

Abstract
Helicobacter pylori infection of the stomach is related to the development of diverse gastric pathologies. The ability of H. pylori to compromise epithelial junctional complexes and to induce proinflammatory cytokines is believed to contribute to pathogenesis. The purpose of this study was to use an in vitro human gastric epithelial model to investigate the ability of H. pylori to affect permeability and the extent and polarity of the host inflammatory response. NCI-N87 monolayers were cocultured with live or heat-killed H. pylori or culture supernatants. Epithelial barrier function was measured by transepithelial electric resistance (TEER) analysis, diffusion of fluorescein isothiocyanate (FITC)-labeled markers, and immunostaining for tight junction proteins. Supernatants from both apical and basolateral chambers were tested for cytokine production by multiplex analysis. H. pylori caused a significant decrease in TEER, an increased passage of markers through the infected monolayer, and severe disruption and mislocalization of ZO-1 and claudin-1 proteins. Cell viability was not altered by H. pylori, indicating that loss of barrier function could be attributed to a breakdown of tight junction integrity. Significantly high levels of cytokine secretion were induced by either viable or heat-killed H. pylori. H. pylori affects monolayer permeability of polarized human gastric epithelial cells. Proinflammatory cytokines were secreted in a polarized manner, mostly basolaterally. Live bacteria are required for disruption of tight junctions but not for the induction of cytokine secretion. The NCI-N87 cell line provides an excellent model for the in vitro study of H. pylori pathogenesis and the epithelial cell host response to infection.
INTRODUCTION
Gastric epithelial barrier function is essential for preventing potentially dangerous organisms present in the lumen from accessing the gastric mucosa. Epithelial cells lining the stomach represent the first line of defense against pathogens. Tight junctions located apically at cell-cell contacts play critical roles in preserving epithelial monolayer barrier integrity and function, cell polarity, and intercellular adhesion. Disruption of tight junction complexes is associated with a variety of human diseases, including cancers of the gastrointestinal tract (1).
Helicobacter pylori is a Gram-negative microaerophilic, spiral bacterium that specifically colonizes the gastric mucosa (2). Over half of the human population is infected with H. pylori. It is usually acquired in childhood and, when left untreated, generally persists for the host's life span (3). Chronic infection causes only superficial or asymptomatic gastritis in most cases, but bacterial interaction with the host can evolve into more serious and acute pathologies such as peptic ulcer disease, mucosa-associated lymphoid tissue (MALT) lymphoma, or gastric adenocarcinoma (4–6). Because epidemiological studies have determined that the attributable risk for gastric cancer conferred by H. pylori is approximately 75%, the World Health Organization (WHO) has classified H. pylori as a type 1 carcinogen (7). The ability of H. pylori to induce superficial gastritis suggests that this organism—or the host inflammatory response to it—could play a pivotal role in the initiation and promotion of gastric neoplasia (8).
H. pylori interacts closely with epithelial cells, eliciting a variety of responses via distinct molecular interactions. The type IV secretion system (T4SS) transfers the CagA oncoprotein into the host cell cytoplasm, where it becomes phosphorylated and influences several distinct cellular processes (9–13). It is the direct contact of the T4SS with the epithelial cell membrane, however, that is believed to be primarily responsible for induction of the secretion of interleukin-8 (IL-8) and other host immune response events (14–17). Epithelial α5β1 integrin has been implicated as one of the host receptors for this activity (18–21). Expression of these integrins, however, is limited to the basolateral membranes. An in vitro epithelial cell monolayer model with tight junction integrity and measurable barrier function would facilitate the study of H. pylori-host cell interactions and allow for detailed examinations of the molecular events that occur apically and basolaterally during H. pylori infection.
A simple monolayer system would also eliminate potential influence from lamina propria cells and/or the enteric nervous system on epithelial cell function and enable the study of host-pathogen interaction in isolation. The lack of human gastric cell lines capable of establishing normal gastric epithelial barrier function has limited the application of a cell culture system as a model of H. pylori-infected gastric mucosa. The NCI-N87 cell line is one of the few human gastric epithelial cell lines demonstrated to form tight and coherent monolayers (22).
The aim of this study was to evaluate the effects of H. pylori on polarized gastric epithelial monolayers with respect to compromising barrier function and also addressing the host cell inflammatory response mechanisms involved. We now report that a coculture system of live H. pylori and polarized NCI-N87 cell monolayers can be used to demonstrate loss of barrier function in viable epithelial cell monolayers through the disruption of barrier proteins ZO-1 and claudin-1 and that this impairment of epithelial barrier integrity is independent of the main individual H. pylori virulence factors. We also report that both live and heat-killed H. pylori bacteria induce an IL-8-dominant inflammatory cytokine response predominantly at the basolateral cell surface and that the ability of heat-killed bacteria to induce strong cytokine production in the absence of transepithelial electric resistance (TEER) reductions suggests that cytokine production and epithelial barrier disassembly are unrelated.
MATERIALS AND METHODS
Cell culture.
The human NCI-N87 gastric cell line was purchased from ATCC (Manassas, VA). Cells were cultured at 37°C in Dulbecco modified Eagle medium (DMEM)-F12K (1:1) medium (Invitrogen, Camarillo, CA) supplemented with 10% fetal bovine serum (FBS). Monolayers were grown to confluence on 1.12-cm2 permeable polyester filters with a 0.4-μm pore size (Corning, Lowell, MA) and tested for barrier formation prior to use.
Bacterial strains and growth conditions.
H. pylori strain 26695 was purchased from ATCC (Manassas, VA). Pathogenicity island-deficient (CagPAI−) and VacA-deficient (VacA−) derivative strains of H. pylori 26695 were obtained from Ellen Beswick (University of New Mexico, Albuquerque, NM). H. pylori strains M5 and HpM5ureB were generated by our laboratory as described previously (23). All H. pylori strains were grown on Columbia blood agar (Difco, Detroit, MI) containing 7% defibrinated horse blood (Hemostat Laboratories, Dixon, CA), amphotericin B (2.5 μg/ml), and the selective antibiotics trimethoprim (20 μg/ml), vancomycin (6 μg/ml), and cefsulodin (16 μg/ml). Antibiotics were purchased from Sigma-Aldrich (St. Louis, MO). Cultures were grown in a designated CO2 incubator with a humidity tray at 37°C and 10% CO2 for 72 to 96 h. In preparation for coculture assays with epithelial cell monolayers, bacteria were transferred to 10 ml Brucella broth (Difco) containing 10% FBS plus antibiotics in 25-cm2 tissue culture flasks overnight. Bacterial density was determined by obtaining readings at an optical density at 450 nm (OD450) and comparing them to a standardized growth curve.
Generation of conditioned media and heat-killed cultures.
Aliquots of overnight precultured H. pylori strain 26695 were grown as liquid cultures to a final OD450 of 0.1. Conditioned media (culture supernatants) were prepared after bacteria were removed by centrifugation. The supernatants were filter sterilized by passage through a 0.22-μm-pore-size filter and used immediately. Heat-killed bacteria were prepared by growing H. pylori 26695 in liquid cultures as described above and placing the culture tubes in a boiling water bath for 30 min.
Measurement of TEER.
Transepithelial electric resistance (TEER) was used to monitor the integrity of the epithelial monolayer and was determined using a Millicell ERS volt-ohm meter (World Precision Instruments, New Haven, CT) according to the manufacturer's instructions. TEER values were calculated as ohms × cm2. Monolayers reaching TEER values between ∼800 to 1,400 Ω · cm2 were considered to have an appropriate barrier function and were used for further study. Cells were gently washed, incubated with DMEM-F12K (1:1) deprived of antibiotics, and allowed to equilibrate at 37°C for 1 to 2 h before bacterial infection. Bacterial suspensions, heat-killed bacteria, or bacterial supernatants were administered apically at an inoculation ratio (multiplicity of infection [MOI]) of 50:1 bacteria/epithelial cell and incubated at 37°C. TEER was measured at 24 and 48 h postinfection.
Cell viability.
The viability of monolayers after infection with H. pylori was assessed using the CytoTox 96 lactate dehydrogenase (LDH) secretion assay (Promega, Madison, WI) to evaluate supernatants according to the manufacturer's instructions. Monolayers were infected with viable, killed, or filtered bacterial cultures, and the supernatants were collected apically after 48 h postinfection. Lysis of the cells with 1% Triton X-100 served as a positive control.
Assessment of NCI-N87 cell monolayer permeability.
The permeability of the gastric cell monolayer was evaluated by measuring the diffusion of fluorescein isothiocyanate (FITC)-dextran and FITC-bovine serum albumin (BSA) with molecular masses of 4.0 and 40 kDa (Sigma), respectively, from the apical to the basal medium compartments. FITC-dextran and FITC-BSA were dissolved in P buffer (10 mM HEPES [pH 7.4], 1 mM sodium pyruvate, 10 mM glucose, 3 mM CaCl2, 145 mM NaCl) or P/EGTA buffer [10 mM HEPES (pH 7.4), 1 mM sodium pyruvate, 10 mM glucose, 145 mM NaCl, 2 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA)]. The apical surface of NCI-N87 cell monolayers was infected with live, heat-killed, or conditioned medium from H. pylori 26695 cultures (MOI of 50:1) for 48 h. The cells were then washed and treated with gentamicin to eliminate extracellular bacteria. In order to measure the paracellular flux, the apical cell culture media were replaced with P buffer containing FITC-dextran (10 mg/ml) or FITC-BSA (10 mg/ml), whereas the basolateral reservoirs were filled with P buffer alone. P/EGTA buffer containing FITC-dextran (10 mg/ml) or FITC-BSA (10 mg/ml) was used as a positive control. After incubation for 4 h, the amounts of FITC-dextran and FITC-BSA in the basolateral media were measured with a fluorometer (excitation at 492 nm, and emission at 520 nm).
Multiplex cytokine assays.
To determine the cytokine response of NCI-N87 cell monolayers cocultured with H. pylori, cells were incubated with bacteria (strain 26695) at an MOI of 50:1. After 48 h of incubation, supernatants in the apical and basolateral compartments were collected and cytokine levels were measured. Cytokines (IL-6, IL-8, IL-10, tumor necrosis factor alpha [TNF-α], IL-12p70, IL-1β, and gamma interferon [IFN-γ]) were quantified using the Human ProInflammatory 7-plex ultra-sensitive kit from MSD (Gaithersburg, MD). MSD plates were analyzed for electrochemiluminescence with an MS2400 imager (MSD). The assay was performed according to the manufacturer's instructions. All standards and samples were measured in duplicate.
Immunostaining.
Uninfected monolayers and H. pylori strain 26695-infected monolayers were washed three times with phosphate-buffered saline (PBS) at 48 h postinfection and fixed in 4% paraformaldehyde (PFA) or in methanol for 20 min at room temperature or −20°C, respectively. Cells were then blocked with 5% normal goat serum in PBS (blocking solution) for 30 min and incubated with primary antibodies diluted in blocking solution overnight at 4°C (anti-ZO-1, 1:100, catalog number 339100; anti-claudin-1, 1:1,000, catalog number 51-9000). Cells were then washed three times with PBS and incubated with tetramethyl rhodamine isocyanate (TRITC)-conjugated or FITC-conjugated secondary antibody (1:100 in blocking solution) at room temperature for 1 h in the dark. Monolayers were washed with PBS, and nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole; 1:1,000 in PBS) solution for 2 min at room temperature. Tissue culture filters housing the epithelial cell monolayers were then carefully detached from their support and mounted on coverslips. Immunostaining was analyzed with a Nikon Eclipse TE2000-E fluorescence microscope.
Antibodies.
Rabbit polyclonal anti-claudin-1 and mouse monoclonal anti-ZO-1 were purchased from Invitrogen (Camarillo, CA). TRITC-conjugated anti-mouse and FITC-conjugated anti-rabbit secondary antibodies were obtained from Sigma.
Statistics.
Data were analyzed with GraphPad (San Diego, CA) software. One-way analysis of variance (ANOVA) multiple pairwise comparisons test and Tukey's posttest were used to determine statistical significance. A P value of <0.05 was significant. Data are expressed as means ± standard errors of the means (SEM) of triplicate samples for all conditions tested.
RESULTS
H. pylori infection induces loss of NCI-N87 cell monolayer transepithelial resistance.
NCI-N87 cells are human gastric epithelial cells that form confluent monolayers. To evaluate their potential for use in coculture studies with H. pylori, we tested their ability to form tight junction complexes and develop barrier function. Cells were allowed to grow on membrane inserts to reach confluence, and TEER was measured at intervals over 2 to 3 days. We observed an increase in TEER over time, suggesting that these cells gradually formed functional tight junctions (data not shown). Immunostaining of confluent monolayers was performed to visualize the tight junction proteins ZO-1 and claudin-1. Both proteins were observed in the typical “chicken wire pattern” at cell-cell contact points (Fig. 1A and C), confirming the ability of NCI-N87 to form a tight monolayer under our growth conditions.
Fig 1.
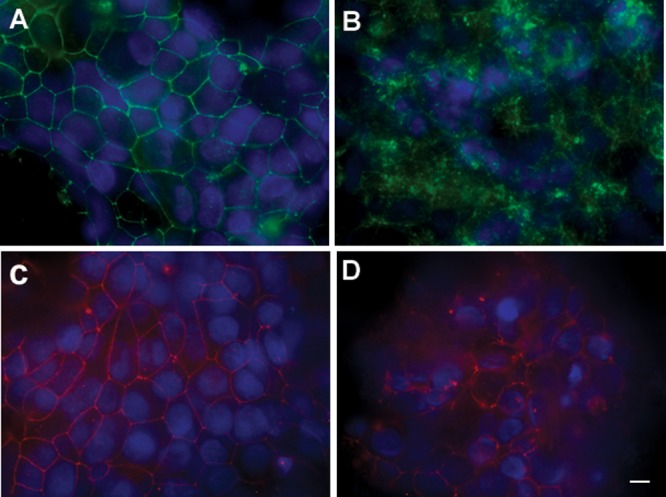
H. pylori disrupts tight junction proteins ZO-1 and claudin-1. Fluorescence microscopy of NCI-N87 monolayers labeled with antibodies specific for claudin-1 (A, B) or ZO-1 (C, D). NCI-N87 polarized monolayers were infected with H. pylori (MOI, 50:1) for 48 h, washed with PBS, fixed, and stained, along with uninfected controls. (A) Claudin-1 staining in uninfected monolayers displayed the characteristic chicken wire patterning. (B) Claudin-1 staining in H. pylori-treated monolayers. Claudin-1 appears completely removed from the cell-cell boundary and is localized in the cytoplasm, visible as punctuate staining. (C) Highly organized ZO-1 chicken wire pattern staining in uninfected monolayers. (D) Disrupted ZO-1 organization in H. pylori-infected monolayer. Bar, 10 μm.
To evaluate the potential effect of H. pylori on barrier function, NCI-N87 monolayers were grown until TEER values reached 800 to 1,400 Ω · cm2. Monolayers were infected via the apical chamber with either the wild-type H. pylori strain 26695 or its VacA− or CagPAI− isogenic mutants or the wild-type M5 or HpM5ureB at a multiplicity of infection (MOI) of 50:1. Bacterial infection caused a progressive decrease in TEER over time (Fig. 2). H. pylori strain 26695 and the VacA− and CagPAI− isogenic mutants (Fig. 2A) induced a significant TEER decrease at both 24 h (691 ± 21.7 Ω · cm2, 742 ± 17.3 Ω · cm2, and 776 ± 25.9 Ω · cm2, respectively; P < 0.0001) and 48 h (436 ± 9.94 Ω · cm2, 469 ± 14.5 Ω · cm2, and 519 ± 7.0 Ω · cm2, respectively; P < 0.0001) postinfection compared to uninfected monolayers (1,110 ± 38.1 Ω · cm2 and 1,096 ± 38.8 Ω · cm2, respectively). Similarly, when NCI-N87 monolayers were infected with H. pylori strain M5 or its isogenic mutant deprived of urease B activity (Fig. 2B), we observed a progressive and significant decrease in TEER, reaching values of 783 ± 14.2 Ω · cm2 and 846 ± 20.3 Ω · cm2, respectively, at 48 h postinfection (P < 0.0001) compared to the uninfected control (1,391 ± 26.0 Ω · cm2). Monolayers incubated with heat-killed H. pylori did not display significantly reduced TEER levels (1,008 ± 22.2 Ω · cm2 compared to uninfected controls (1,096 ± 38.8 Ω · cm2) at 48 h postinfection (Fig. 2A). No effect on TEER was observed when culture supernatant from strain 26695 was applied to NCI-N87 cell monolayers (data not shown) as well. Our data show that infection with live H. pylori and all mutant strains applied leads to a significant TEER loss at both 24 h and 48 h postinfection, showing not only that viable bacteria were required in order to induce barrier function alterations but also demonstrating that these changes in epithelial barrier integrity were independent of the H. pylori main virulence effectors, such as VacA, urease B, or the entire pathogenicity island.
Fig 2.

H. pylori-induced decreases in transepithelial electric resistance (TEER) of NCI-N87 cell monolayers. (A) Monolayers were incubated with live or heat-killed H. pylori strain 26695 or the VacA− or CagPAI− isogenic mutant at an MOI of 50:1 for 48 h. (B) Monolayers incubated with live H. pylori M5, isogenic M5:UreB−, or 26695 as a control at an MOI of 50:1 for 48 h. Data are expressed as means ± SEM of triplicate samples for all conditions tested. Statistical comparison with uninfected control at the same time point: **, P < 0.01; ***, P < 0.0001.
NCI-N87 gastric cell monolayer viability is not affected by infection with H. pylori.
In order to establish whether TEER loss was the result of alterations in the viability of the epithelial cell monolayer upon bacterial infection, we analyzed NCI-N87 cell viability by measuring the release of LDH. The LDH assay measures LDH activity present in the supernatants due to cells whose cell membrane integrity has been seriously compromised. There was no increase in LDH activity in monolayers incubated with conditioned medium, heat-killed H. pylori (not shown), or viable H. pylori strain 26695 compared to unstimulated cells (Fig. 3A). Similarly, no LDH increase was observed on monolayers infected with H. pylori 26695 isogenic mutants or with M5 or HpM5ureB. Therefore, H. pylori infection did not significantly alter cell viability, indicating that its effect on cellular permeability is likely the result of modulation of tight junction barrier function.
Fig 3.

Coculture of NCI-N87 monolayers with H. pylori does not decrease cell viability. (A) LDH release from NCI-N87 monolayers treated with viable H. pylori strain 26695 or heat-killed bacteria (MOI, 50:1) or an equivalent volume of bacterial culture supernatants for 48 h. (B) LDH release was measured in monolayers treated with H. pylori strain 26695 and its VacA− and CagPAI− isogenic mutants and strain M5 and its M5:UreB isogenic mutant (MOI, 50:1) for 48 h. Apical media was collected at 48 h postinfection for LDH release measurement. Results are shown as percentage of those for the positive control (Triton X-100-treated monolayers).
H. pylori infection increases paracellular permeability through modulation of the tight junctional complex.
Tight junctions play a critical role in preventing the leakage of compounds across the epithelial layer. They are essential for the tight sealing of the cellular sheets, thus controlling paracellular ion flux and maintaining tissue homeostasis. To confirm that the altered function of tight junctions upon infection with H. pylori is responsible for reductions in TEER, we analyzed monolayer paracellular permeability by measuring the apical-to-basolateral flux of 4-kDa FITC-dextran and 40-kDa FITC-BSA (Fig. 4A and B). In uninfected, heat-killed, or conditioned medium-treated NCI-N87 cells, we observed only a very small amount of paracellular diffusion of these tracers across the monolayer. When live bacteria were apically applied to the cells, a considerably larger amount of both dextran (P < 0.05) and BSA (P < 0.0001) diffused across the monolayer than the uninfected controls, thus confirming the involvement of the paracellular pathway during H. pylori infection of NCI-N87 cell monolayers.
Fig 4.
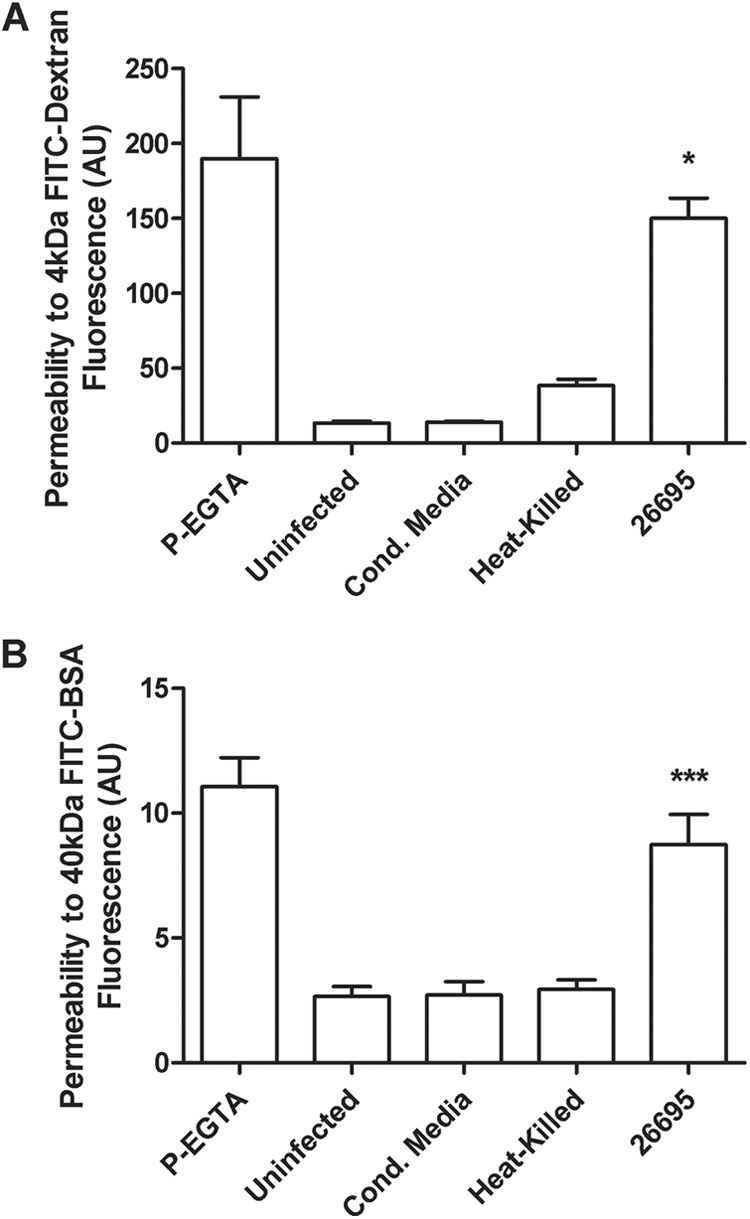
Paracellular permeability in NCI-N87 cell monolayer increases after infection with H. pylori strain 26695. H. pylori strain 26695, heat-killed bacteria (MOI, 50:1), or an equivalent volume of culture supernatant was used to inoculate NCI-N87 cells for 48 h. FITC-labeled markers were added to the apical medium reservoir. After 3 h, aliquots of the basolateral medium were removed and the amount of labeled markers that passed through the monolayer was determined. (A) FITC-dextran (4 kDa) net transport after infection with H. pylori. (B) FITC-BSA (40 kDa) net transport after infection with H. pylori. Calcium-free medium supplemented with EGTA to disrupt TJs served as a positive control. Results are expressed as means ± SEM of triplicate samples for each condition. Statistical comparison with uninfected control: *, P < 0.05; ***, P < 0.001.
The dysregulation of tight junction proteins concomitant with the severe effects of H. pylori on cellular permeability was further demonstrated by immunofluorescence of infected monolayers. As shown in Fig. 1A and C, in uninfected controls, both claudin-1 and ZO-1, respectively, display a typical chicken wire pattern of distribution and are localized at the cell boundary. Upon infection with H. pylori, the distribution of both proteins is severely disrupted. Claudin-1 association with cell membranes is completely lost, and the protein is mostly observed in the cytoplasm in a punctuate distribution pattern (Fig. 1B). Regarding ZO-1, although it appears still associated with the plasma membrane in a few sections of the cell boundary, most of its highly organized distribution is severely disrupted with large cell-cell contact areas where ZO-1 localization is completely misplaced (Fig. 1D). These data show that H. pylori strongly affects the highly organized structure of tight junctions.
H. pylori elicits the release of cytokines in NCI-N87 gastric cell monolayers.
The secretion of immune mediators by NCI-N87 gastric epithelial cells was analyzed by measuring the secretion level of six proinflammatory cytokines (IL-8, IL-6, TNF-α, IFN-γ, IL-12p70, and IL-1β) and one anti-inflammatory cytokine (IL-10) in media collected from both the apical and basolateral compartments of untreated monolayers and of viable or heat-killed H. pylori strain 26695- or culture supernatant-treated monolayers. The results are shown in Fig. 5. Cytokine production was uniformly higher in the basolateral compartment, although significant secretion was measured in the apical side as well. Even though similar profiles were observed for all seven cytokines/chemokines, the dominant cytokine was IL-8, with over 2,000 pg/ml secreted protein. H. pylori-infected cells exhibited an approximately 4-fold increase in production of IL-8 compared to uninfected cells (P < 0.0001) in the basal side. Interestingly, heat-killed bacteria induced the highest release of IL-8 and other cytokines in the two compartments. In the basolateral compartment, the level of IL-8 measured following treatment with heat-killed bacteria was about 50% higher than for live-H. pylori-infected cells. In NCI-N87 monolayer supernatants, we detected measurable levels of all cytokines included in the 7-plex assay, although they were considerably lower than that of IL-8. Nevertheless, the level of cytokines detected in H. pylori-infected samples was significantly higher than the control for all cytokines tested and followed the same pattern of IL-8, with heat-killed bacteria eliciting the highest response in both compartments. Immune responses triggered by heat-killed bacteria were statistically significantly higher than those triggered by wild-type H. pylori for IL-8 (P < 0.01) and IL-6 (P < 0.05) in the apical side and for IL-8, IL-6, and TNF-α (P < 0.0001) and IL-10 and IL-1β (P < 0.05) in the basolateral compartment. Our data show that NCI-N87 epithelial cells secrete immune mediators when infected with H. pylori and that heat-inactivated bacteria are capable of eliciting the strongest release of inflammatory molecules.
Fig 5.

H. pylori triggers cytokine secretion from NCI-N87 monolayers into basolateral culture supernatants. H. pylori strain 26695, heat-killed bacteria (MOI, 50:1), or an equivalent volume of culture supernatant was used to inoculate NCI-N87 cells. Media were collected to quantify the amounts of IL-8, IL-12p70, TNF, IFN, IL-6, IL-10, and IL-1 in the apical and basolateral compartments. Results are representative of means ± SEM of triplicate samples for each condition. Statistical comparison with uninfected control: ***, P < 0. 0001; **, P < 0.001; *, P < 0.05.
DISCUSSION
In the present study, we demonstrated polarized secretion of IL-8 and other proinflammatory cytokines in response to H. pylori infection using a gastric epithelial cell monolayer model that forms a functional barrier with high TEER values. We demonstrated disruption of tight-junction (TJ) integrity following coculture with live H. pylori. This was evidenced by significant increases in monolayer permeability within 24 h by a drop in TEER values and increased diffusion of a 40-kDa FITC-BSA conjugate. The increased permeability was associated with severe disruption in the distribution of both claudin-1 and ZO-1 proteins and it was not the result of epithelial cell death.
Several epithelial cell culture systems have been used to demonstrate the ability of H. pylori to increase monolayer permeability and to describe the molecular mechanisms involved. In vitro studies to assess the relationship of H. pylori to gastric epithelial cells have relied predominantly on the AGS human gastric cancer epithelial cell line (24–27). Although injected CagA has been demonstrated to associate with the tight junction scaffolding protein ZO-1 in these cells, AGS cells do not form tight junctions or form a functional barrier (28). Nonhuman cell culture systems employing MDCK cells, or a nontumorigenic duodenal epithelial cell line (SCBN), have been used as alternatives since they do form tight junctions and establish TEER sufficient to form functional barriers (28, 29).
More recently, Wroblewski et al. utilized the MKN28 human gastric epithelial cell line to establish a useful polarized monolayer system (30). They showed that coculture with H. pylori increased permeability through a bacterial urease-dependent mechanism that dysregulated TJs and increased phosphorylation of myosin light chain (MLC) through the activation of MLC kinase. The cellular inflammatory response was not evaluated, however, and although TEER values were prolonged and consistent, they averaged approximately 100 Ω · cm2. Our studies demonstrate that the NCI-N87 cell line routinely generated TEER values of 800 to 1,400 Ω · cm2 above background, although, in general, these cells grew slowly. These monolayers could be used to analyze the cytokine profile and the polarity of the response upon coculture with H. pylori, and therefore, these monolayers serve as a useful model for the study of the host inflammatory responses to H. pylori.
The measurement of IL-8 and other cytokines in the supernatants of AGS cells cocultured with H. pylori has been well demonstrated and has been important in assessing the cellular response to virulence factors such as the type IV secretion system (T4SS) (9–13). An evaluation of the host immune response in polarized human gastric epithelial cells, however, has not been previously performed, largely due to the lack of suitable cells with the ability to establish TJ integrity in monolayers. Jung et al. employed the NCI-N87 cell line to form monolayers and evaluate H. pylori-associated reductions in TEER values in a limited study using H. pylori strain 60190 (31). IL-8 production was significantly increased, but the relative amounts between apical and basolateral supernatants were difficult to discern, and the amount produced was less than a third of the IL-8 produced in response to IL-1α. As described above, gastric MKN28 cells have been used to describe the ability of H. pylori to increase membrane permeability via myosin II activation and cytoskeletal rearrangement through the activity of bacterial urease (30). More recently, Lapointe et al. employed the HGE-20 human gastric epithelial cell line to describe the role of Rho kinase activation and IL-1R1 phosphorylation in disrupting TJs as measured by the distribution of claudin-4 within the cells (32). The HGE-20 cell line is a derivative of the NCI-N87 cells employed in the present study (33). Although HGE-20 cells have been shown previously to be capable of forming polarized monolayers, no analysis was performed to demonstrate polarization and the establishment of transmembrane electrical resistance. Additionally, while conditioned media from H. pylori HGE-20 cocultures were used as a potential source of IL-1α, IL-1β, and IL-18, these cytokines could not be detected.
The present study demonstrates that H. pylori-induced IL-8 production is consistent with studies in AGS cells but that apical and basolateral production can be differentiated. IL-8 is the most prevalent cytokine, and there was an almost 4-fold-higher basal secretion than apical secretion. This phenomenon is consistent with the role of IL-8 in recruiting white blood cells to the lamina propria of the mucosa. Interestingly, whereas heat-killed H. pylori failed to increase monolayer permeability, it induced twice as much IL-8 production and as significantly high levels of IL-6, TNF-α, and Il-1β as did live H. pylori in basal supernatant.
The ability of heat-killed H. pylori to induce the highest levels of cytokine production in basolateral supernatants in the absence of TEER reduction demonstrates that cytokine production is independent of TJ dysregulation. Additionally, the reduced amounts of cytokine that resulted when live H. pylori that significantly reduced TEER values was used indicate that whatever dysregulation H. pylori induces in TJ proteins, such events do not contribute to cytokine production beyond what is induced simply through the contact of H. pylori components with the epithelial cell membrane.
The mechanisms by which heat-killed H. pylori induces higher levels of cytokine than the live organism are unknown. These observations are consistent with previous studies demonstrating that certain T4SS proteins having contact with epithelial cell membranes are primarily responsible for inducing IL-8 production (14–17). H. pylori strain 26695 is a potent inducer of IL-8, and the T4SS proteins responsible for this biological effect may be present in a functional state in the heat-killed H. pylori preparations. It is also possible that Toll-like receptor (TLR) ligands such as H. pylori peptidoglycans may be released or more accessible as a result of heat killing. TLR2 and NOD1 have been shown to be primarily responsible for the IL-8 production that results from H. pylori infection (27, 34).
Relatively small amounts of IL-8 and other cytokines were measured in the apical media. Whether this represents actual apical secretion or “leakage” due to H. pylori-induced increases in monolayer permeability remains to be determined; however, the presence of apical IL-8 when using heat-killed H. pylori that fails to compromise TEER values suggests the former. Since TLR expression is generally limited on the apical surface of gastrointestinal epithelial cells, the predominant expression at the basolateral surface would be expected. There is also limited utility to the host in secreting IL-8 into the lumen unless such a function contributes to crypt abscess formation, a phenomenon that is not unusual with H. pylori infection of the gastric mucosa. Significant reductions in TEER values were observed at 24 h. Since our cytokine analysis was performed on supernatants collected at 48 h, it is possible that the presence of these cytokines at the apical surface is due to increased monolayer permeability. Indeed, the diffusion of FITC-labeled dextran and BSA indicates that cytokines should be leaking readily into the apical supernatant. The dominant presence of IL-8 and the other cytokines in the basolateral supernatant reinforces the possibility that the basolateral surface is the primary site of cytokine release.
Moreover, we have demonstrated that H. pylori infection of NCI-N87 gastric cell monolayers affects paracellular permeability most likely via dysregulation of TJs and independent of the individual effectors VacA and urease B and other effectors, such as CagA and the T4SS encoded by the Cag pathogenicity island. We could not establish if these factors operate in concert to affect barrier function because of a lack of a CagPAI/VacA/UreB deletion mutant. Also, if this dysregulation is directly induced by still-unidentified bacterial pathogenic compounds, it is not known at present. We have proven that H. pylori induces polarized IL-8 secretion from epithelial cells that is independent from the microorganism viability. Conversely, we have demonstrated that H. pylori causes TJ disassembly independently of virulence factors, such as VacA, urease B, and CagA, previously described by other investigators to be involved in this mechanism. Taken together, these data indeed suggest that these responses are rather host driven than microorganism induced.
Our hypothesis is further supported by our recent data showing that unrelated pathogens such as Shigella (35) and Salmonella M. Fiorentino, K. M. Lammers, M. M. Levine, S. B. Sztein, and A. Fasano, submitted for publication induce similar responses in epithelia from different regional districts of the gastrointestinal tract (small intestine and colon). It is well established that an increased permeability in the presence of pathogenic microorganisms is an attempt of the host to “fight the enemy,” i.e., to dilute harmful toxins secreted by pathogens or flush them away (i.e., cholera toxin [36] and/or Shigella [35] in the intestine). Similarly, increased paracellular permeability might also lead to neutrophil efflux into the lumen and/or to the host luminal secretion of antimicrobial compounds, actions that epithelial cells, as the first line of defense against harmful microorganisms, would take, aiming at preventing pathogens in the lumen from gaining access to the lamina propria and eventually causing a systemic disease. It is plausible to assume that pathogens are apically sensed by specific receptors that, through basolateral IL-8 secretion, attract immune cells in the lamina propria, getting them ready to face a possible passage of the pathogen through the epithelial barrier. At the same time, the increased gastrointestinal epithelial permeability and polarized apical IL-8 secretion could lead to neutrophil transmigration from the lamina propria to the gastrointestinal lumen, a phenomenon already described for other enteric pathogens (37, 38).
The establishment of this cell model will be useful for additional studies designed to evaluate the mechanisms of induction of cytokines, chemokines, and other immune mediators (e.g., IL-8), the breadth of the inflammatory response, the impact of H. pylori bacteria and bacterial factors when exposed to the apical and basolateral membranes, and the relationship of H. pylori-activated epithelial cells to granulocytes, monocytes, and lymphocytes when added to the basal chamber. H. pylori remains an important and widespread bacterial pathogen. The use of NCI-N87 cells to form polarized monolayers with high TEER values will provide a means of studying the potential contribution of increased monolayer permeability to H. pylori-induced inflammatory events and increase our understanding of bacterial pathogenesis.
ACKNOWLEDGMENTS
These studies were funded by NIAID (NIH, DHHS) grant U19 AI082655 (Cooperative Center for Translational Research in Human Immunology and Biodefense; CCHI) to M.B.S. and A.F. and by NIDDK (NIH, DHHS) grant RO1 DK 45461 to S.J.C.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases, the National Institute of Diabetes and Digestive and Kidney Diseases, or the National Institutes of Health.
We declare no conflicting financial interests.
Footnotes
Published ahead of print 7 January 2013
REFERENCES
- 1. Turner JR. 2006. Molecular basis of epithelial barrier regulation: from basic mechanisms to clinical application. Am. J. Pathol. 169: 1901–1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marshall BJ, Warren JR. 1984. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet i: 1311–1315 [DOI] [PubMed] [Google Scholar]
- 3. Everhart JE. 2000. Recent developments in the epidemiology of Helicobacter pylori. Gastroenterol. Clin. North Am. 29: 559–578 [DOI] [PubMed] [Google Scholar]
- 4. NIH Consensus Conference 1994. Helicobacter pylori in peptic ulcer disease. JAMA 272: 65–69 [PubMed] [Google Scholar]
- 5. Parsonnet J, Hansen S, Rodriguez L, Gelb AB, Warnke RA, Jellum E, Orentreich N, Vogelman JH, Friedman GD. 1994. Helicobacter pylori infection and gastric lymphoma. N. Engl. J. Med. 330: 1267–1271 [DOI] [PubMed] [Google Scholar]
- 6. Peek RM, Jr, Blaser MJ. 2002. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2: 28–37 [DOI] [PubMed] [Google Scholar]
- 7. World Health Organization 1994. Infection with Helicobacter pylori, p 177–241 In Schistosomes, liver flukes and Helicobacter pylori, vol 61 International Agency for Research on Cancer, Lyon, France [Google Scholar]
- 8. Correa P. 1992. Human gastric carcinogenesis: a multistep and multifactorial process—First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 52: 6735–6740 [PubMed] [Google Scholar]
- 9. Asahi M, Azuma T, Ito S, Ito Y, Suto H, Nagai Y, Tsubokawa M, Tohyama Y, Maeda S, Omata M, Suzuki T, Sasakawa C. 2000. Helicobacter pylori CagA protein can be tyrosine phosphorylated in gastric epithelial cells. J. Exp. Med. 191: 593–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Backert S, Ziska E, Brinkmann V, Zimny-Arndt U, Fauconnier A, Jungblut PR, Naumann M, Meyer TF. 2000. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell. Microbiol. 2: 155–164 [DOI] [PubMed] [Google Scholar]
- 11. Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R. 2000. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287: 1497–1500 [DOI] [PubMed] [Google Scholar]
- 12. Segal ED, Cha J, Lo J, Falkow S, Tompkins LS. 1999. Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc. Natl. Acad. Sci. U. S. A. 96: 14559–14564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stein M, Rappuoli R, Covacci A. 2000. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc. Natl. Acad. Sci. U. S. A. 97: 1263–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fischer W, Puls J, Buhrdorf R, Gebert B, Odenbreit S, Haas R. 2001. Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol. Microbiol. 42: 1337–1348 [DOI] [PubMed] [Google Scholar]
- 15. Foryst-Ludwig A, Naumann M. 2000. p21-activated kinase 1 activates the nuclear factor kappa B (NF-kappa B)-inducing kinase-Ikappa B kinases NF-kappa B pathway and proinflammatory cytokines in Helicobacter pylori infection. J. Biol. Chem. 275: 39779–39785 [DOI] [PubMed] [Google Scholar]
- 16. Rieder G, Einsiedl W, Hatz RA, Stolte M, Enders GA, Walz A. 2001. Comparison of CXC chemokines ENA-78 and interleukin-8 expression in Helicobacter pylori-associated gastritis. Infect. Immun. 69: 81–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rieder G, Hatz RA, Moran AP, Walz A, Stolte M, Enders G. 1997. Role of adherence in interleukin-8 induction in Helicobacter pylori-associated gastritis. Infect. Immun. 65: 3622–3630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jimenez-Soto LF, Kutter S, Sewald X, Ertl C, Weiss E, Kapp U, Rohde M, Pirch T, Jung K, Retta SF, Terradot L, Fischer W, Haas R. 2009. Helicobacter pylori type IV secretion apparatus exploits beta1 integrin in a novel RGD-independent manner. PLoS Pathog. 5: e1000684 doi:10.1371/journal.ppat.1000684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, Misselwitz R, Berger J, Sewald N, Konig W, Backert S. 2007. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 449: 862–866 [DOI] [PubMed] [Google Scholar]
- 20. Saha A, Backert S, Hammond CE, Gooz M, Smolka AJ. 2010. Helicobacter pylori CagL activates ADAM17 to induce repression of the gastric H, K-ATPase alpha subunit. Gastroenterology 139: 239–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tegtmeyer N, Hartig R, Delahay RM, Rohde M, Brandt S, Conradi J, Takahashi S, Smolka AJ, Sewald N, Backert S. 2010. A small fibronectin-mimicking protein from bacteria induces cell spreading and focal adhesion formation. J. Biol. Chem. 285: 23515–23526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Basque JR, Chenard M, Chailler P, Menard D. 2001. Gastric cancer cell lines as models to study human digestive functions. J. Cell. Biochem. 81: 241–251 [PubMed] [Google Scholar]
- 23. McGovern KJ, Blanchard TG, Gutierrez JA, Czinn SJ, Krakowka S, Youngman P. 2001. γ-Glutamyltransferase is a Helicobacter pylori virulence factor but is not essential for colonization. Infect. Immun. 69: 4168–4173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chang YT, Wu MS, Chang YJ, Chen CC, Lin YS, Hsieh T, Yang PC, Lin JT. 2006. Distinct gene expression profiles in gastric epithelial cells induced by different clinical isolates of Helicobacter pylori—implication of bacteria and host interaction in gastric carcinogenesis. Hepatogastroenterology 53: 484–490 [PubMed] [Google Scholar]
- 25. Moese S, Selbach M, Brinkmann V, Karlas A, Haimovich B, Backert S, Meyer TF. 2007. The Helicobacter pylori CagA protein disrupts matrix adhesion of gastric epithelial cells by dephosphorylation of vinculin. Cell. Microbiol. 9: 1148–1161 [DOI] [PubMed] [Google Scholar]
- 26. Oliveira MJ, Costa AC, Costa AM, Henriques L, Suriano G, Atherton JC, Machado JC, Carneiro F, Seruca R, Mareel M, Leroy A, Figueiredo C. 2006. Helicobacter pylori induces gastric epithelial cell invasion in a c-Met and type IV secretion system-dependent manner. J. Biol. Chem. 281: 34888–34896 [DOI] [PubMed] [Google Scholar]
- 27. Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, Athman R, Memet S, Huerre MR, Coyle AJ, DiStefano PS, Sansonetti PJ, Labigne A, Bertin J, Philpott DJ, Ferrero RL. 2004. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 5: 1166–1174 [DOI] [PubMed] [Google Scholar]
- 28. Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. 2003. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 300: 1430–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fedwick JP, Lapointe TK, Meddings JB, Sherman PM, Buret AG. 2005. Helicobacter pylori activates myosin light-chain kinase to disrupt claudin-4 and claudin-5 and increase epithelial permeability. Infect. Immun. 73: 7844–7852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wroblewski LE, Shen L, Ogden S, Romero-Gallo J, Lapierre LA, Israel DA, Turner JR, Peek RM., Jr 2009. Helicobacter pylori dysregulation of gastric epithelial tight junctions by urease-mediated myosin II activation. Gastroenterology 136: 236–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jung YJ, Lee KL, Kim BK, Kim JW, Jeong JB, Kim SG, Kim JS, Jung HC, Song IS. 2006. Usefulness of NCI-N87 cell lines in Helicobacter pylori infected gastric mucosa model. Korean J. Gastroenterol. 47: 357–362. (In Korean.) [PubMed] [Google Scholar]
- 32. Lapointe TK, O'Connor PM, Jones NL, Menard D, Buret AG. 2010. Interleukin-1 receptor phosphorylation activates Rho kinase to disrupt human gastric tight junctional claudin-4 during Helicobacter pylori infection. Cell. Microbiol. 12: 692–703 [DOI] [PubMed] [Google Scholar]
- 33. Chailler P, Menard D. 2005. Establishment of human gastric epithelial (HGE) cell lines exhibiting barrier function, progenitor, and prezymogenic characteristics. J. Cell. Physiol. 202: 263–274 [DOI] [PubMed] [Google Scholar]
- 34. Rad R, Ballhorn W, Voland P, Eisenacher K, Mages J, Rad L, Ferstl R, Lang R, Wagner H, Schmid RM, Bauer S, Prinz C, Kirschning CJ, Krug A. 2009. Extracellular and intracellular pattern recognition receptors cooperate in the recognition of Helicobacter pylori. Gastroenterology 136: 2247–2257 [DOI] [PubMed] [Google Scholar]
- 35. Faherty C, Harper JM, Shea-Donohue T, Barry EM, Kaper JB, Fasano A, Nataro JP. 2012. Chromosomal and plasmid-encoded factors of shigella flexneri induce secretogenic activity ex vivo. PLoS One 7: e49980 doi:10.1371/journal.pone.0049980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. El Asmar R, Panigrahi P, Bamford P, Berti I, Not T, Coppa GV, Catassi C, Fasano A. 2002. Host-dependent zonulin secretion causes the impairment of the small intestine barrier function after bacterial exposure. Gastroenterology 123: 1607–1615 [DOI] [PubMed] [Google Scholar]
- 37. Boll EJ, Struve C, Sander A, Demma Z, Krogfelt KA, McCormick BA. 2012. Enteroaggregative Escherichia coli promotes transepithelial migration of neutrophils through a conserved 12-lipoxygenase pathway. Cell. Microbiol. 14: 120–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mumy KL, Bien JD, Pazos MA, Gronert K, Hurley BP, McCormick BA. 2008. Distinct isoforms of phospholipase A2 mediate the ability of Salmonella enterica serotype Typhimurium and Shigella flexneri to induce the transepithelial migration of neutrophils. Infect. Immun. 76: 3614–3627 [DOI] [PMC free article] [PubMed] [Google Scholar]