

Abstract
MicroRNAs (miRNAs) are small noncoding molecules that control gene expression posttranscriptionally, with microRNA-155 (miR-155) one of the first to be implicated in immune regulation. Here, we show that miR-155-deficient mice are less able to eradicate a mucosal Citrobacter rodentium infection than wild-type C57BL/6 mice. miR-155-deficient mice exhibited prolonged colonization associated with a higher C. rodentium burden in gastrointestinal tissue and spread into systemic tissues. Germinal center formation and humoral immune responses against C. rodentium were severely impaired in infected miR-155-deficient mice. A similarly susceptible phenotype was observed in μMT mice reconstituted with miR-155-deficient B cells, indicating that miR-155 is required intrinsically for mediating protection against this predominantly luminal bacterial pathogen.
INTRODUCTION
MicroRNAs (miRNAs) are small RNA molecules of about 22 nucleotides in length found within all eukaryotic cells that control gene expression posttranscriptionally (1), a process mediated by targeted degradation of mRNA. MicroRNA-155 (miR-155) has received a great deal of interest because it has been implicated in the development of a number of different immune functions and cancer. The loci encoding miR-155, c-bic, was originally identified during a study investigating integration sites of the avian leukosis virus during B cell lymphoma generation (2). miR-155 was subsequently found to be highly expressed in a variety of human B cell lymphomas, such as Hodgkin, primary mediastinal, and diffuse large B cell lymphomas. Furthermore, transgenic mice engineered to overexpress miR-155 in B cells develop high-grade lymphoma.
miR-155 transcripts are expressed in both T and B lymphocytes as well as in monocytes/macrophages after activation with both bacterial or viral stimuli (3, 4). Additionally, miR-155-deficient mice exhibit impairments in a number of immune response pathways, such as antigen presentation, T and B cell immunity, germinal center formation, and the generation of class-switched plasma cells (5–7). Relatively little is known about the role of miR-155 in protection against infection or in mucosa-associated immunity (8).
Citrobacter rodentium is a predominantly mucosal enteric murine pathogen that exhibits pathogenic traits in common with enteropathogenic (EPEC) and enterohemorrhagic (EHEC) Escherichia coli. As with EPEC and ETEC, C. rodentium colonization of the gastrointestinal epithelium is influenced by the formation of attaching and effacing (A/E) lesions, which are characterized by localized destruction of the brush border microvilli, formation of pedestal-like structures on the apical cell surface, and intimate bacterial adhesion to the host cell plasma membrane. A/E lesion formation by C. rodentium can trigger substantial epithelial cell proliferation, crypt dilation, and thickening of the colonic mucosa referred to as colonic crypt hyperplasia (9, 10). Such infections are associated with robust humoral, Th1, and Th17 immune responses (11–16). Cells of the adaptive immune system, such as CD4+ T cells and B cells, make important contributions to protection against C. rodentium infection, and mice depleted of either cell type have an impaired ability to clear the infection (17–20). Thus, C. rodentium is a useful agent for probing the host mucosal immune response.
The aim of this study was to explore the role of miR-155 in controlling a mucosal C. rodentium infection in the context of the overall immune response. Here, we demonstrate that miR-155-deficient mice are highly susceptible to a primary C. rodentium infection and that this is associated with defects in B cell function.
MATERIALS AND METHODS
Mice.
Female and male 6- to 8-week-old C57BL/6 mice from Charles River, United Kingdom, were used in all animal experiments. miR-155-deficient mice were obtained from the Wellcome Trust Sanger Institute breeding colonies. At no point were the mice cohoused. All animals were given food and water ad libitum. Mice were sacrificed by cervical dislocation. Animal husbandry and experimental procedures were conducted according to the United Kingdom Animals (Scientific Procedures) Act of 1986. All procedures involving live mice were approved by the United Kingdom Home Office under the United Kingdom Animal (Scientific) Procedures Act and institutional ethical committees.
Bacterial and oral infection of mice.
C. rodentium ICC180 was used in this study (21). Bacterial inoculums were prepared by culturing bacteria overnight at 37°C in 100 ml of Luria Bertani (LB) broth supplemented with nalidixic acid (100 μg/ml), with shaking (220 rpm). Cultures were harvested by centrifugation and resuspended in a 1:10 volume of Dulbecco's phosphate-buffered saline (D-PBS). Mice were orally inoculated under anesthesia by using a gavage needle with 200 μl of the bacterial suspension. The viable count of the inocula was determined by retrospective plating on LB agar supplemented with nalidixic acid (100 μg/ml).
454 pyrosequencing analysis of intestinal microbiota 16S rRNA genes.
DNA was extracted from frozen fecal pellets using the FastDNA SPIN kit for soil (MP Biomedicals, United Kingdom). 16S rRNA gene PCR amplicons were generated for Lib-L 454 Titanium sequencing using barcoded primers targeting the V3 to V5 regions of the 16S rRNA gene. PCR products were generated using AccuPrime Taq DNA polymerase high fidelity (Invitrogen). PCR cycling conditions were as follows: 94°C for 2 min followed by 20 cycles of 94°C for 30 s, 53°C for 30 s, and 68°C for 2 min. Barcoded PCR products were then quantified individually using a Qubit 2.0 fluorometer (Invitrogen) and combined into an equimolar mastermix prior to sequencing.
After sequencing, raw sequences were processed using the mothur software package Schloss SOP (22) to remove poor-quality reads, cluster sequences into operational taxonomic units (OTUs) at 97% similarity, and assign taxonomic classifications to each OTU based on the RDP database (23). After processing and subsequent manual removal of suspect OTUs, 61,252 sequences remained, which were split into 457 OTUs overall. The median number of sequences per sample was 2,042 (range, 664 to 3,753). Overall bacterial community structures were compared between each sample by calculating cluster dendrograms (with the Bray-Curtis calculator) in mothur (22) and visualized using the iTOL Web package (24). The Metastats program (25), as implemented in mothur (22), was used to determine whether or not the OTU corresponding to C. rodentium was significantly differentially abundant between the wild-type and miR-155-deficient mice at day 14 postinfection.
Measurement of C. rodentium burden.
At regular time points postinfection, fecal samples from individual mice were collected in separate sterile Eppendorf tubes. Fecal samples were weighed, and for every 0.01 g of feces, 100 μl of sterile PBS was added (example, 0.02 g feces, add 200 μl PBS). Fecal samples were homogenized on a vortex and serially diluted. The number of viable bacteria was determined by viable count on LB agar containing nalidixic acid (100 μg/ml).
At selected time points postinfection, mice were killed by cervical dislocation and surface sterilized with 70% ethanol. Colons, ceca, livers, spleens, and mesenteric lymph nodes (mLNs) were removed aseptically. The terminal 6 cm of colon was removed, and the colon was weighed after removal of fecal pellets. Ceca were flushed with PBS before further processing, and the flowthrough was collected in a 15-ml falcon tube to determine the number of bacteria not intimately attached to the epithelium. Colons, ceca, livers, spleens, and mLNs were homogenized in 5 ml of sterile double-distilled water using a Seward Stomacher 80 (Seward, London, United Kingdom) for 2 min at high speed. The number of viable bacteria in organ homogenates was determined by serial dilution and viable count on LB agar supplemented with 100 μg/ml nalidixic acid.
Histology.
The terminal 0.5 cm of colon was removed aseptically, placed in 5% formaldehyde, and incubated overnight at room temperature for subsequent histological analysis. Tissues were processed using a Shandon excelsior tissue processor (Thermo Fisher Scientific) and then embedded in paraffin wax. Five-micrometer sections were cut using a Leica RM2125 microtome and transferred to Superfrost Plus slides (VWR International). Sections were deparaffinized by incubating sections twice in Histoclear II solution for 10 min. Sections were then rehydrated by incubation in 100%, 90%, and 70% ethanol for 5 min, followed by two 5-min washes in PBS. Sections were stained first in Mayer's hematoxylin and second in eosin. Sections were then dehydrated and mounted before assessment.
Analysis of humoral immune responses.
On days 7, 14, and 21 postchallenge (p.c.), 0.2 ml of blood was collected from the tail vein. The serum was collected and stored at −20°C until analyzed. For analysis of C. rodentium-specific antibody responses, wells of microtiter plates (Maxisorp, Nunc, Denmark) were coated overnight at 4°C with 50 μl carbonate buffer (pH 9.6) containing recombinant EspA (2 μg/ml). After incubation, antigen/carbonate buffer solution was removed by plate inversion, and plates were washed once with PBS containing 0.01% Tween 20 (wash buffer). Plates were blocked by the addition of 100 μl of 3% bovine serum albumin (BSA) in PBS (blocking solution) at room temperature for 1 h. Plates were then washed once with wash buffer before sera from individual mice were added as follows: a 1:100 dilution of serum was added to the top well and then serially diluted 5-fold down the plate. Each plate contained control wells with naive serum and PBS alone. The plates were incubated for 1 h at 37°C. For the determination of total immunoglobulin (Ig), IgG, IgG1, IgG2a, IgM, or IgA antibody titers, plates were washed with wash buffer, before the addition of 100 μl of Ig-, IgG-, IgG1-, IgG2a-, IgM-, or IgA-specific antibody conjugated to horseradish peroxidase (HRP) (Becton, Dickinson) diluted 1:1,000 in antibody buffer. Plates were then incubated for 1 h at 37°C. Finally, after being washed, bound antibody was detected by the addition of 50 μl of o-phenylenediamine substrate (Sigma) and incubation for 15 min at room temperature. The reaction was stopped by adding 25 μl 3 M sulfuric acid. Absorbances were read at 490 nm, and titers were determined arbitrarily as the reciprocal serum dilution corresponding to an optical density of 0.3.
Recombinant proteins.
EspA was cloned from EPEC strain E2348/69, expressed as a His-tagged fusion protein in E. coli, and purified by nickel affinity chromatography as previously described (26).
Immunohistology.
mLNs were removed aseptically, placed into separate Corning external thread cryogenic vials tubes (Corning), and immediately snap-frozen in liquid nitrogen. Five-micrometer sections were cut using a Shandon cryostat. Sections were transferred to slides, allowed to dry for 1 h, and subsequently fixed in 100% acetone for 5 min. Following three 5-min washes in PBS, sections were blocked with PBS supplemented with 10% normal goat serum, 5% fish gelatin, 0.01% sodium azide, 0.1% BSA, and 0.01% Tween 20 (IHC blocking solution) for 45 min at room temperature. Serum was tapped off, and relevant antibodies were added. CD45R/B220 fluorescein isothiocyanate (FITC) (BD)- and PNA rhodamine (Vector Laboratories)-conjugated antibodies were diluted 1:100 with IHC blocking solution. For control sections, antibody was omitted. Slides were incubated for 1 h at room temperature in darkness. After incubation, sections were washed twice in PBS at room temperature for 15 min, in total darkness. Finally, slides were mounted with ProLong Gold (Invitrogen) and left to air dry in the dark for 4 to 5 h before visualization using a Zeiss LSM510 confocal microscope.
Electron microscopy.
Colons/ceca were cut into 5-mm open segments and fixed in 2.5% glutaraldehyde and 2% paraformaldehyde in 0.01 M phosphate buffer (pH 7.5) at room temperature for 15 min and at 4°C for the remainder of 1 h. Samples were then rinsed thoroughly in 0.1 M sodium cacodylate buffer (pH 7.4) 3 times and fixed again in 1% buffered osmium tetroxide for 1 h at room temperature. At this point, samples were separated for either transmission electron microscopy (TEM) or scanning electron microscopy (SEM) analysis. For SEM, the samples were further impregnated with 1% aqueous thiocarbohydrazide and osmium tetroxide separated by sodium cacodylate washes by following the protocol for OTOTO (modified thiocarbohydrazide procedure for scanning electron microscopy: routine use for normal, pathological, or experimental tissues) (27). Tissues were then dehydrated in an ethanol series and critical point dried in a Bal-Tec CPD030 before being mounted onto aluminum stubs with conducting araldite. Finally, they were sputter coated with a 2-nm gold layer in a Bal-Tec SCD050 and examined on a Hitachi S-4800 SEM. For TEM, the samples were mordanted in 1% tannic acid for an hour, rinsed in 1% sodium sulfate, dehydrated through an ethanol series (staining en bloc with saturated uranyl actetate at the 30% stage), and embedded in TAAB 812 resin. Semi-thin sections, cut on a Leica EM UC6 ultramicrotome, were collected onto glass slides and stained with 1% toluidine blue for analysis by light microscopy (Zeiss Axiovert; 200 M) prior to TEM examination, and selected regions of interest were then ultrathin sectioned and contrasted with uranyl acetate and lead citrate. Images were recorded on an FEI 120kV Spirit Biotwin with an F415 Tietz charge-coupled-device (CCD) camera.
RNA isolation.
Cecal patches and colons were removed aseptically, and a 5-mm2 piece of each was placed immediately into 5 ml of RNAlater solution (Ambion). Samples were stored at 4°C overnight and then either processed immediately or placed at −70°C for long-term storage. Surfaces and equipment were thoroughly cleaned with 70% ethanol followed by RNaseZap and DNaseZap (Ambion) before use. Pieces of tissue were individually removed from RNAlater solution and placed into a 50-ml falcon tube containing double the volume of Qiazol lysis reagent recommended in the Qiagen miRNeasy minikit (Qiagen) handbook. Cecal patch and colon tissues were homogenized using a homogenizer. Total RNA was then isolated using the Qiagen miRNeasy minikit (Qiagen) as per the manufacturer's instructions. RNA was stored at −70°C until required. All RNA samples were quantified and quality checked using a Thermo Scientific NanoDrop 1000 spectrophotometer (Thermo Scientific) and Agilent 2100 bioanalyzer (Agilent Technologies), respectively, as per the manufacturer's instructions.
Microarray expression profiling.
Gene expression profiling was performed on the Illumina whole-genome gene expression direct hybridization assay system. Genes with an adjusted P value of <0.05 and a fold difference of ≥1.5 were selected for further analysis. InnateDB is a comprehensive database and analysis platform that enables testing for overrepresentation of differentially expressed genes in greater than 2,500 known innate immune response pathways, sourced from several of the publicly available pathway databases (28). Additionally, it provides a useful tool for conducting comprehensive network analysis for the identification of signaling cascades and functionally relevant subnetworks involving molecular interactions between differentially expressed genes and their nondifferentially expressed interacting partners. Using network analysis, it is possible to identify key regulators of gene expression, which exert their effects through protein modification or other nontranscriptional mechanisms.
Real-time PCR.
Total RNA was reverse transcribed using Qiagen Quantitect or miScript reverse transcription kits. Quantitative real-time PCR was performed with Quantitect primer assays (Mmp3, QT00107751 product code); Bcl6, QT01057196; Cd86, QT01055250; Mmp10, Qt00115521; Cd19, Qt00108801; and miR-155, MS00001701) and Quantitect SYBR green PCR kit (Qiagen), using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) for normalization across samples. For the miR-155 expression, 2 samples were taken for each time point (0, 3, 9, 16, and 27 days postinfection), and expression values for miR-155 were normalized against the housekeeping gene GADPH [ΔCT = CT(miR-155) − CT(GADPH)]. The fold change between the normalized expression values for miR-155 at each time point and the value in the uninfected mouse (day 0) were then calculated [ΔΔCT = ΔCT(time point x) − ΔCT(time point 0)], and the average from the two samples was plotted. For the difference in gene expression between wild-type and mutant mice, we calculated fold change in expression as ΔΔCT = average ΔCT mutant − average ΔCT wild type. Plotted on the graph is log102−fold change, which is equivalent to −fold change/log210.
Generation of chimeric mice.
miR-155-deficient mice were previously described (5). For the generation of mixed chimeras, μMT mice were irradiated (5.0 Gy) and reconstituted with a mixture of 80% bone marrow cells of μMT origin and 20% wild-type or miR-155-deficient cells. Blood samples from tail bleeds were analyzed by fluorescence-activated cell sorting (FACS) to confirm reconstitution of cells.
Statistical analysis.
Statistical analysis was performed with a two-tailed Student t test with GraphPad Prism 5.
Microarray data accession number:
The microarray data identified in this study have been deposited in the ArrayExpress database under accession number E-MTAB-1301.
RESULTS
miR-155-deficient mice are more susceptible to oral C. rodentium infection and develop more severe colitis.
Prior to investigating the ability of miR-155-deficient mice to control C. rodentium infection, we measured expression levels of miR-155 RNA in colonic tissues of wild-type C.57BL/6 mice infected with this pathogen (see Fig. S1a in the supplemental material). miR-155 RNA was detected in colonic tissues prior to the infection peak, but the relative abundance increased early in infection and then steadily decreased from this early peak as the infection was cleared (see Fig. S1a). Next, miR-155-deficient or control C57BL/6 mice were orally challenged with 109 C. rodentium, and the levels of morbidity and mortality were monitored. Although there were no obvious disease-related mortalities among either group, miR-155-deficient mice took significantly longer (∼20 days) to resolve infection (P value of <0.0001; Fig. 1a and b). To ascertain if the delayed clearance was accompanied by an increase in the C. rodentium burden, mice were sacrificed at various time points postchallenge (p.c.), and the numbers of C. rodentium in the cecal patch, cecum and colon, liver, spleen, and mLNs were determined. miR-155-deficient mice harbored higher numbers of C. rodentium in the cecal patch, cecum, and colon on day 20 p.c. (see Fig. S1b) than similarly infected wild-type mice. Furthermore, on day 26 p.c., when all C57BL/6 mice were clear, C. rodentium was still present in gastrointestinal tissues of miR-155-deficient mice. Interestingly, low levels of C. rodentium (>103 CFU/organ) were recovered from the spleen, liver, and mLNs of miR-155-deficient mice on day 20 p.c. (see Fig. S1b), in stark contrast to wild-type infected C57BL/6 mice. Due to the predominantly noninvasive nature of C. rodentium, in immunocompetent mice, colonization is typically restricted to the luminal surface of the gut epithelium. Thus, this would suggest that the miR-155-deficient mice have an impaired ability to control aspects of both local and systemic C. rodentium infection.
Fig 1.

C. rodentium infection in miR-155-deficient (red bars and red squares) and control wild-type C57BL/6 (blue bars and blue circles) mice. Mice were orally gavaged with approximately 1 × 109 CFU of C. rodentium. (a) Viable C. rodentium bacteria enumerated from fecal samples, n = 6 mice per group; the dashed line indicates the detection level of the assay used. (b) Time (days) taken to resolve C. rodentium infection (±standard errors of the means [SEM]), n = 5 mice per group. *** indicates P value of <0.0001 by a Student t test. (c) Histopathological analyses (H&E staining; original magnification, ×5) of colon sections from miR-155-deficient and control wild-type C57BL/6 mice on day 14 postinoculation; images are representative of 5 mice per group. (d) Average crypt length (±SEM) was determined from naïve and infected mice; 10 sections from n = 5 mice per group. *** indicates P value of <0.001 by a Student t test. (e and f) Representative scanning electron micrographs from distal colons of infected miR-155-deficient and wild-type mice, respectively.
The degree of hyperplasia in the colon of mice can be determined by measuring increases in crypt length and tissue weight. Morphometric analysis of the distal colons of mice on day 14 p.c. revealed that crypt lengths were significantly increased in C. rodentium-infected miR-155-deficient mice compared to those of similarly infected controls (P value of <0.0001; Fig. 1c and d). miR-155-deficient mice had increased cellular infiltrate into the lamina propria and, following closer examination with scanning electron microscopy, showed more obvious areas of surface damage, with bacteria penetrating the luminal cell surface (Fig. 1c, e, f; see also Fig. S2 in the supplemental material).
The indigenous intestinal microbiota can potentially influence both the levels of colonization of and the immune response to C. rodentium. Consequently, we investigated the composition of the microbiota in miR-155-deficient and wild-type mice. Fecal samples were collected from miR-155-deficient and control C57BL/6 mice immediately prior to C. rodentium challenge (day 0) and at 4 and 14 days p.c.; 16S rRNA gene-based sequence analysis was then performed on extracted DNA using 454 pyrosequencing. Although C. rodentium was not particularly dominant within the microbiota at any time point, the proportional abundance of the pathogen was significantly higher at day 14 in the miR-155-deficient mice than in the control cohort (P < 0.001) (Fig. 2a). The 16S rRNA sequencing results therefore confirmed the findings from the culture-based analysis and indicate that miR-155-deficient mice show impaired clearance of C. rodentium at day 14 postinfection. When looking at global differences in microbiota structure, at day 14 postinfection, the microbial communities of both miR-155-deficient and control C. rodentium-infected mouse cohorts clustered separately from those collected at days 0 and 4, which were largely indistinguishable from each other (Fig. 2b). The proportional abundance of the Bacteroidetes phylum was reduced in both cohorts at day 14 (control mice, P = 0.021; miR-155-deficient mice, P = 0.019), indicating that although C. rodentium infection may have some impact on the wider microbiota, it occurs regardless of the host background.
Fig 2.
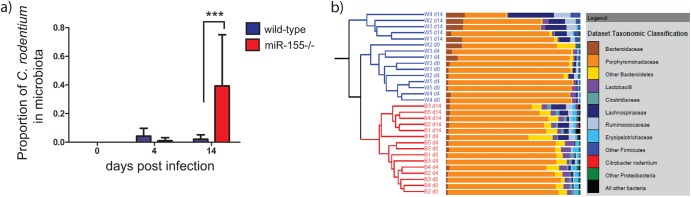
Analysis of microbiota from miR-155-deficient and control wild-type C57BL/6 mice orally infected with approximately 1 × 109 CFU of C. rodentium. (a) Proportional abundance of C. rodentium in wild-type and miR-155-deficient mice as a percentage of the total microbiota. *** indicates P value of <0.001 as calculated by the Metastats program within the mothur software package. (b) Dendrogram illustrating similarity in overall microbiota community structure between wild-type and miR-155-deficient mice prior to and following C. rodentium infection. W1 to W5, wild-type mice; B1 to B5, miR-155-deficient mice; d0, immediately prior to infection; d4 and d14, day 4 and day 14 postinfection, respectively. Adjacent bars show microbiota composition of each sample. Brown/orange/yellow sections are members of the Bacteroidetes phylum; blue/purple/gray sections are members of the Firmicutes phylum.
Impaired humoral immune responses and germinal center (GC) formation in the absence of miR-155.
Immunoglobulin G (IgG) antibodies play a key role in mediating protection against C. rodentium (18), as mice lacking B cells or serum IgG antibodies are less able to prevent mucosal infection and systemic dissemination of C. rodentium (19). Given that miR-155 has been implicated as a key player in the production of switched antibodies (6, 7), we investigated whether humoral immune responses are affected in miR-155-deficient mice infected with C. rodentium. Groups of miR-155-deficient and wild-type C57BL/6 mice were orally challenged with C. rodentium, and the subsequent serum Ig responses were measured against the C. rodentium surface protein, EspA (26). miR-155-deficient mice produced greatly reduced levels of EspA-specific serum IgG and IgA antibodies compared with those of wild-type equivalents (Fig. 3a). No anti-EspA titers were detected in prebleeds for the immunized mice. In addition, EspA-specific IgA titers in fecal homogenates were considerably lower in C. rodentium-infected miR-155-deficient mice (Fig. 3a).
Fig 3.

Antibody titers and germinal center response in miR-155-deficient (red bars and red squares) and control wild-type (blue bars and blue circles) mice. Mice were orally infected with approximately 1 × 109 CFU of C. rodentium. (a) Anti-EspA-specific antibody titers were measured 21 days later; ** indicates P value of <0.005 by a Student t test. (b) Immunohistochemistry was performed on mesenteric lymph node sections from wild-type and miR-155-deficient mice to detect germinal centers (B220+ cells, green; and PNA+, orange) on day 14 p.i., magnification ×20. (c) Number of GCs (±SEM) was determined from stained sections. ** indicates P value of <0.0038 by a Student t test, n = 5 mice per group. (d) Average germinal center size (±SEM) calculated from sections in panel b; ** indicates P value of <0.0033 by a Student t test, n = 5 mice per group.
GCs are regions within primary lymphoid follicles that support the generation of memory B cells and plasma cells capable of producing specific class-switched antibodies. After infection with C. rodentium, GCs rapidly form in gut-associated lymphoid tissue (GALT) such as the mLNs. Two recent studies have shown a requirement for miR-155 in GC responses and generation of class-switched plasma cells to model antigens (6, 7). Therefore, we examined GC formation in the lymphoid tissue (cecal patch and mLNs) of miR-155-deficient and control mice following oral challenge with C. rodentium. Examination of lymph tissue from naive miR-155-deficient mice revealed no obvious abnormalities in the development of primary B cell follicles compared to that of uninfected wild-type mice (data not shown). However, on day 14 after C. rodentium challenge, there were significantly fewer GCs in the lymphoid tissue of miR-155-deficient mice than in that of wild-type mice (Fig. 3b and c), and the few GCs produced were considerably smaller in size than those present in similar C57BL/6 mice (Fig. 3d). Taken together, these results suggest that the susceptibility of miR-155-deficient mice to a primary C. rodentium infection is potentially associated with impaired GC formation and significantly reduced production of class-switched, pathogen-specific antibody.
Transcriptional profiling of C. rodentium-infected cecal patches reveals B cell function is affected in the absence of miR-155.
The primary mode of action of miR-155 is through specific mRNA targeting (1). Consequently, we performed microarray analysis on C. rodentium-infected cecal patch tissues from groups of wild-type and miR-155-deficient mice, as this tissue is targeted early in C. rodentium infection. To gain a greater insight, we analyzed our gene expression data using Innate DB, a pathway analysis tool annotated for innate immune genes (www.innatedb.ca; see Materials and Methods). Mice were orally challenged with C. rodentium, and on days 4 and 14 p.c., transcriptional responses in the cecal patch lymphoid tissues were analyzed using Illumina gene chips. mRNAs were considered significant only if they were altered by at least 1.5-fold and had an adjusted P value of less than 0.05. Using these strict criteria, we found that 314 genes (229 up and 85 down) on day 4 and 8 genes (7 up and 1 down) on day 14 pc were differentially expressed in the cecal patches of miR-155-deficient mice compared with control C57BL/6 mice (see Table S1 in the supplemental material). Thus, relatively major changes in gene expression were detected at day 4 compared to day 14, with most genes being upregulated. Preliminary analysis of these genes identified many related to cell metabolism that may be associated with the increased cell proliferation and epithelium cell damage observed in the miR-155-deficient mice. Additionally, of the 85 genes downregulated in the miR-155-deficient cecal patches, on day 4 p.c., ∼17% have a reported immune function (Table 1). Furthermore, we noted that many of these genes are involved, at some stage, in the differentiation and/or function of B cells. For example, CD19 has a role in GC formation, B cell homing, and apoptosis. Similar to miR-155-deficient mice, CD19-null (CD19−/−) mice have decreased mitogenic responses, low GC formation, and decreased humoral immune responses to T cell-independent type 1 and T cell-dependent antigens (29, 30). Additionally, Bcl6 is essential for the differentiation of GC B cells, since Bcl6-null (Bcl6−/−) mice have severely impaired germinal GC formation (31, 32).
Table 1.
Gene expression profiling in tissues of C. rodentium-infected miR-155-deficient micea
| Day p.c. | Target ID | Symbol | Fold change | Adjusted P value | GO biological process |
|---|---|---|---|---|---|
| 4 | scl37307.8.1_29-S | Mmp3 | −3.79 | 0.00669 | Peptidoglycan metabolic process (GO:0000270), proteolysis (GO:0006508), collagen catabolic process (GO:0030574) |
| scl48501.9_645-S | Cd86 | −1.68 | 0.0127 | Defense response (GO:0006952) | |
| scl38155.8_378-S | Tnfaip3 | −1.99 | 0.0136 | Ubiquitin cycle (GO:0006512), apoptosis (GO:0006915) | |
| scl25940.13_350-S | Lat2 | −1.92 | 0.0144 | Immune response (GO:0006955), B cell activation (GO:0042113), mast cell degranulation (GO:0043303), B cell receptor signaling pathway (GO:0050853) | |
| scl0003597.1_94-S | Mmp3 | −2.45 | 0.0152 | Peptidoglycan metabolic process (GO:0000270), proteolysis (GO:0006508), collagen catabolic process (GO:0030574) | |
| scl37305.10.1_6-S | Mmp10 | −2.02 | 0.0187 | Peptidoglycan metabolic process (GO:0000270), proteolysis (GO:0006508), collagen catabolic process (GO:0030574) | |
| scl48625.8_280-S | Bcl6 | −1.93 | 0.0341 | Negative regulation of transcription from R-polymerase II promoter (GO:0000122), cell morphogenesis (GO:0000902), negative regulation of cell-matrix adhesion (GO:0001953), germinal center formation (GO:0002467), negative regulation of T-helper 2 type immunoresponse (GO:0002829), transcription (GO:0006350), regulation of transcription, D dependent (GO:0006355), Rho protein signal transduction (GO:0007266), spermatogenesis (GO:0007283), protein localization (GO:0008104), negative regulation of cell proliferation (GO:0008285), actin cytoskeleton organization and biogenesis (GO:0030036), B cell differentiation (GO:0030183), positive regulation of B cell proliferation (GO:0030890), regulation of Rho GTPase activity (GO:0032319), negative regulation of mast cell cytokine production (GO:0032764), negative regulation of Rho protein signal transduction (GO:0035024), T-helper 2 type immune response (GO:0042092), regulation of cell proliferation (GO:0042127), negative regulation of apoptosis (GO:0043056), regulation of memory T cell differentiation (GO:0043380), negative regulation of cell differentiation (GO:0045596), negative regulation of T-helper 2 cell differentiation (GO:0045629), negative regulation of isotype switching to IgE isotypes (GO:0048294), erythrocyte development (GO:0048821), regulation of inflammatory response (GO:0050727), positive regulation of cell motility (GO:0051272) | |
| scl30689.14.1_1-S | Cd19 | −1.98 | 0.0377 | ||
| scl0012514.2_91-S | Cd68 | −1.56 | 0.038 | ||
| scl41469.8_427-S | Tnfrsf13b | −2.18 | 0.0401 | B cell homeostasis (GO:0001782), immune response (GO:0006955), negative regulation of B cell proliferation (GO:0030889) | |
| scl38756.3.1_44-S | Vpreb3 | −2.27 | 0.0404 | ||
| scl21758.4.1_5-S | Cd2 | −1.67 | 0.0404 | Protein amino acid phosphorylation (GO:0006468), defense response (GO:0006952), cell adhesion (GO:0007155) | |
| scl44840.4_185-S | Cd83 | −2.26 | 0.0449 | ||
| scl44948.10_526-S | Irf4 | −1.6 | 0.0458 | Transcription (GO:0006350), regulation of transcription, D dependent (GO:0006355), myeloid dendritic cell differentiation (GO:0043011) | |
| 14 | scl53984.2_610-S | Cxcr3 | −1.52 | 0.0361 | Chemotaxis (GO:0006935), defense response (GO:0006952), signal transduction (GO:0007165), G-protein coupled receptor protein signaling pathway (GO:0007186) |
Immunomodulatory genes and genes involved in B cell differentiation and/or function (highlighted in gray) that were significantly downregulated in miR-155-deficient cecal patches on day 4 and day 14 p.c. compared with control mice.
We further analyzed the day 4 p.c. cecal patch expression data using InnateDB, and the B cell receptor (BCR) signaling pathway was identified as being significantly associated with downregulated genes. It is worth noting that the most significantly downregulated gene in the cecal patch on day 4 p.c. was stomelysin-1 (matrix metalloproteinase-3 [mmp3]). Mmp3 is a member of the matrix metalloproteinase family, a group of molecules which are involved in mediating matrix remodelling and cell migration during tissue injury and repair. Mmp3 has previously been shown to be important during infection with C. rodentium, as mice lacking mmp3 (mmp3−/−) show delayed clearance of bacteria from the colon (33). We selected five genes of interest that were differentially expressed in infected miR-155-deficient mice and reanalyzed them using real-time quantitative reverse transcription-PCR (qRT-PCR) utilizing SYBR green. Real-time qRT-PCR was performed on cDNA synthesized from the same total RNA used as templates for the cDNA probes hybridized to the arrays. For all five genes, levels of expression changed in the same relative direction as with the microarray analysis (see Fig. S3 in the supplemental material). Interestingly, we found no significant enrichment of miR-155 complementary sequences in these differentially expressed genes. This suggests that secondary effects, such as differential recruitment and activation of cells, were occurring upstream of the direct miR-155 effects.
The C. rodentium susceptibility phenotype is observed in miR-155-deficient, μMT-deficient chimeras.
Our data strongly suggested that BCR signaling and B cell function is adversely affected in miR-155-deficient mice. To explore this possibility further, chimeric mice were created by transferring 20% of either wild-type or miR-155-deficient bone marrow cells together with 80% of μMT-deficient bone marrow cells into sublethally irradiated μMT mice as previously described (34), and these mice were challenged orally with C. rodentium. The μMT mutation prevents the innate generation of B cells, and thus the 20/80 ratio favors reconstitution of B cells from the donor wild-type or miR-155-deficient mice and all other hematopoietic lineages from the μMT mice which express functional miR-155 RNA. Both groups of reconstituted chimeras had similar proportions and numbers of B, CD4+, and CD8+ T cells (data not shown).
As with miR-155-deficient mice, miR-155/μMT-deficient chimeric mice completely resolved infection, but again the time to clearance was significantly delayed (∼9 days) compared with that of wild-type/μMT-deficient chimeras (P value of <0.01; Fig. 4a and b). Bacterial counts from harvested organs indicated that the miR-155/μMT-deficient chimeras have a prolonged time to clearance from gastric tissue and show breakthrough to the systemic tissue similar to that observed in challenged miR-155-deficient mice (Fig. 4c). Additionally, miR-155/μMT-deficient chimeras produced significantly fewer mLN GCs (Fig. 5a and b) and reduced levels of EspA-specific serum IgG at day 21 p.c. compared with wild-type/μMT-deficient chimeras (Fig. 5c). These data support the hypothesis that impaired responses in miR-155-deficient mice is linked to a lack of miR-155 in B cells.
Fig 4.

C. rodentium infection in miR-155/μMT-deficient chimeric (red bars and red squares) and control (blue bars and blue circles) mice. Mice were orally infected with approximately 1 × 109 CFU of C. rodentium. (a) Viable C. rodentium bacteria enumerated from fecal samples, n = 3 mice per group; (b) time (days) taken to resolve C. rodentium infection (±SEM), n = 3 mice per group; * indicates P value of <0.01 by a Student t test; (c) enumeration of C. rodentium in organ cultures from miR-155/μMT-deficient chimeric (red bars) and control wild-type (blue bars) mice at day 21 postinfection (±SEM), n = 4 mice per group. The dashed lines indicate the levels of detection in each assay.
Fig 5.

(a) Immunohistochemistry was performed on mesenteric lymph node sections from wild-type and miR-155/μMT-deficient chimeric mice to detect germinal centers (B220+ cells, green; and PNA+, orange) on day 14 postinfection (p.i.); magnification, ×20. (b) Number of GCs (±SEM) was determined from stained sections. * indicates P value of <0.05 by a Student t test, n = 3 mice per group. Average germinal center size (±SEM) calculated from stained sections in panel b, n = 3 mice per group. (c) Anti-EspA-specific antibody titers were measured in the serum of infected mice 21 days p.i.
DISCUSSION
Here, we investigate the role of miR-155 in protection and mucosa-associated immunity to the luminal bacterial pathogen C. rodentium. Although miR-155-deficient mice are able to resolve a C. rodentium infection, the time to achieve complete clearance is significantly increased, taking on average 20 days longer than control C57BL/6 mice. In addition, infected miR-155-deficient mice exhibit a higher pathogen burden in their gastrointestinal tissues, such as the cecal patch, cecum, and colon. The greater numbers of C. rodentium colonizing the colon of miR-155-deficient mice consequently drove gross colonic hyperplasia, typified by significant increases in tissue weight and crypt length. We were also able to recover low numbers (>103 CFU/organ) of viable C. rodentium from systemic tissues such as the liver, spleen, and mesenteric lymph nodes of infected miR-155-deficient mice, thus providing evidence that the systemic immune system was also impaired. Infected miR-155-deficient mice presented with significantly fewer and smaller GCs than those observed in controls, and associated C. rodentium-specific serum IgG and IgA antibodies were severely reduced. The diminished levels of C. rodentium-specific antibody is likely to be responsible at least in part for the prolonged infection resolution time, increased tissue pathogen burden, and systemic spread of C. rodentium. There were no obvious global differences in the microbiota between infected miR-155 and wild-type mice to explain the phenotype. Thus, the results presented here strongly suggest that humoral immune responses are drastically impaired in infected miR-155-deficient mice following a luminal bacterial challenge.
miRNAs can play a key role in regulating the absolute levels of mRNAs in cells and corresponding tissues (1). Therefore, we set out to examine the global gene expression profile in miR-155-deficient C. rodentium-infected cecal patches with the aim of identifying key immune genes and pathways that are affected in vivo by the loss of miR-155. Initially we expected to find a significant correlation between the differentially expressed genes and the predicted miR-155 targets. However, we found little enrichment of miR-155 complementary sequences in the genes that were differentially expressed. A possible explanation for this discrepancy is that many of the transcripts altered may not represent direct targets of miR-155, but the changes were actually associated with different migratory cell populations and activation levels. In principle, the mRNAs could be indirectly targeted if they are part of an miRNA-mediated signaling cascade controlled by the direct miRNA targets (1).
Although a number of different immunomodulatory genes were differentially expressed in the day 4 p.c. miR-155-deficient cecal patches, we observed that an overrepresentative number, approximately 17%, of the downregulated genes are known to be involved in B cell development and/or function. Most notably, Bcl6 and CD19 are critically important during GC formation. The downregulation of these genes during infection could explain the impaired humoral immune response in C. rodentium miR-155-deficient mice. When the expression data were analyzed further using the InnateDB data analysis and pathway overrepresentation tools, the BCR signaling pathway was highlighted as being significantly associated with downregulated genes, thus suggesting that a lack of miR-155 impairs signaling through the BCR. We propose that during a mucosal C. rodentium infection in wild-type mice, B cells residing within the cecal patch encounter bacterial antigens and are subsequently activated to begin proliferation and differentiation into plasma B cells capable of producing antigen-specific antibodies. However, in miR-155-deficient mice, B cells are incapable of being sufficiently stimulated to begin differentiation since the loss of miR-155 adversely affects the BCR signaling pathway. The results presented here are consistent with previously published reports that have shown the importance of miR-155 for the regulation of GC responses and the generation of immunoglobulin class-switched plasma cells (6, 7).
In addition, we found that mmp3 and CXCR3 were downregulated in miR-155-deficient cecal patches on day 4 and 14 p.c., respectively. Mmp3- is known to mediate cell migration during tissue injury and repair, and C. rodentium-infected Mmp3-deficient mice exhibit delayed clearance of bacteria from the colon; the prolongation in clearance time has been attributed to a delayed migration of CD4+ T cells into the lamina propria of these mice (33). CXCR3 is known to be involved not only in the recruitment of leukocytes to sites of inflammation but also in mediating the homing of murine IgG-secreting plasma cells. A recently published report has shown that a deficiency of CXCR3 in mice results in a considerable delay in the induction of IgG antibodies after infection with C. rodentium, and it is the lack of C. rodentium-specific antibody which consequently leads to increased bacterial loads and systemic dissemination in these mice (35). Moreover, 2 weeks after infection, CXCR3-deficient mice develop modestly increased submucosal and mucosal infiltration of inflammatory cells into the colon. Based on these findings, we hypothesize that leukocyte trafficking may also be affected in miR-155-deficient mice.
Since our data strongly suggest that BCR signaling and B cell function is adversely affected in miR-155-deficient mice, we investigated whether the defect was intrinsic to B cells. Accordingly, we infected miR-155/μMT-deficient chimeras with C. rodentium to establish if the phenotypic alterations observed in miR-155-deficient germ line mice would be replicated in chimeric mice possessing only miR-155-deficient B cells. Analogous to germ line miR-155-defective mice, miR-155/μMT-deficient chimeras took significantly longer than control chimeric mice to resolve infection. Additionally, we found that the miR-155/μMT-deficient chimeras produced not only significantly less but considerably smaller mLN germinal centers compared with wild-type/μMT-deficient chimeras, and consequently levels of C. rodentium-specific serum IgG and IgA antibodies were reduced. We realize that the defect is only partially recapitulated in the chimeric mice, which suggests that miR-155 has an effect via alternative immune mechanisms. We and others have previously shown that miR-155 plays an important role in T-cell function and that this may in some way contribute to the phenotype observed. For example T cells may be recruited by some of the chemokines shown to be downregulated in the RNA analysis performed in this study (5, 8).
In summary, we have presented evidence that miR-155 plays a limited but significant role in the mucosal immune response to the enteric pathogen C. rodentium. We find that after infection with C. rodentium, the absence of miR-155 results in impaired BCR signaling in vivo. As a result, insufficient signaling through the BCR leads to impaired GC formation and defective humoral immune responses. Most importantly, we conclusively show that miR-155 is fundamentally required for proper B cell function.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Wellcome Trust grants (098051 and 076964).
We thank RSF staff for maintenance of the animal breeding colonies and in particular Cordelia Brandt for technical assistance with animal experiments.
Footnotes
Published ahead of print 21 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00969-12.
REFERENCES
- 1. Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM. 2005. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433:769–773 [DOI] [PubMed] [Google Scholar]
- 2. Tam W, Ben-Yehuda D, Hayward WS. 1997. bic, a novel gene activated by proviral insertions in avian leukosis virus-induced lymphomas, is likely to function through its noncoding RNA. Mol. Cell. Biol. 17:1490–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. O'Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. 2007. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. U. S. A. 104:1604–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B, Fabbri M, Alder H, Liu CG, Calin GA, Croce CM. 2007. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol. 179:5082–5089 [DOI] [PubMed] [Google Scholar]
- 5. Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA, Vetrie D, Okkenhaug K, Enright AJ, Dougan G, Turner M, Bradley A. 2007. Requirement of bic/microRNA-155 for normal immune function. Science 316:608–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL, Schmidt-Supprian M, Rajewsky N, Yancopoulos G, Rao A, Rajewsky K. 2007. Regulation of the germinal center response by microRNA-155. Science 316:604–608 [DOI] [PubMed] [Google Scholar]
- 7. Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, Das PP, Miska EA, Rodriguez A, Bradley A, Smith KG, Rada C, Enright AJ, Toellner KM, Maclennan IC, Turner M. 2007. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity 27:847–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Oertli M, Engler DB, Kohler E, Koch M, Meyer TF, Muller A. 2011. MicroRNA-155 is essential for the T cell-mediated control of Helicobacter pylori infection and for the induction of chronic gastritis and colitis. J. Immunol. 187:3578–3586 [DOI] [PubMed] [Google Scholar]
- 9. Barthold SW, Coleman GL, Jacoby RO, Livestone EM, Jonas AM. 1978. Transmissible murine colonic hyperplasia. Vet. Pathol. 15:223–236 [DOI] [PubMed] [Google Scholar]
- 10. Schauer DB, Falkow S. 1993. The eae gene of Citrobacter freundii biotype 4280 is necessary for colonization in transmissible murine colonic hyperplasia. Infect. Immun. 61:4654–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dougan G, Ghaem-Maghami M, Pickard D, Frankel G, Douce G, Clare S, Dunstan S, Simmons C. 2000. The immune responses to bacterial antigens encountered in vivo at mucosal surfaces. Philos. Trans. R. Soc. Lond. B Biol. Sci. 355:705–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eckmann L. 2006. Animal models of inflammatory bowel disease: lessons from enteric infections. Ann. N. Y. Acad. Sci. 1072:28–38 [DOI] [PubMed] [Google Scholar]
- 13. Higgins LM, Frankel G, Douce G, Dougan G, MacDonald TT. 1999. Citrobacter rodentium infection in mice elicits a mucosal Th1 cytokine response and lesions similar to those in murine inflammatory bowel disease. Infect. Immun. 67:3031–3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. MacDonald TT, Frankel G, Dougan G, Goncalves NS, Simmons C. 2003. Host defences to Citrobacter rodentium. Int. J. Med. Microbiol. 293:87–93 [DOI] [PubMed] [Google Scholar]
- 15. Mundy R, MacDonald TT, Dougan G, Frankel G, Wiles S. 2005. Citrobacter rodentium of mice and man. Cell Microbiol. 7:1697–1706 [DOI] [PubMed] [Google Scholar]
- 16. Wiles S, Pickard KM, Peng K, MacDonald TT, Frankel G. 2006. In vivo bioluminescence imaging of the murine pathogen Citrobacter rodentium. Infect. Immun. 74:5391–5396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bry L, Brigl M, Brenner MB. 2006. CD4+-T-cell effector functions and costimulatory requirements essential for surviving mucosal infection with Citrobacter rodentium. Infect. Immun. 74:673–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maaser C, Housley MP, Iimura M, Smith JR, Vallance BA, Finlay BB, Schreiber JR, Varki NM, Kagnoff MF, Eckmann L. 2004. Clearance of Citrobacter rodentium requires B cells but not secretory immunoglobulin A (IgA) or IgM antibodies. Infect. Immun. 72:3315–3324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Simmons CP, Clare S, Ghaem-Maghami M, Uren TK, Rankin J, Huett A, Goldin R, Lewis DJ, MacDonald TT, Strugnell RA, Frankel G, Dougan G. 2003. Central role for B lymphocytes and CD4+ T cells in immunity to infection by the attaching and effacing pathogen Citrobacter rodentium. Infect. Immun. 71:5077–5086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spahn TW, Ross M, von Eiff C, Maaser C, Spieker T, Kannengiesser K, Domschke W, Kucharzik T. 2008. CD4+ T cells transfer resistance against Citrobacter rodentium-induced infectious colitis by induction of Th1 immunity. Scand. J. Immunol. 67:238–244 [DOI] [PubMed] [Google Scholar]
- 21. Wiles S, Clare S, Harker J, Huett A, Young D, Dougan G, Frankel G. 2004. Organ specificity, colonization and clearance dynamics in vivo following oral challenges with the murine pathogen Citrobacter rodentium. Cell Microbiol. 6:963–972 [DOI] [PubMed] [Google Scholar]
- 22. Schloss PD. 2009. A high-throughput DNA sequence aligner for microbial ecology studies. PLoS One 4:e8230 doi:10.1371/journal.pone.0008230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. 2009. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37:D141–D145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Letunic I, Bork P. 2007. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23:127–128 [DOI] [PubMed] [Google Scholar]
- 25. White JR, Nagarajan N, Pop M. 2009. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 5:e1000352 doi:10.1371/journal.pcbi.1000352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Knutton S, Rosenshine I, Pallen MJ, Nisan I, Neves BC, Bain C, Wolff C, Dougan G, Frankel G. 1998. A novel EspA-associated surface organelle of enteropathogenic Escherichia coli involved in protein translocation into epithelial cells. EMBO J. 17:2166–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malick LE, Wilson RB. 1975. Modified thiocarbohydrazide procedure for scanning electron microscopy: routine use for normal, pathological, or experimental tissues. Stain Technol. 50:265–269 [DOI] [PubMed] [Google Scholar]
- 28. Lynn DJ, Winsor GL, Chan C, Richard N, Laird MR, Barsky A, Gardy JL, Roche FM, Chan TH, Shah N, Lo R, Naseer M, Que J, Yau M, Acab M, Tulpan D, Whiteside MD, Chikatamarla A, Mah B, Munzner T, Hokamp K, Hancock RE, Brinkman FS. 2008. InnateDB: facilitating systems-level analyses of the mammalian innate immune response. Mol. Syst. Biol. 4:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carter RH, Myers R. 2008. Germinal center structure and function: lessons from CD19. Semin. Immunol. 20:43–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang Y, Carter RH. 2005. CD19 regulates B cell maturation, proliferation, and positive selection in the FDC zone of murine splenic germinal centers. Immunity 22:749–761 [DOI] [PubMed] [Google Scholar]
- 31. Fukuda T, Yoshida T, Okada S, Hatano M, Miki T, Ishibashi K, Okabe S, Koseki H, Hirosawa S, Taniguchi M, Miyasaka N, Tokuhisa T. 1997. Disruption of the Bcl6 gene results in an impaired germinal center formation. J. Exp. Med. 186:439–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jardin F, Ruminy P, Bastard C, Tilly H. 2007. The BCL6 proto-oncogene: a leading role during germinal center development and lymphomagenesis. Pathol. Biol. (Paris) 55:73–83 [DOI] [PubMed] [Google Scholar]
- 33. Li CK, Pender SL, Pickard KM, Chance V, Holloway JA, Huett A, Goncalves NS, Mudgett JS, Dougan G, Frankel G, MacDonald TT. 2004. Impaired immunity to intestinal bacterial infection in stromelysin-1 (matrix metalloproteinase-3)-deficient mice. J. Immunol. 173:5171–5179 [DOI] [PubMed] [Google Scholar]
- 34. Huntington ND, Xu Y, Puthalakath H, Light A, Willis SN, Strasser A, Tarlinton DM. 2006. CD45 links the B cell receptor with cell survival and is required for the persistence of germinal centers. Nat. Immunol. 7:190–198 [DOI] [PubMed] [Google Scholar]
- 35. Spehlmann ME, Dann SM, Hruz P, Hanson E, McCole DF, Eckmann L. 2009. CXCR2-dependent mucosal neutrophil influx protects against colitis-associated diarrhea caused by an attaching/effacing lesion-forming bacterial pathogen. J. Immunol. 183:3332–3343 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.