal of ovaries affects the UPEC pathogenic cycle, we generated ovariectomized (OVX) mice, which exhibited reduced estrogen levels, and sham-operated controls (SHAM). We then infected the bladders of adult OVX and SHAM mice with UPEC and monitored the progress of the UTI by measuring the shedding of bacteria into the urine (bacteriuria) for 2 weeks (schema in Fig. 1). We found that OVX mice exhibited significantly higher and more sustained bacteriuria, evidenced by higher bacterial load at three to 10 days postinfection (dpi), than SHAM mice, who cleared the infection by day 3 (Fig. 2A). This finding indicates that removal of ovaries results in a prolonged UTI.
Fig 1.
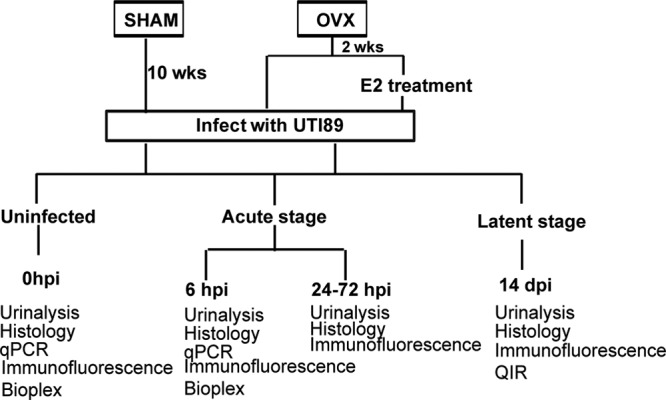
Experimental design: diagram illustrating experimental strategy.
Fig 2.
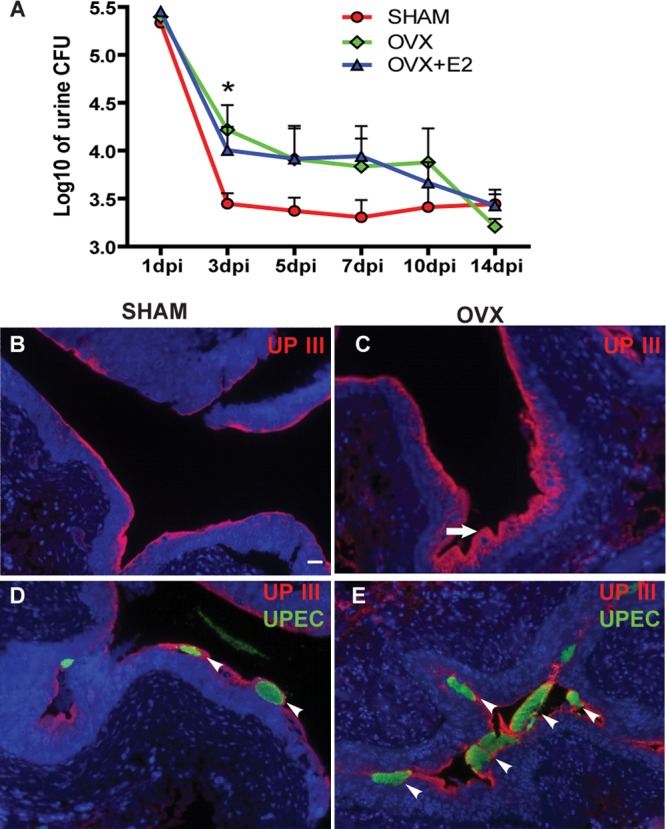
Ovariectomized mice exhibit prolonged infection. (A) CFU counts of bacteriuria over a time course in SHAM, OVX, and OVX+E2 mice plotted as means ± standard errors of the means (SEM) of the log10 value (n = 6 to 12 mice/time point/group in two experiments). *, P < 0.05 (between SHAM and OVX mice by two-way ANOVA with a Bonferroni posttest). (B to E) Immunofluorescence (IF) analysis reveals more UPIII+ (red) superficial cells in OVX mice than SHAM mice at 24 hpi (B and C), as well as more intracellular UPEC communities (green) (D and E). Bar = 20 μm.
Ovariectomy decreases exfoliation of superficial urothelial cells.
One of the first host responses to UPEC infection in the bladder is exfoliation of the superficial urothelial cells containing IBCs into the urine. Although the majority of superficial cells were sloughed into the urine in SHAM mice at 24 h postinfection (hpi) (Fig. 2B), a relatively unperturbed superficial layer was evident in OVX mice (arrow in Fig. 2C; note the thickness of the superficial cell layer marked by expression of uroplakin III [UPIII]). Additionally, UPEC staining of the bladders indicated that more IBCs remained in the intact superficial cells in OVX mice (arrowheads in Fig. 2E) than in SHAM mice (Fig. 2D) at this time point. Thus, ovary removal resulted in decreased exfoliation of infected superficial cells at the acute stage (24 hpi), which might contribute to the prolonged UPEC infection.
Estrogen-deficient mice display a severe proinflammatory response upon UPEC infection.
In addition to the sloughing of superficial cells, the host responds to the pathogen by inducing proinflammatory cytokines, including IL-6 (33), and recruiting immune cells, such as neutrophils, to the infected site. Cytological analysis of urine samples of infected mice revealed greater extent of neutrophil infiltration in the urine at 24 and 72 hpi in OVX mice than in SHAM mice (Fig. 3A). Next, we performed cytokine assays using Bioplex bead arrays on sera to determine whether increased influx of neutrophils was associated with high production of cytokines in OVX mice. We observed significantly higher levels of IL-6 in sera from infected OVX mice than from SHAM mice at 6 hpi (Fig. 3B), suggesting an overall enhanced systemic and luminal proinflammatory response to infection in OVX mice.
Fig 3.

Ovariectomized mice display a severe proinflammatory response upon UPEC infection. (A) Urine inflammation scores are higher in UPEC-infected OVX mice than SHAM mice at 24 and 72 hpi. *, P < 0.05 (by Mann-Whitney U test). (B) Bioplex cytokine assay of sera reveals more IL-6 secreted in UPEC-infected OVX mice than SHAM mice at 6 hpi, reversible upon E2 supplementation. *, P < 0.05; **, P < 0.01 (by one-way ANOVA followed by Tukey's multiple-comparison posttest). (C) Inflammation scoring of bladder tissue at 24 hpi reveals higher scores in OVX mice than SHAM mice, reversible upon E2 supplementation. *, P < 0.05 (by Mann-Whitney U test). (D to F) Hematoxylin and eosin staining of bladders from SHAM, OVX, and OVX+E2 mice at 24 hpi depicts greatest inflammation (arrow points to neutrophils) and edema (arrowhead) in OVX mice. Bar = 40 μm.
To determine whether bladder tissue of OVX mice was prone to greater inflammation, we examined bladders from SHAM and OVX mice at 24 hpi and quantified the level of tissue inflammation. We found that bladders from UPEC-infected OVX mice exhibited higher inflammation scores than SHAM mice (Fig. 3C). Histopathological analyses revealed more severely inflamed bladder tissue in OVX mice with greater immune/inflammatory influx including neutrophils (arrow in Fig. 3E) and severe edema (arrowhead in Fig. 3E) than bladders from SHAM mice (Fig. 3D). Together, these results suggest that OVX mice mount a more robust and prolonged proinflammatory response to UPEC infection than SHAM mice.
Ovariectomization affects USC niche response to UPEC infection.
Superficial urothelial cell loss and associated inflammation induced by UPEC infection cause damage to urothelial barriers that are critical for bladder function. To restore homeostasis, the urothelium activates a regeneration response fueled by activation of the urothelial stem cell (USC) niche and proliferation of the basal urothelial layer. Niche activation and terminal differentiation into mature superficial urothelial cells is dependent on the Bmp4 signaling pathway (16, 34). We sought to determine whether estrogen deficiency affected the USC response to infection. Because the predominant estrogen receptor in the bladder, ERβ, is mainly expressed in the basal USC layer (35), we speculated that the basal USC layer is most likely to be affected by estrogen deficiency. We found that OVX mice exhibited a thickened CK5+ (a marker for basal cells) (16) cell layer at 14 dpi than SHAM mice and, correspondingly, a thinner UPIII+ superficial cell layer (Fig. 4A and B).
Fig 4.

Estrogen deficiency leads to aberrant urothelial regeneration and increased bacterial reservoir formation. (A) CK5 and UPIII staining of bladders at 14 dpi depicting thickened CK+ (green) cell layer in the OVX urothelium. (B) IF analysis depicts reduced UPIII+ (red) staining in OVX bladders at 14 dpi. Dotted lines demarcate epithelium from bladder stroma. Bar = 20 μm. (C) p27kip1 staining of bladders at 14 dpi indicates fewer p27kip1+ cells in OVX urothelia. Bar = 40 μm. (D and E) qRT-PCR analysis reveals Bmp4 (D) and p27kip1 (E) gene expression is downregulated upon UPEC infection at 6 hpi in OVX mice. (F) Quantification of p27kip1+ nuclei reveals reduced numbers in OVX bladders at 14 dpi. Bars represent means ± SEM. *, P < 0.05 (by unpaired two-tailed t test). (G) BrdU counts at the indicated times after infection (n = 3 to 6 mice/time point/condition). Bars represent means ± SEM. (H) Representative image depicting a quiescent intracellular reservoir (QIR). Bar = 10 μm. (I) Quantification of QIRs in bladders at 14 dpi reveals a highly significant increased number in OVX mice, reversible upon E2 supplementation (n = 6 sections/bladder, 10 to 15 mice/group; n = 3 experiments). *, P < 0.05; **, P < 0.01 (by one-way ANOVA followed by Tukey's multiple-comparison posttest).
The thickened basal cell layer in OVX mice could be a result of increased and sustained USC proliferation or a defect in terminal differentiation. To determine whether there was a block in terminal differentiation, we assessed expression of Bmp4 pathway components. qPCR analysis revealed that expression of Bmp4 and its downstream target p27kip1 were both downregulated in OVX mice after UPEC infection (Fig. 4D and E). We also found that bladders from OVX mice had fewer p27kip1-positive nuclei than SHAM mice (Fig. 4C and F), suggesting that these cells were not exiting the cell cycle to terminally differentiate. We assessed proliferation by BrdU labeling and found no significant differences in proliferative activity between SHAM and OVX mice (Fig. 4G), indicating that the thickened basal cell layer in OVX mice was not due to increased proliferation. Together, our findings suggest that the Bmp4 pathway was downregulated, and the differentiation of basal cells to superficial cells was blocked by ovariectomy, thus impairing the USC niche adaptation to infection.
Estrogen deficiency enhances UPEC persistence.
Even after epithelial exfoliation and proinflammatory responses eliminate the majority of intracellular bacteria, a subset of UPEC can survive and establish long-term reservoirs, termed quiescent intracellular reservoirs (QIRs), which serve as seeds for recurrent infection (19). We thus examined SHAM and OVX bladders at 14 dpi to determine whether enhanced bacterial colonization in the bladders of OVX mice at the acute stage of infection was associated with increased establishment of QIRs. Because UPEC can establish reservoirs containing small numbers of bacteria that are below the detectable limit in bladder titers (19), we investigated QIR formation by immunostaining bladder sections with antibodies to UPEC and Lamp-1, a marker for the vesicles in which QIRs are found (17, 19) (Fig. 4H). Bladders of OVX mice harbored significantly more QIRs in Lamp1-positive vesicles than bladders of SHAM mice (Fig. 4I). Thus, estrogen deficiency is associated with increased establishment of persistent bacterial reservoirs.
Estrogen replacement therapy overall restores bladder response to UPEC to premenopausal state.
Ovariectomy results in decreases in both estrogen and progesterone levels. Thus, to establish whether the effects of OVX we observed were mediated exclusively by estrogen, we supplemented a cohort of OVX mice with 17β-estradiol (OVX+E2) for 8 weeks before induction of a UTI and followed the pathogenic cycle as described above. Exogenous administration of E2 did not significantly affect the bacterial loads in the urine (Fig. 2A). We next asked whether the immunological responses of the OVX mice to infection were rescued by estrogen supplementation. We observed a slight, but not significant, decrease in neutrophil infiltration in the urine at 24 and 72 h after UPEC infection (Fig. 3A). However, we observed significantly lower IL-6 serum levels (Fig. 3B) and low inflammation levels in the bladder mucosa (Fig. 3C; compare panel F to E) in OVX+E2 mice than OVX mice. Additionally, we did not observe a thickening of the CK5+ basal cell layer in OVX+E2 mice (Fig. 4A). Together, these findings suggest that the increased and sustained inflammation and tissue damage observed in OVX mice is, at least in part, due to estrogen deficiency. Finally, we asked whether estrogen supplementation could reduce the number of latent QIRs that formed in OVX mice. We found that bladders from OVX mice that received E2 supplementation contained fewer QIRs than mice that were ovariectomized but did not receive E2 supplementation (Fig. 4I). In fact, at 14 dpi, OVX mice receiving E2 before infection harbored similar numbers of QIRs as SHAM mice, indicating that the enhanced QIR formation we observed in OVX mice was largely due to estrogen deficiency.
DISCUSSION
Here, we employ a murine model of surgical menopause by ovariectomy to demonstrate that estrogen plays a protective role in regulating the host response to UPEC infection. We show that ovariectomization results in prolonged and more severe infection that is associated not only with increased and sustained bacteriuria but also with elevated inflammation and a significantly greater number of persistent bacterial reservoirs. We further demonstrate that estrogen deficiency is associated with an aberrant tissue regenerative response wherein restoration of urothelial barriers is delayed considerably. Thus, our model has shown that critical aspects of disease pathogenesis are under estrogenic control; namely, tissue restoration and regeneration following infection-induced injury, inflammatory response to UPEC, and, importantly, UPEC persistence in the bladder wall. We propose that our model lays the groundwork for exploring the mechanisms of estrogenic action and for testing hormone therapy efficacy.
Estrogen and immune response.
High estrogen levels have potent anti-inflammatory functions, including repression of proinflammatory gene transcription and cytokine production such as IL-6 (36–39). The anti-inflammatory effects of estrogens have been observed in several disease models, including autoimmunity, atherosclerosis, arthritis, inflammatory bowel disease, asthma, and influenza (40–42). Our data are consistent with an inhibitory role of estrogen on IL-6 production (38, 43) and the presence of systemic elevated IL-6 levels in menopausal women (39). A heightened IL-6 response may play a role in the pathogenesis of UTIs in menopausal patients similar to other chronic inflammatory diseases such as rheumatoid arthritis (39). Our data suggest that estrogen might contribute to the functional integrity of the bladder barrier by quenching the inflammatory response associated with UTI. Excessive IL-6 or production of other proinflammatory cytokines can result in urothelial damage that may affect disease outcome. It has been previously shown that elevated and sustained levels of serum cytokines, including IL-6, along with IL-5, granulocyte colony-stimulating factor (G-CSF), and keratinocyte-derived chemokine (KC) (biomarkers of local and systemic acute inflammation), precede the development of chronic cystitis and that the early immune events serve as a “checkpoint” for predicting infection outcomes (32). The elevated early IL-6 response in OVX mice may predispose the mice to the prolonged high-titer UPEC urine load noted. This window of time may be the key in setting the stage for disease outcome and/or UTI recurrence. Our data, together with other studies in postmenopausal women (44), suggest that IL-6 levels may be a major biomarker of chronic inflammatory activity in a postmenopausal state.
Estrogen, urothelial stem cell niche activation, and recurrent UTIs.
UPEC can persist indefinitely as quiescent reservoirs within the immature basal cells of the bladder and can reemerge to seed recurrent UTIs (19). The basal cells are the early progenitor cells and serve as a protective niche in which UPEC can escape immune detection and evade exfoliation. Here, we showed that hypoestrogenization induces a thickened basal cell layer due to the disrupted differentiation process of basal cells to superficial cells. OVX mice not only displayed low estrogen levels, but the expression of Bmp4 and its downstream target p27kip1 were downregulated in these mice. Because the Bmp4 pathway is required for USC niche activation and differentiation (16), this result may explain why the basal cell layer was thicker in OVX mice than in SHAM mice after UPEC infection. The mechanisms by which Bmp4 signaling is modulated by estrogen in response to UPEC infection in the bladder are unclear, but one possibility is that Bmp4 signaling may be modulated by the glycosaminoglycan (GAG) layer covering the urothelium. GAGs are large linear polysaccharides with a high degree of structural heterogeneity mediated by GAG biosynthetic enzymes (45). GAGs are known to modulate growth factors (46–48). We have previously shown that levels of a GAG-sulfating enzyme, HS6ST1, a key modulator of Bmp4 signaling, are increased upon infection (49). Additionally, we have shown that estrogen plays a key role in influencing the GAG thickness and the increased expression of GAG sulfation enzymes over the course of UPEC infection (29). Together, this leads us to speculate that increased sulfation of GAGs may modulate the downregulation of Bmp4 signaling and thereby the USC niche response to infection.
UTI recurrence may depend on UPEC's ability to manipulate differentiation and proliferation of USCs. Thus, the thickened USC layer may provide more protective niches for QIRs to form. These QIRs may hide in the bladder longer due to the slower turnover rate from basal cells to superficial cells. Traditional antibiotic therapies are not effective against bacteria sequestered in QIRs and can also increase the risk of driving the pathogens into quiescence (50). As these reservoirs are a source of recurrent UTIs, any reduction in their establishment and greater understanding of the interplay between latency and estrogen signaling will have great significance for studying infectious disease in aging female populations.
Estrogen therapy and UTIs.
Our results suggest that hormone therapy is beneficial to OVX mice with UTI, and this beneficial effect may be due to the downregulation of the proinflammatory response and the homeostasis of urothelium promoted by the presence of exogenous estrogen. While further studies using multiple UPEC strains would be valuable, we propose that our findings provide an explanation for why menopausal women may be at greater risk for recurrent UTIs. In postmenopausal women, efficacy of estrogen supplementation in UTI prevention has relied upon alterations in bacteriuria loads as a measure of success. Our findings showing that estrogen supplementation does not affect bacteriuria may offer an explanation for the contradictory reports and are consistent with those of Curran and coworkers, who did not observe changes in bacterial load in the bladder (22), as well as another study demonstrating that estrogen's effect on disease outcome was independent of influenza viral load (40). Although testing different levels of E2 supplementation and the short-term and long-term consequences of therapy remain to be determined, our work suggests that estrogen therapy may be beneficial to women with recurrent UTIs and could have implications for reducing the burden of this infectious disease in aging populations. For example, longer-term estrogen therapy with transiently increased doses during an acute episode of UTI might be beneficial. Our data may have significant clinical implications for understanding the etiology of recurrent UTIs in menopausal women and warrant further studies for potential usage of estrogen as a therapeutic intervention.
ACKNOWLEDGMENTS
We thank members of our laboratory and Jason Mills, Congxing Lin, and Rodney Newberry for comments.
This work was supported by a pilot grant from the Center for Women's Infectious Disease Research at Washington University (IUM) and by the Multiplex Gene Analysis Core of the Siteman Cancer Center (supported in part by National Cancer Institute grant P30 CA91842).
Footnotes
Published ahead of print 21 December 2012
REFERENCES
- 1. Dielubanza EJ, Schaeffer AJ. 2011. Urinary tract infections in women. Med. Clin. North Am. 95: 27–41 [DOI] [PubMed] [Google Scholar]
- 2. Raz R. 2011. Urinary tract infection in postmenopausal women. Korean J. Urol. 52: 801–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Foxman B. 1990. Recurring urinary tract infection: incidence and risk factors. Am. J. Public Health 80: 331–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hextall A, Cardozo L. 2001. The role of estrogen supplementation in lower urinary tract dysfunction. Int. Urogynecol. J. Pelvic Floor Dysfunct. 12: 258–261 [DOI] [PubMed] [Google Scholar]
- 5. Foxman B. 1999. Urinary tract infection in postmenopausal women. Curr. Infect. Dis. Rep. 1: 367–370 [DOI] [PubMed] [Google Scholar]
- 6. Ikaheimo R, Siitonen A, Heiskanen T, Karkkainen U, Kuosmanen P, Lipponen P, Makela PH. 1996. Recurrence of urinary tract infection in a primary care setting: analysis of a 1-year follow-up of 179 women. Clin. Infect. Dis. 22: 91–99 [DOI] [PubMed] [Google Scholar]
- 7. Foxman B, Brown P. 2003. Epidemiology of urinary tract infections: transmission and risk factors, incidence, and costs. Infect. Dis. Clin. North Am. 17: 227–241 [DOI] [PubMed] [Google Scholar]
- 8. Foxman B, Gillespie B, Koopman J, Zhang L, Palin K, Tallman P, Marsh JV, Spear S, Sobel JD, Marty MJ, Marrs CF. 2000. Risk factors for second urinary tract infection among college women. Am. J. Epidemiol. 151: 1194–1205 [DOI] [PubMed] [Google Scholar]
- 9. Foxman B, Somsel P, Tallman P, Gillespie B, Raz R, Colodner R, Kandula D, Sobel JD. 2001. Urinary tract infection among women aged 40 to 65: behavioral and sexual risk factors. J. Clin. Epidemiol. 54: 710–718 [DOI] [PubMed] [Google Scholar]
- 10. Hass MA, Nichol P, Lee L, Levin RM. 2009. Estrogen modulates permeability and prostaglandin levels in the rabbit urinary bladder. Prostaglandins Leukot. Essent. Fatty Acids 80: 125–129 [DOI] [PubMed] [Google Scholar]
- 11. Hung CS, Dodson KW, Hultgren SJ. 2009. A murine model of urinary tract infection. Nat. Protoc. 4: 1230–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Garofalo CK, Hooton TM, Martin SM, Stamm WE, Palermo JJ, Gordon JI, Hultgren SJ. 2007. Escherichia coli from urine of female patients with urinary tract infections is competent for intracellular bacterial community formation. Infect. Immun. 75: 52–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rosen DA, Hooton TM, Stamm WE, Humphrey PA, Hultgren SJ. 2007. Detection of intracellular bacterial communities in human urinary tract infection. PLoS Med. 4: e329 doi:10.1371/journal.pmed.0040329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hannan TJ, Totsika M, Mansfield KJ, Moore KH, Schembri MA, Hultgren SJ. 2012. Host-pathogen checkpoints and population bottlenecks in persistent and intracellular uropathogenic Escherichia coli bladder infection. FEMS Microbiol. Rev. 36: 616–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nielubowicz GR, Mobley HL. 2010. Host-pathogen interactions in urinary tract infection. Nat. Rev. Urol. 7: 430–441 [DOI] [PubMed] [Google Scholar]
- 16. Mysorekar IU, Isaacson-Schmid M, Walker JN, Mills JC, Hultgren SJ. 2009. Bone morphogenetic protein 4 signaling regulates epithelial renewal in the urinary tract in response to uropathogenic infection. Cell Host Microbe 5: 463–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang C, Mendonsa GR, Symington JW, Zhang Q, Cadwell K, Virgin HW, Mysorekar IU. 2012. Atg16L1 deficiency confers protection from uropathogenic Escherichia coli infection in vivo. Proc. Natl. Acad. Sci. U. S. A. 109: 11008–11013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang C, Symington JW, Mysorekar IU. 2012. ATG16L1 and pathogenesis of urinary tract infections. Autophagy 8: 1693–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mysorekar IU, Hultgren SJ. 2006. Mechanisms of uropathogenic Escherichia coli persistence and eradication from the urinary tract. Proc. Natl. Acad. Sci. U. S. A. 103: 14170–14175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brown JS, Vittinghoff E, Kanaya AM, Agarwal SK, Hulley S, Foxman B. 2001. Urinary tract infections in postmenopausal women: effect of hormone therapy and risk factors. Obstet. Gynecol. 98: 1045–1052 [DOI] [PubMed] [Google Scholar]
- 21. Eriksen B. 1999. A randomized, open, parallel-group study on the preventive effect of an estradiol-releasing vaginal ring (Estring) on recurrent urinary tract infections in postmenopausal women. Am. J. Obstet. Gynecol. 180: 1072–1079 [DOI] [PubMed] [Google Scholar]
- 22. Curran EM, Tassell AH, Judy BM, Nowicki B, Montgomery-Rice V, Estes DM, Nowicki S. 2007. Estrogen increases menopausal host susceptibility to experimental ascending urinary-tract infection. J. Infect. Dis. 195: 680–683 [DOI] [PubMed] [Google Scholar]
- 23. Oliveria SA, Klein RA, Reed JI, Cirillo PA, Christos PJ, Walker AM. 1998. Estrogen replacement therapy and urinary tract infections in postmenopausal women aged 45–89. Menopause 5: 4–8 [PubMed] [Google Scholar]
- 24. Orlander JD, Jick SS, Dean AD, Jick H. 1992. Urinary tract infections and estrogen use in older women. J. Am. Geriatr. Soc. 40: 817–820 [DOI] [PubMed] [Google Scholar]
- 25. Raz R, Colodner R, Rohana Y, Battino S, Rottensterich E, Wasser I, Stamm W. 2003. Effectiveness of estriol-containing vaginal pessaries and nitrofurantoin macrocrystal therapy in the prevention of recurrent urinary tract infection in postmenopausal women. Clin. Infect. Dis. 36: 1362–1368 [DOI] [PubMed] [Google Scholar]
- 26. Raz R, Stamm WE. 1993. A controlled trial of intravaginal estriol in postmenopausal women with recurrent urinary tract infections. N. Engl. J. Med. 329: 753–756 [DOI] [PubMed] [Google Scholar]
- 27. Perrotta C, Aznar M, Mejia R, Albert X, Ng CW. 2008. Oestrogens for preventing recurrent urinary tract infection in postmenopausal women. Obstet. Gynecol. 112: 689–690 [DOI] [PubMed] [Google Scholar]
- 28. Stamm WE. 2007. Estrogens and urinary-tract infection. J. Infect. Dis. 195: 623–624 [DOI] [PubMed] [Google Scholar]
- 29. Anand M, Wang C, French J, Isaacson-Schmid M, Wall LL, Mysorekar IU. 2012. Estrogen affects the glycosaminoglycan layer of the murine bladder. Female Pelvic Med. Reconstr. Surg. 18: 148–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stemler KM, Crock LW, Lai HH, Mills JC, Gereau RW, IV, Mysorekar IU. 2013. Protamine sulfate induced bladder injury protects from distention induced bladder pain. J. Urol. 189: 343–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim SH, Roth KA, Moser AR, Gordon JI. 1993. Transgenic mouse models that explore the multistep hypothesis of intestinal neoplasia. J. Cell Biol. 123: 877–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hannan TJ, Mysorekar IU, Hung CS, Isaacson-Schmid ML, Hultgren SJ. 2010. Early severe inflammatory responses to uropathogenic E. coli predispose to chronic and recurrent urinary tract infection. PLoS Pathog. 6: e1001042 doi:10.1371/journal.ppat.1001042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hunstad DA, Justice SS, Hung CS, Lauer SR, Hultgren SJ. 2005. Suppression of bladder epithelial cytokine responses by uropathogenic Escherichia coli. Infect. Immun. 73: 3999–4006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miyazaki Y, Oshima K, Fogo A, Ichikawa I. 2003. Evidence that bone morphogenetic protein 4 has multiple biological functions during kidney and urinary tract development. Kidney Int. 63: 835–844 [DOI] [PubMed] [Google Scholar]
- 35. Imamov O, Yakimchuk K, Morani A, Schwend T, Wada-Hiraike O, Razumov S, Warner M, Gustafsson JA. 2007. Estrogen receptor beta-deficient female mice develop a bladder phenotype resembling human interstitial cystitis. Proc. Natl. Acad. Sci. U. S. A. 104: 9806–9809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu H, Liu K, Bodenner DL. 2005. Estrogen receptor inhibits interleukin-6 gene expression by disruption of nuclear factor kappaB transactivation. Cytokine 31: 251–257 [DOI] [PubMed] [Google Scholar]
- 37. Ray P, Ghosh SK, Zhang DH, Ray A. 1997. Repression of interleukin-6 gene expression by 17 beta-estradiol: inhibition of the DNA-binding activity of the transcription factors NF-IL6 and NF-kappa B by the estrogen receptor. FEBS Lett. 409: 79–85 [DOI] [PubMed] [Google Scholar]
- 38. Galien R, Garcia T. 1997. Estrogen receptor impairs interleukin-6 expression by preventing protein binding on the NF-kappaB site. Nucleic Acids Res. 25: 2424–2429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Straub RH. 2007. The complex role of estrogens in inflammation. Endocr. Rev. 28: 521–574 [DOI] [PubMed] [Google Scholar]
- 40. Robinson DP, Lorenzo ME, Jian W, Klein SL. 2011. Elevated 17beta-estradiol protects females from influenza A virus pathogenesis by suppressing inflammatory responses. PLoS Pathog. 7: e1002149 doi:10.1371/journal.ppat.1002149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jansson L, Olsson T, Holmdahl R. 1994. Estrogen induces a potent suppression of experimental autoimmune encephalomyelitis and collagen-induced arthritis in mice. J. Neuroimmunol. 53: 203–207 [DOI] [PubMed] [Google Scholar]
- 42. Kim S, Liva SM, Dalal MA, Verity MA, Voskuhl RR. 1999. Estriol ameliorates autoimmune demyelinating disease: implications for multiple sclerosis. Neurology 52: 1230–1238 [DOI] [PubMed] [Google Scholar]
- 43. Deshpande R, Khalili H, Pergolizzi RG, Michael SD, Chang MD. 1997. Estradiol down-regulates LPS-induced cytokine production and NFkB activation in murine macrophages. Am. J. Reprod. Immunol. 38: 46–54 [DOI] [PubMed] [Google Scholar]
- 44. Kim OY, Chae JS, Paik JK, Seo HS, Jang Y, Cavaillon JM, Lee JH. 2012. Effects of aging and menopause on serum interleukin-6 levels and peripheral blood mononuclear cell cytokine production in healthy nonobese women. Age (Dordr.) 34: 415–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Taylor KR, Gallo RL. 2006. Glycosaminoglycans and their proteoglycans: host-associated molecular patterns for initiation and modulation of inflammation. FASEB J. 20: 9–22 [DOI] [PubMed] [Google Scholar]
- 46. Lamanna WC, Kalus I, Padva M, Baldwin RJ, Merry CL, Dierks T. 2007. The heparanome—the enigma of encoding and decoding heparan sulfate sulfation. J. Biotechnol. 129: 290–307 [DOI] [PubMed] [Google Scholar]
- 47. Esko JD, Lindahl U. 2001. Molecular diversity of heparan sulfate. J. Clin. Invest. 108: 169–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Garcia-Garcia MJ, Anderson KV. 2003. Essential role of glycosaminoglycans in Fgf signaling during mouse gastrulation. Cell 114: 727–737 [DOI] [PubMed] [Google Scholar]
- 49. Mysorekar IU, Mulvey MA, Hultgren SJ, Gordon JI. 2002. Molecular regulation of urothelial renewal and host defenses during infection with uropathogenic Escherichia coli. J. Biol. Chem. 277: 7412–7419 [DOI] [PubMed] [Google Scholar]
- 50. Blango MG, Mulvey MA. 2010. Persistence of uropathogenic Escherichia coli in the face of multiple antibiotics. Antimicrob. Agents Chemother. 54: 1855–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]