

Abstract
Campylobacter jejuni is a major cause of bacterial diarrheal disease worldwide. The organism is characterized by a diversity of polysaccharide structures, including a polysaccharide capsule. Most C. jejuni capsules are known to be decorated nonstoichiometrically with methyl phosphoramidate (MeOPN). The capsule of C. jejuni 81-176 has been shown to be required for serum resistance, but here we show that an encapsulated mutant lacking the MeOPN modification, an mpnC mutant, was equally as sensitive to serum killing as the nonencapsulated mutant. A nonencapsulated mutant, a kpsM mutant, exhibited significantly reduced colonization compared to that of wild-type 81-176 in a mouse intestinal colonization model, and the mpnC mutant showed an intermediate level of colonization. Both mutants were associated with higher levels of interleukin 17 (IL-17) expression from lamina propria CD4+ cells than from cells from animals infected with 81-176. In addition, reduced levels of Toll-like receptor 4 (TLR4) and TLR2 activation were observed following in vitro stimulation of human reporter cell lines with the kpsM and mpnC mutants compared to those with wild-type 81-176. The data suggest that the capsule polysaccharide of C. jejuni and the MeOPN modification modulate the host immune response.
INTRODUCTION
Campylobacter jejuni is one of the major causes of bacterial diarrhea worldwide. The organism is unusual among enteric pathogens in that it expresses a polysaccharide capsule (CPS) that contributes to serum resistance, invasion of intestinal epithelial cells in vitro, and virulence in ferret and Galleria mellonella larvae models of disease (1–3). CPS is the major serodeterminant of the Penner serotyping scheme of C. jejuni (4), in which there are 47 serotypes, a reflection of the diversity of polysaccharide capsular structures in C. jejuni. In addition to variation in sugar composition, the CPS can be modified with ethanolamine, glycerol, and O-methyl phosphoramidate (MeOPN). The MeOPN modification, which is found on about 75% of C. jejuni CPSs, has been shown to modulate cytokine release from mouse dendritic cells and to be a key determinant in virulence in the moth larva model of disease (2, 3). Both CPS expression itself (1) and expression of the modifications are phase variable due to slip strand mismatch repair (5–7). Thus, reversible phase variations in multiple genes result in mixed populations of wild-type cells, some of which express CPS and others that do not (1). Similarly, the levels of the MeOPN modifications found on the CPS are present in nonstoichiometric amounts because of phase variation in genes encoding the enzymes involved in transfer of these groups to specific sugars (8).
We have shown that a polysaccharide conjugate vaccine composed of the capsule of strain 81-176 conjugated to carrier protein CRM197 showed significant protection against diarrheal disease in a nonhuman primate model of diarrhea, also suggesting a role for CPS in virulence (9). Here, we further demonstrate that the CPS and the MeOPN modification both play significant roles in modulation of several aspects of the immune response, including serum resistance, activation of NF-κB, and cytokine induction in vivo.
MATERIALS AND METHODS
Bacterial strains and media.
C. jejuni strain 81-176, its motile, isogenic kpsM mutant, and the complement of that mutant have been described previously (1). Bacteria were routinely cultivated microaerobically on Mueller-Hinton (MH) agar supplemented with antibiotics as appropriate. For serum resistance assays, strains were grown in biphasic MH cultures for 18 to 20 h at 37°C. For mouse infection studies, strains were inoculated in MH broth to an optical density at 600 nm (OD600) of ∼0.01 to 0.05 and incubated with shaking under microaerobic conditions at 37°C for 18 h.
Mutation and complementation of a gene for biosynthesis of MeOPN in C. jejuni 81-176.
A region of the CPS locus of the 81-176 chromosome carrying genes for MeOPN synthesis (8) was cloned as a PCR fragment into BamHI-digested pBluescript. The primers used were pg08.90 (CGGGATCCGGAATGCCTGCTGTTATAGGAGTTGGA) in CJJ81176_1417 (labeled mpnA in Fig. 1) and pg08.91 (CGGGATCCCATCGAAGCATCATCTTCAACTTGAGC) in CJJ81176_1413 (kpsC). Both primers introduced a BamHI site at the 5′ ends, indicated by the underlining. The resulting plasmid was subjected to transposon mutagenesis using an in vitro Tn5-based transposition system (Epicentre, Madison, WI) with a chloramphenicol resistance (Cmr; cat) cassette, and the insertion points were identified by sequencing individual insertions with primers within the cassette, as previously described (10–12). A clone with a nonpolar insertion into gene CJJ81176_1415 (mpnC in Fig. 1) was identified; the insertion was 472 bp into the 762-bp gene. This plasmid was used to electroporate C. jejuni 81-176 to Cmr, and the resulting mutant was confirmed to have undergone a double crossover by PCR with primers that bracketed the insertion point of the transposon. The mutant was complemented by cloning a wild-type allele of mpnC into pRY107/28, which is pRY107 (13) containing the σ28 promoter of flaA cloned between the XbaI and BamHI sites. The wild-type mpnC gene was PCR amplified using high-fidelity polymerase (Clontech) and primers pg08.155 (5′-CGGGATCCGGTATAATGTGGCATATTGAAAGAG-3′) and pg08.150 (5′-CCGCTCGAGCTCTTAACTCATCTCCATCGAGATAAATAAG-3′), which introduced BamHI and XhoI sites, respectively. This fragment was cloned into BamHI-XhoI-digested pRY107/28. The plasmid complement was introduced into 81-176 mpnC by conjugal transfer from E. coli DH5α containing RK212.2, as previously described, with selection on kanamycin (14).
Fig 1.

(A) Diagram of the capsule locus of strain 81-176. The locus is organized, like other class 2 capsule loci, into conserved regions 1 and 3, encoding proteins involved in capsule assembly and transport, and the variable region 2, encoding proteins involved in polysaccharide synthesis. The MeOPN biosynthesis genes (mpnA to -D) are found within region 2 but are highly conserved among strains if present and correspond to CJ1415 to CJ1418 in NCTC 11168 (8). Genes corresponding to CJJ81176_1418 and CJJ81176_1419 are also highly conserved among strains expressing MeOPN, but mutational analyses have failed to demonstrate a role for these genes in MeOPN synthesis (16). CJJ81176_1420 is annotated as a putative MeOPN transferase. There are 15 additional genes within region 2 of 81-176. (B) 31P NMR of CPSs from wild-type 81-176 (blue), the mpnC mutant (green), and the mpnC mutant complemented in trans (red).
31P NMR spectrometry.
Preparations containing the CPSs were dissolved in D2O, and 31P nuclear magnetic resonance (NMR) was performed on a 400-MHz Bruker NMR instrument. Orthophosphoric acid was used as the external reference (δ 0.00).
Serum survival assays.
Cultures (18 h old) of C. jejuni grown in MH biphasic media were washed and adjusted to an OD600 of 0.1 in minimal essential medium (MEM). Aliquots (100 μl) were added to wells of a 24-well plate containing 900 μl of prewarmed MEM supplemented with 10% normal human serum (Sigma; NHS) and incubated under microaerobic conditions at 37°C. The percentage of survivors was determined by serial dilution onto MH agar plates. Assays were run in duplicate 3 or 4 times.
Mouse infection experiments.
BALB/c mice (7 to 8 weeks old; Jackson Laboratories, Bar Harbor, ME) were housed in groups of 10 with access to food and water ad libitum. For infection with C. jejuni, 1 liter of broth culture was harvested by centrifugation and resuspended in phosphate-buffered saline (PBS). The inocula were normalized by OD600 to ∼1011 CFU/ml, and animals were inoculated intragastrically with 100 μl of the cell suspension. The inoculum doses were validated on MH agar plates prior to and immediately after infection of the animals. All animal experiments were conducted in compliance with the Animal Welfare Act and in accordance with the principles set forth in the Guide for the Care and Use of Laboratory Animals (15).
Assessment of colonization.
Fecal collections were performed by allowing individual mice to defecate in clean, empty shoebox cages prior to returning to group housing. Feces were collected using forceps into 5-ml Falcon snap-cap tubes and then diluted 1:10 by weight into PBS. Various stool dilutions were plated onto campylobacter selective media (CVA plates; Remel) and incubated under microaerobic conditions at 42°C for 2 days.
Lymphocyte isolation.
At various time points following oral challenge, mice were sacrificed and small intestines were removed to recover lamina propria lymphocytes (LPLs) as described previously (16), with some modifications. In brief, Peyer's patches (PP) were removed from the intestines and intestines were cleared of contents using forceps, opened longitudinally, and then cut into ∼5-mm sections. Intraepithelial lymphocytes (IELs) were removed from these intestinal sections by placing the tissue in a solution of 1 mM dithiothreitol (DTT) and 1 mM EDTA at 37°C for two 20-min incubations. After each incubation, the supernatant was removed and replaced with fresh DTT-EDTA. To isolate LPLs, the remaining intestinal pieces were digested with collagenase D (Roche; 1 mg/ml) and DNase I (Sigma; 40 μg/ml) for two 1-h incubations at 37°C. The supernatant was removed following each incubation and replaced with fresh medium. Following the digestion of small intestinal tissue sections, cells were pelleted by centrifugation and LPLs were isolated using a discontinuous (80 to 40%) Percoll gradient.
Intracellular cytokine staining.
LPLs were cultured in vitro for 4 to 6 h in the presence of medium alone or phorbol-12-myristate-13-acetate (PMA) (Sigma; 20 ng/ml) and ionomycin (Sigma; 0.5 μg/ml). Medium was Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, nonessential amino acids, 50 μM 2-mercaptoethanol, penicillin (100 IU/ml), and streptomycin (100 μg/ml). Protein transport was inhibited with the addition of brefeldin A (10 μg/ml). Following culture, LPLs were stained with CD4 allophycocyanin (APC). Cells were fixed in 4% formaldehyde prior to permeabilization with 0.1% saponin (in PBS and 1% FBS). Intracellular staining was performed using anti-mouse gamma interferon (IFN-γ) fluorescein isothiocyanate (FITC) (eBioscience) or anti-mouse interleukin 17 (IL-17) phycoerythrin (PE) (eBioscience). Cells were then washed and resuspended in 1% formaldehyde prior to analysis on a Becton, Dickinson FACScan equipped with red and blue lasers (i.e., 5-color capability). Data were analyzed using FlowJo software (TreeStar).
TLR signaling assay using whole bacteria.
The following cell lines were purchased from InvivoGen: HEK-Blue-hTLR4, HEK-Blue-hTLR2, and THP1-XBlueTM-MD2-CD14. Human epithelial kidney (HEK) 293 cells are stably transfected with either human Toll-like receptor 4 (hTLR4), MD2, and CD14 (HEK-Blue-hTLR4) or hTLR2 and CD14 (HEK-Blue-hTLR2). THP1-XBlueTM-MD2-CD14 cells are derived from the human monocytic THP-1 cell line and are stably transfected with MD2 and CD14. These HEK-Blue and THP1-XBlue clones also stably express secreted embryonic alkaline phosphatase (SEAP) under the control of a promoter inducible by NF-κB and activator protein 1 (AP-1). Thus, stimulation of Toll-like receptors will result in an amount of extracellular SEAP in the supernatant that is proportional to the level of NF-κB induction. The HEK cell lines were maintained in standard DMEM with 10% heat-inactivated FBS (Gibco) supplemented with 4.5 g/liter of glucose, 2 mM l-glutamine, 50 U/ml of penicillin, 50 μg/ml of streptomycin, 100 μg/ml of Normocin (InvivoGen), and 1× HEK-Blue selection (InvivoGen) in a 5% saturated CO2 atmosphere at 37°C. The THP-1 cell line was maintained in standard RPMI 1640 medium with 10% heat-inactivated FBS (Gibco) supplemented with 4.5 g/liter of glucose, 2 mM l-glutamine, 1.5 g/liter of sodium bicarbonate, 10 mM HEPES, 1 mM sodium pyruvate, 50 U/ml of penicillin, 50 μg/ml of streptomycin, 100 μg/ml of Normocin (InvivoGen), 200 μg/ml of Zeocin (InvivoGen), and 250 μg/ml of G418 (InvivoGen) in a 5% saturated CO2 atmosphere at 37°C.
The induction of TLR signaling in HEK-Blue-hTLR4, HEK-Blue-hTLR2, and THP1-XBlueTM-MD2-CD14 clones was assessed by measuring SEAP activity using the QUANTI-Blue colorimetric assay (InvivoGen). The assays were performed according to the manufacturer's protocols. Briefly, cells were seeded in a 96-well plate in triplicate (2.5 × 104 cells/well for HEK-Blue-hTLR4, 5 × 104 cells/well for HEK-Blue-hTLR2, and 1 × 105 cells/well for THP1-XBlueTM-MD2-CD14). Whole bacterial cells were grown on Mueller-Hinton agar for 16 h, collected from the plate, washed, and resuspended in sterile PBS. Serial dilutions (10-fold) were prepared based on OD600 to yield the number of bacteria inoculated into each well. CFU were confirmed by plating serial dilutions on MH agar. Erythromyin (50 μg/ml) was included to prevent bacterial growth during incubation, and other antibiotics used in the media for cell propagation were omitted in the assay. After 18 h of incubation, supernatants (20 μl) were transferred to a 96-well plate and incubated at 37°C with QUANTI-Blue (180 μl). SEAP activity was measured by reading the OD655 with a Synergy Mx multimode microplate reader (BioTek).
Statistical analyses.
Differences in mouse colonization level, as assessed by the number of organisms shed (log10 CFU/gram of feces), were compared using a repeated-measures analysis of variance with the C. jejuni strain as the between-animal factor (i.e., wild type, kpsM mutant, and mpnC mutant) and collection time points as the repeated factor. The covariance structure was modeled using a first-order antedependence model. A Tukey adjustment was utilized to control the type I error rate. Comparisons of the proportions of mice infected by strains over time were made using a Cox proportional-hazards model. These analyses were conducted with SAS, version 9.2, for Windows (SAS Institute, Inc., Cary, NC), using a two-tailed alpha of 0.05.
Statistical analyses of complement killing, intracellular cytokine expression, and TLR assays were conducted using Student's t test. Differences were considered significant at a P value of <0.05.
RESULTS
Construction of a mutant in the MeOPN biosynthetic pathway of 81-176.
McNally et al. (8) identified the genes in C. jejuni strain NCTC 11168 (CJ1415 to CJ1418) that were responsible for MeOPN synthesis, as well as two distinct MeOPN transferases that were responsible for attachment of MeOPN to two different sites in the CPS of this strain. CJ1415 to CJ1418 are highly conserved among C. jejuni strains, while the transferases are more variable based on differences in attachment of the MeOPN to sugars. The genes corresponding to CJ1415 to CJ1418 in 81-176 are CJJ81176_1414 to CJJ81176_1417. Since the function of genes has been established in NCTC 11168, we have named the genes for MeOPN synthesis mpnA to -D, shown in Fig. 1A, for clarity in discussing these conserved genes in different strains. A mutant with a mutation in mpnC in 81-176 was shown to lack MeOPN by 31P NMR, as predicted based on the NCTC 11168 data (8), and MeOPN was restored when the mutant was complemented (Fig. 1). The mpnC mutant produced CPS as determined by both NMR and immunoblotting with rabbit polyclonal antibody to whole cells of 81-176 (data not shown).
MeOPN contributes to serum resistance.
There have been several reports demonstrating that nonencapsulated mutants of C. jejuni are more sensitive to normal human serum than wild-type strains (1, 17). Data comparable to those published for 81-176 and its isogenic kpsM mutant (1) are shown in Fig. 2. Surprisingly, the mpnC mutant, expressing the polysaccharide CPS lacking MeOPN, displayed the same pattern of serum killing as the kpsM mutant, which lacked all CPS (Fig. 2). The mpnC mutant was significantly more sensitive than the wild type to complement at both 60 min (P < 0.001) and 120 min (P < 0.005). When the mpnC mutant was complemented in trans, serum resistance returned to levels comparable to those of the wild type (Fig. 2).
Fig 2.
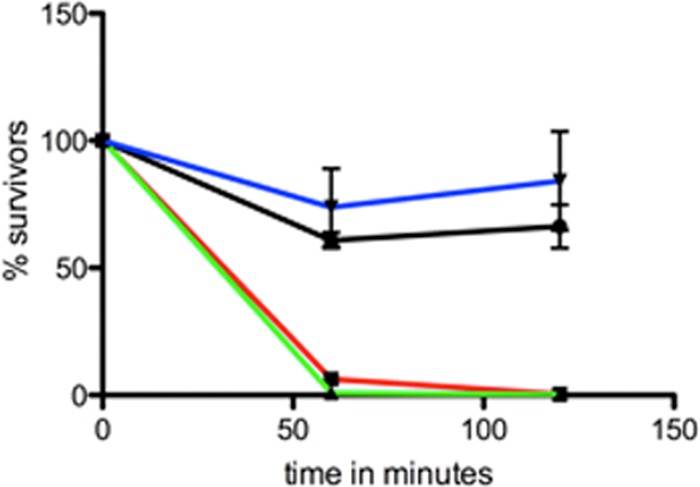
Sensitivities of 81-176 and mutants to complement killing by NHS. The percentages of survivors after 60 and 120 min of incubation with 10% NHS are shown. Black, 81-176; red, kpsM mutant; green, mpnC mutant; blue, mpnC mutant complemented in trans.
Capsule is required for prolonged mouse colonization.
The abilities of the kpsM and mpnC mutants to colonize mice were compared to that of the wild type in a series of experiments. Animals were intragastrically infected with C. jejuni, and colonization was monitored postinfection by fecal shedding. Following infection, wild-type C. jejuni 81-176 colonized mice on average at levels exceeding 106 CFU/g of feces (Fig. 3A). This high level of colonization was maintained for more than 15 days before counts began to drop below the initial colonization levels. Mice infected with the kpsM mutant generally had early colonization levels similar to that of the wild type (Fig. 3A). In addition, the kpsM-infected mice had a shorter duration of colonization than did those infected with the wild type (P = 0.06). The majority of kpsM-infected mice cleared the infection by day 18, in contrast to the wild-type-infected mice, which remained colonized at some level through day 28 postinfection (the last day tested) (Fig. 3A). Thus, despite the similar levels of colonization seen early postinfection, by day 14, there was a statistically significant difference in colonization levels of mice infected with kpsM compared to those infected with the wild type (P < 0.01).
Fig 3.
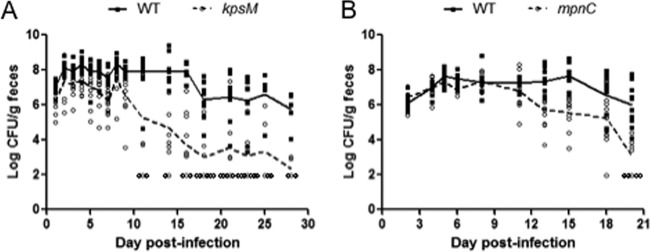
Colonization of BALB/c mice by C. jejuni strain 81-176 and various CPS mutants. The log CFU/g of feces shed by the wild type and either a kpsM mutant (A) or an mpnC mutant (B) are shown over the course of infection. Groups of 10 mice were intragastrically infected with ∼1010 CFU. Each data point represents an individual mouse infected with the wild type (squares) or a mutant (circles), and the group mean is displayed as a connected line (solid for the wild type and dashed for mutants). The limit of detection was 102 CFU/g of feces. These data are representative of 3 or 4 independent experiments.
In parallel experiments, the colonization capacity of the mpnC mutant was compared to that of the wild type. Upon infection with the mpnC mutant, mice shed numbers of C. jejuni organisms in their stool similar to those shed by mice infected with the wild type (Fig. 3B). In fact, there were no significant differences in stool counts between the mpnC mutant and the wild type during the first 10 days postinfection, and only later in infection did the mpnC mutant demonstrate a significant reduction in the level of colonization compared to that of the wild type (Fig. 3B; P = 0.02 for day 20). Thus, although not directly compared, the colonization ability of the mpnC mutant appeared intermediate in nature compared to those of the wild type and the kpsM mutant (Fig. 3).
IL-17 expression from intestinal T cells is modulated by the CPS.
To determine if CPS had a role on immune responses in vivo, mice were orally infected with wild-type 81-176 and the mpnC or kpsM strain. At selected times postinfection, T cells were isolated from small intestine Peyer's patches, the epithelium, and the lamina propria. Following an ex vivo restimulation, the levels of expression of IL-17 and IFN-γ were determined using intracellular cytokine staining and flow cytometry. Figure 4A and B show representative histograms for CD4 LPLs and dot plots demonstrating gating strategies for intracellular cytokine staining. Following infection with either the kpsM or wild-type strain, CD4 cells from the lamina propria of kpsM mutant-infected mice exhibited significantly higher percentages (P < 0.05) of CD4+ cells that expressed IL-17 at day 7 and day 21 (Fig. 4C). No difference was observed in IFN-γ expression of CD4+ LPLs isolated from kpsM mutant- or wild-type-infected mice at either time point. Differences in cytokine expression were not observed from PP cells or IELs (data not shown).
Fig 4.
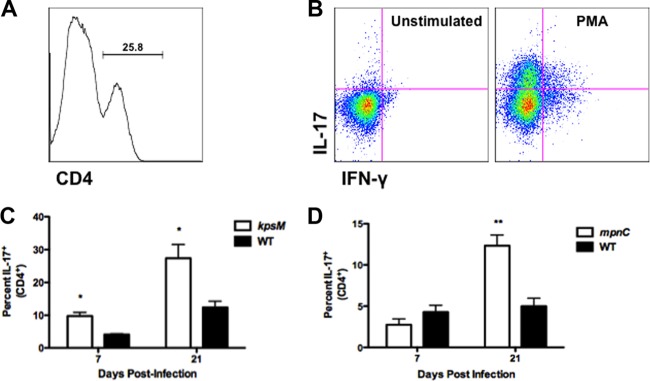
IL-17 expression is reduced in small intestinal CD4+ LPLs from mice infected with wild-type C. jejuni 81-176. BALB/c mice (4 or 5/group) were orally infected with ∼1010 CFU of C. jejuni. At days 7 and 21 postinfection, small intestines were removed and processed to isolate LPLs. LPLs were restimulated in vitro with PMA (20 ng/ml) and ionomycin (500 ng/ml) for 4 to 6 h. Protein transport was inhibited by addition of brefeldin A (10 μg/ml). Intracellular cytokine staining for IL-17 and IFN-γ was performed on cells and analyzed by flow cytometry. (A) Representative histograms demonstrating the percentages of CD4+ and CD8+ cells isolated from mouse small intestines. (B) Representative dot plots demonstrating intracellular staining for IL-17 and IFN-γ in unstimulated and stimulated CD4+ LPLs. (C) Percent expression of IL-17 in CD4+ LPLs from mice infected with kpsM mutant or wild-type C. jejuni. (D) IL-17 expression in CD4+ LPLs from mice infected with mpnC mutant or wild-type C. jejuni. Data represent the means ± standard errors of the means. *, P < 0.05; **, P < 0.01 (Student's t test). Data are representative of 2 or 3 independent experiments.
Next, the MeOPN modification on CPS was examined for its role in modulating immune responses in vivo. CD4+ LPLs from mpnC-infected mice did not express significantly higher percentages of IL-17+ cells on day 7 than did LPLs isolated from mice orally challenged with wild-type 81-176 (Fig. 4D). However, on day 21 postinfection, mice colonized by mpnC exhibited significantly higher percentages (P < 0.01) of CD4+ LPLs expressing IL-17 than animals infected with the wild type. In addition, no significant differences were seen in other lymphocyte subsets or in IFN-γ expression patterns (data not shown).
Effects of CPS on TLR signaling.
To determine the impact of CPS production and modification on TLR activation, we performed reporter cell signaling assays with whole bacteria. The kpsM mutant exhibited significantly higher activation than the wild type from 104 to 107 CFU for hTLR4 activation and from 105 to 108 CFU for hTLR2 activation (Fig. 5). Although similar results were seen with the mpnC mutant, the lack of complete complementation confounds these results (data not shown).
Fig 5.
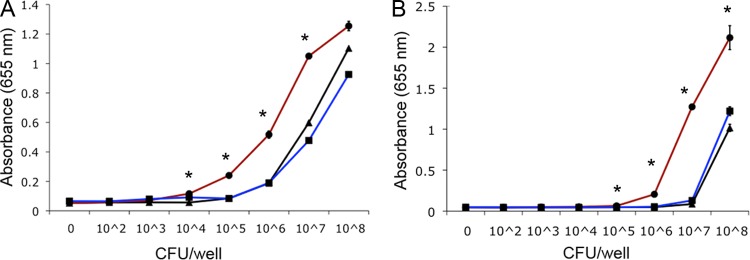
Activation of TLR4 in HEK 293 cells transfected with human TLR4-MD2-CD14 (A) and TLR2 in HEK 293 cells stably transfected with human TLR2-CD14 (B). TLR activation was monitored colorimetrically using a SEAP reporter gene placed under the control of an NF-κB inducible promoter. Tenfold serial dilutions of whole bacterial cells of the indicated strains of C. jejuni were added to each well in triplicate. Values represent the means and standard deviations of one experiment assayed in triplicate. The graphs are representative of three independent experiments. Asterisks indicate a P value of <0.005 compared to wild-type 81-176 or the complement. Black lines, wild-type 81-176; red lines, the isogenic kpsM mutant; blue lines, the kpsM mutant complemented in trans.
We subsequently compared the overall TLR activation of kpsM and mpnC mutants using a human monocytic reporter line that expresses several TLRs, including TLR1, TLR2, TLR4, TLR6, TLR8, NOD1, and NOD2. Significant increases (P < 0.005) in signaling were observed for both mutant strains compared to the wild type and their complements (Fig. 6). For the kpsM mutant, these differences were observed from 104 to 107 CFU, and for the mpnC mutant, the differences were observed between 105 and 107 CFU.
Fig 6.
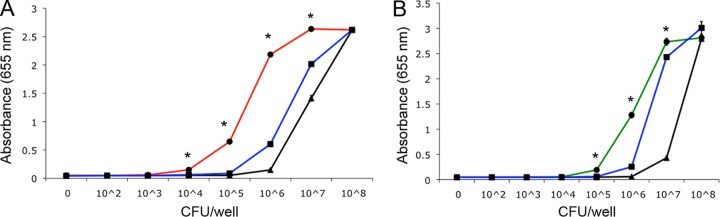
Activation of Toll-like receptors using a human monocytic (THP-1) reporter cell line by kpsM (A) and mpnC (B) mutants compared to wild-type 81-176 and complements of each mutant. Bacteria were added to cells as described in the legend to Fig. 5. Values represent the means and standard deviations of one experiment assayed in triplicate. The graphs are representative of three independent experiments. Asterisks indicate a P value of <0.005 compared to wild-type 81-176 or the complement. Black lines, 81-176; red line, the isogenic kpsM mutant; green line, the isogenic mpnC mutant; blue lines, the complement of each mutant.
DISCUSSION
C. jejuni remains a poorly understood pathogen, in part because of the absence of small-animal models that mimic human disease. Following orogastric infection with C. jejuni, adult, immunocompetent mice can become colonized for variable lengths of time, but without the disease symptoms seen in immunodeficient mice (18–23). Despite the lack of disease, the mouse model can provide information on traits required for colonization, the first step in pathogenesis (23). Here, we show that wild-type 81-176 colonizes BALB/c mice better than either an isogenic mutant lacking capsule (kpsM mutant) or a mutant expressing CPS without MeOPN (mpnC mutant). Interestingly, a reduction in colonization ability of an MeOPN-negative mutant of 81-176 (in the gene corresponding to mpnA) compared to wild-type 81-176 was also reported with MyD88-defective mice (24). In our studies, BALB/c mice that were colonized with wild-type 81-176 remained colonized for the duration of the experiments (>21 days). The kpsM mutant showed similar colonization levels for about 9 days before colonization levels dropped. The mpnC mutant colonized at levels that were generally lower than those of the wild type, although statistical significance was reached only at day 20. Thus, expression of CPS by C. jejuni facilitated colonization in the mouse model. CPS has also been shown to play a role in C. jejuni colonization of chickens (25, 26).
Following restimulation, IL-17 production by CD4+ LPLs was reduced in mice colonized by C. jejuni 81-176 compared to both mutant strains. Mice colonized by either the kpsM or mpnC mutant possessed higher levels of IL-17+ CD4+ cells in the small intestine than did mice colonized by the wild type at day 21, and this increased IL-17 production was associated with a reduction in colonization levels. However, despite the fact that both the kpsM mutant and the mpnC mutant were associated with higher frequencies of IL-17-producing CD4+ cells in the small intestine than was wild-type 81-176, the kpsM mutant appeared to show a greater reduction in colonization capacity than the mpnC mutant, suggesting that the presence of the CPS, even without the MeOPN modification, affords some protection against the immune response in the intestine. Although in vivo cytokine responses were not measured directly in this study, the data suggest that CPS expression, and more specifically, the MeOPN modification on the wild-type capsule, may affect the generation of IL-17 responses in the gut mucosa. Future studies are needed to determine the specificity of the IL-17 response against C. jejuni in the intestine.
T helper 17 (Th17) responses have come into focus due to their roles in maintaining intestinal homeostasis (27, 28) and protective immune responses against enteric pathogens (28–31). The gut Th17 response is composed of both innate and adaptive immune system components. Innate Th17 (iTh17) responses are induced by segmented filamentous bacteria (SFB) that colonize the gut (32, 33) and maintain a symbiotic balance between the microbiota and host (28). Specific animal vendors supply mice that are either colonized with SFB or not (Jackson Laboratories, SFB−; Taconic, SFB+) (33). These models can be exploited to evaluate innate or adaptive Th17 responses. Whereas iTh17 responses can be induced relatively quickly by cytokine signals (29, 30), adaptive Th17 responses occur later (days to weeks) (34) and are antigen-specific responses. In our present study, the Th17 responses likely represented an adaptive immune response, since Jackson Laboratory mice were used. Key to Th17 responses are the cytokines IL-17 and IL-22 and the upstream cytokines that lead to their expression, such as IL-1, IL-6, and IL-23 (34–36). IL-17 is primarily thought to be effective against extracellular pathogens by inducing inflammation and recruiting neutrophils to sites of infection (reviewed in reference 35). IL-22 exerts its protective effects by inducing epithelial cells to produce antibacterial molecules (37). Recently, Th17 responses have been demonstrated to have protective roles against Salmonella and Citrobacter infections in mice (29, 30). To date, only limited data exist regarding Th17 responses and Campylobacter infection. Edwards et al. (38) showed that cytokines involved in Th17 responses were induced in colon biopsy tissues following coculture with C. jejuni and that the addition of exogenous IL-17 reduced C. jejuni invasion into an intestinal epithelial cell line. However, additional work must be performed to determine the precise role of Th17 immune responses to C. jejuni.
Consistent with these data, we have also shown that the presence of the CPS on wild-type 81-176 resulted in reduced activation of both TLR2 and TLR4 using HEK cells expressing each receptor. Our data are consistent with those of Rose et al. (3), who showed that mutants of NCTC 11168 lacking CPS or MeOPN induced higher levels of IL-6, tumor necrosis factor alpha (TNF-α), and IL-10 from mouse dendritic cells than did the wild-type strain. Using dendritic cells from TLR4−/− mice, they also showed that some of these differences in cytokines were due to TLR4 signaling. Similar downregulation of the immune response has been observed for other bacterial capsules (39–44). In some cases, this inhibition may be due to shielding of the bacterial surface by the capsule and prevention of TLR stimulation. However, the CPS of Neisseria meningitidis actively inhibits TLR2 activation by binding CD14 (45). The MeOPN modification on two distinct CPS structures of C. jejuni has now been shown to modulate cytokine responses and TLR signaling (3), suggesting an active role for this unusual structure. Similarly, the C. jejuni CPS may inhibit binding of complement activators and components to the surface of the bacterial cell, but the fact that the mpnC mutant was as sensitive as the kpsM mutant to complement killing also suggests an active role for the MeOPN group. In contrast to C. jejuni, modification of Haemophilus influenzae lipopolysaccharide with phosphorycholine, which is also under phase-variable expression, enhances sensitivity to complement killing (46). The mechanism by which MeOPN interacts with components of the complement cascade is under investigation.
Collectively, these data indicate that CPS of 81-176 and the MeOPN modification modulate the immune response to this pathogen and are consistent with previous observations suggesting a stealth strategy by which C. jejuni may avoid the immune response. It has been known for some time that C. jejuni flagellin is unable to induce TLR5 because of structural changes to the monomeric subunit protein that are reflected in changes in filament formation (47, 48). C. jejuni also expresses altered linkages of hydroxyacyl chains on lipid A that reduce TLR4 activation (49), and there is evidence that the N-linked glycan on proteins and certain lipooligosaccharide glycoforms can downregulate IL-6 induction (50). Previous work has shown that the CPS of NCTC 11168, and specifically the MeOPN modification on this CPS, reduced cytokine production from mouse dendritic cells in culture (3). Here, we have demonstrated that a second C. jejuni CPS and the MeOPN modification modulate the immune response at multiple levels, including resistance to complement killing and cytokine induction via NF-κB signaling. The ability to avoid the immune response of the host provides an advantage in establishing colonization by C. jejuni, be it as a commensal in animals or as a pathogen in humans. Moreover, asymptomatic infection by C. jejuni is common among children in the developing world, and acute infections are frequently followed by periods of asymptomatic shedding (51–53), which may be due, at least in part, to the ability of this pathogen to avoid the host immune response. Similarly, recrudescence of infection following appropriate antibiotic treatment in an immunocompetent adult has been reported (54).
One of the hallmark characteristics of C. jejuni is its ability to undergo phase variation of surface antigens by slip strand mismatch repair (1, 7, 55–58). In terms of the CPS, this phase variation occurs at two levels. One is the high frequency on/off reversible expression that was originally described to occur in strain 81-176, such that a culture grown in vitro is a mixed population of encapsulated and unencapsulated variants (1). The other level of phase variation affects CPS structure and is best understood in terms of the MeOPN modification. Thus, all MeOPN transferases that have been described to date are subject to phase variation at homopolymeric tracts of bases, resulting in nonstoichiometric amounts of this modification. The reason for this variability in both CPS expression and structure is not understood, but the data presented here suggest that the CPS, with and without MeOPN, modulates the host immune response at multiple levels. Since C. jejuni produces an inflammatory diarrhea, phase variation during replication in vivo may also modulate the severity of illness and, at least in part, explain variability in severity of symptoms seen with this pathogen.
ACKNOWLEDGMENTS
Work at NMRC was funded by NIAID R56 AI080593 (to P.G.) and Navy Work Unit 6000.RAD1.DA3.A0308. Work at UT Austin was funded by NIAID grants AI064184 and AI76322 and ARO grant 61789-MA-MUR (to M.S.T.). Work at the University of Guelph was funded by NSERC.
We thank Dawn Pattarini for technical assistance.
The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, or the U.S. government. P.G. and C.K.P. are employees of the U.S. government. This work was prepared as part of their official duties.
Footnotes
Published ahead of print 17 December 2012
REFERENCES
- 1.Bacon DJ, Szymanski CM, Burr DH, Silver RP, Alm RA, Guerry P. 2001. A phase variable capsule is involved in virulence of Campylobacter jejuni 81-176. Mol. Microbiol. 40:769–777 [DOI] [PubMed] [Google Scholar]
- 2.Champion OL, Karlyshev AV, Senior NJ, Woodward M, La Ragione R, Howard SL, Wren BW, Titball RW. 2010. Insect infection model for Campylobacter jejuni reveals that O-methyl phosphoramidate has insecticidal activity. J. Infect. Dis. 201:776–782 [DOI] [PubMed] [Google Scholar]
- 3.Rose A, Kay E, Wren BW, Dallman MJ. 2012. The Campylobacter jejuni NCTC 11168 capsule prevents excessive cytokine production by dendritic cells. Med. Microbiol. Immunol. 201:137–144 [DOI] [PubMed] [Google Scholar]
- 4.Karlyshev AV, Linton D, Gregson NA, Lastovica AJ, Wren BW. 2000. Genetic and biochemical evidence of a Campylobacter jejuni capsular polysaccharide that accounts for Penner serotype specificity. Mol. Microbiol. 35:529–541 [DOI] [PubMed] [Google Scholar]
- 5.Guerry P, Szymanski CM. 2008. Campylobacter sugars sticking out. Trends Microbiol. 16:428–435 [DOI] [PubMed] [Google Scholar]
- 6.Karlyshev AV, Champion OL, Churcher C, Brisson J-R, Jarrell HC, Gilbert M, Brochu D, St Michael F, Li J, Wakarchuk WW, Goodhead I, Sanders M, Stevens K, White B, Parkhill J, Wren BW, Szymanski CM. 2005. Analysis of Campylobacter jejuni capsular loci reveals multiple mechanisms for the generation of structural diversity and the ability to form heptoses. Mol. Microbiol. 55:90–103 [DOI] [PubMed] [Google Scholar]
- 7.Parkhill J, Wren BW, Mungall K, Ketley JM, Churcher C, Basham D, Chillingworth T, Davies RM, Feltwell T, Holroyd S, Jagels K, Karlyshev AV, Moule S, Pallen MJ, Penn CW, Quail MA, Rajandream MA, Rutherford KM, van Vliet AHM, Whitehead S, Barrell BG. 2000. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable tracts. Nature 403:665–668 [DOI] [PubMed] [Google Scholar]
- 8.McNally DJ, Lamoureux MP, Karlyshev AV, Fiori LM, Li J, Thacker G, Coleman RA, Khieu NH, Wren BW, Brisson JR, Jarrell HC, Szymanski CM. 2007. Commonality and biosynthesis of the O-methyl phosphoramidate capsule modification in Campylobacter jejuni. J. Biol. Chem. 282:28566–28576 [DOI] [PubMed] [Google Scholar]
- 9.Monteiro MA, Baqar S, Hall ER, Chen Y-H, Porter CK, Bentzel DE, Applebee L, Guerry P. 2009. A capsule polysaccharide conjugate vaccine against diarrheal disease caused by Campylobacter jejuni. Infect. Immun. 77:1128–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ewing CP, Andreishcheva E, Guerry P. 2009. Functional characterization of flagellin glycosylation in Campylobacter jejuni 81-176. J. Bacteriol. 191:7086–7093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guerry P, Ewing CP, Hickey TE, Prendergast MM, Moran AP. 2000. Sialylation of lipooligosaccharide cores affects immunogenicity and serum resistance of Campylobacter jejuni. Infect. Immun. 68:6656–6662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanipes MI, Tan X, Akelaitis A, Li J, Rockabrand D, Guerry P, Monteiro MA. 2008. Genetic analysis of lipooligosaccharide core biosynthesis in Campylobacter jejuni 81-176. J. Bacteriol. 190:1568–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yao R, Alm RA, Trust TJ, Guerry P. 1993. Construction of new Campylobacter cloning vectors and a new mutational cat cassette. Gene 130:127–130 [DOI] [PubMed] [Google Scholar]
- 14.Guerry P, Ewing CP, Schirm M, Lorenzo M, Kelly J, Pattarini D, Majam G, Thibault P, Logan SM. 2006. Changes in flagellin glycosylation affect Campylobacter autoagglutination and virulence. Mol. Microbiol. 60:299–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Institute of Laboratory Animals Resources, National Research Council 1966. Guide for the care and use of laboratory animals. National Academy Press, Washington, DC [Google Scholar]
- 16.Lefrançois L, Lycke N. 2001. Isolation of mouse small intestinal intraepithelial lymphocytes, Peyer's patch and lamina propria cells. Curr. Protoc. Immunol. 17:3.19.1–3.19.16 doi:10.1002/0471142735.im0319s17. [DOI] [PubMed] [Google Scholar]
- 17.Keo T, Collins J, Kunwar P, Blaser MJ, Iovine NM. 2011. Campylobacter capsule and lipooligosaccharide confer resistance to serum and cationic antimicrobials. Virulence 2:30–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berndtson E, Danielsson-Tham ML, Engvall A. 1994. Experimental colonization of mice with Campylobacter jejuni. Vet. Microbiol. 41:183–188 [DOI] [PubMed] [Google Scholar]
- 19.Blaser MJ, Duncan DJ, Warren GH, Wang WLL. 1983. Experimental Campylobacter jejuni infection of adult mice. Infect. Immun. 39:908–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fox JG. 1992. In vivo models of enteric campylobacteriosis: natural and experimental infections, p 131–138. In Nachamkin I, Blaser MJ M. J., Tompkins LS. (ed), Campylobacter jejuni: current status and future trends. American Society for Microbiology, Washington, DC [Google Scholar]
- 21.Hodgson AE, McBride BW, Hudson MJ, Hall G, Leach S. 1998. Experimental campylobacter infection and diarrhoea in immunodeficient mice. J. Med. Microbiol. 47:799–809 [DOI] [PubMed] [Google Scholar]
- 22.Mansfield LS, Bell JA, Wilson DL, Murphy AJ, Elsheikha HM, Rathinam VAK, Fierro BR, Linz JE, Young VB. 2007. C57BL/6 and congenic interleukin-10-deficient mice can serve as models of Campylobacter jejuni colonization and enteritis. Infect. Immun. 75:1099–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naito M, Frirdich E, Fields JA, Pryjma M, Li J, Cameron A, Gilbert M, Thompson SA, Gaynor EC. 2010. Effects of sequential Campylobacter jejuni 81-176 lipooligosaccharide core truncations on biofilm formation, stress survival and pathogenesis. J. Bacteriol. 192:182–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watson RO, Novik V, Hofreuter D, Lara-Tejero M, Galan JE. 2007. A MyD88-deficient mouse model reveals a role for Nramp1 in Campylobacter jejuni infection. Infect. Immun. 75:1994–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bachtiar BM, Coloe PJ, Fry BN. 2007. Knockout mutagenesis of the kpsE gene of Campylobacter jejuni 81116 and its involvement in bacterium-host interactions. FEMS Immunol. Med. Microbiol. 49:149–154 [DOI] [PubMed] [Google Scholar]
- 26.Grant AJ, Coward C, Jones MA, Woodall CA, Barrow PA, Maskell DJ. 2005. Signature-tagged transposon mutagenesis studies demonstrate the dynamic nature of cecal colonization of 2-week old chickens by Campylobacter jejuni. Appl. Environ. Microbiol. 71:8031–8041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hooper LV, Littman DR, Macpherson AJ. 2012. Interactions between the microbiota and the immune system. Science 336:1268–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, Shibata N, Grunberg S, Sinha R, Zahm AM, Tardif MR, Sathaliyawala T, Kubota M, Farber DL, Collman RG, Shaked A, Fouser LA, Weiner DB, Tessier PA, Friedman JR, Kiyono H, Bushman FD, Chang KM, Artis D. 2012. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science 336:1321–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geddes K, Rubino SJ, Magalhaes JG, Streutker C, LeBourhis L, Cho JH, Robertson SJ, Kim CJ, Kaul R, Philpott DJ, Girardin SE. 2011. Identification of an innate T helper type 17 response to intestinal bacterial pathogens. Nat. Med. 17:837–844 [DOI] [PubMed] [Google Scholar]
- 30.Mayuzumi H, Inagaki-Ohara K, Uyttenhouve C, Okamoto Y, Matsuzaki G. 2010. Interleukin 17-A is required to suppress invasion of Salmonella enterica serovar Typhimurium to enteric mucosa. Immunology 131:377–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song X, Zhu S, Shi P, Liu Y, Shi Y, Levin SD, Qian Y. 2011. IL-17RE is the functional receptor for IL-17C and mediates mucosal immunity to infection with intestinal pathogens. Nat. Immunol. 12:1151–1158 [DOI] [PubMed] [Google Scholar]
- 32.Gaboriau-Routhiau V, Rakotobe S, Lecuyer E, Mulder I, Lan A, Bridonneau C, Rochet V, Pisi A, De Paepe M, Brandi G, Eberl G, Snel J, Kelly D, Cerf-Bensussan N. 2009. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 31:677–689 [DOI] [PubMed] [Google Scholar]
- 33.Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, Littman DR. 2009. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139:485–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rubino SJ, Geddes K, Girardin SE. 2012. Innate IL-17 and IL-22 responses to enteric bacterial pathogens. Trends Immunol. 33:112–118 [DOI] [PubMed] [Google Scholar]
- 35.Korn T, Bettelli E, Oukka M, Kuchroo VK. 2009. IL-17 and Th17 cells. Annu. Rev. Immunol. 27:485–517 [DOI] [PubMed] [Google Scholar]
- 36.Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. 2006. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 203:2271–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W. 2008. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 14:282–289 [DOI] [PubMed] [Google Scholar]
- 38.Edwards LA, Nistala K, Mills DC, Stephenson HN, Zilbauer M, Wren BW, Dorrell N, Lindley KJ, Wedderburn LR, Bajaj-Elliott M. 2010. Delineation of the innate and adaptive T-cell immune outcome in the human host in response to Campylobacter jejuni infection. PLoS One 5:e15398 doi:10.1371/journal.pone.0015398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dongari-Bagtzoglou AI, Ebersole JL. 1996. Production of inflammatory mediators and cytokines by human gingival fibroblasts following bacterial challenge. J. Periodontal Res. 31:90–98 [DOI] [PubMed] [Google Scholar]
- 40.Raffatellu M, Chessa D, Wilson RP, Tukel C, Akcelik M, Baumler AJ. 2006. Capsule-mediated immune evasion: a new hypothesis explaining aspects of typhoid fever pathogenesis. Infect. Immun. 74:19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raffatellu M, Chessa D, Wilson RP, Dusold R, Rubino S, Baumler AJ. 2005. The Vi capsular antigen of Salmonella enterica serotype typhi reduces Toll-like receptor-dependent interleukin-8 expression in the intestinal mucosa. Infect. Immun. 73:3367–3374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raffatellu M, Santos RL, Chessa D, Wilson RP, Winter SE, Rossetti CA, Lawhon SD, Chu H, Lau T, Bevins CL, Adams LG, Baumler AJ. 2007. The capsule encoding the viaB locus reduces interleukin-17 expression and mucosal innate responses in the bovine intestinal mucosa during infection with Salmonella enterica serotype Typhi. Infect. Immun. 75:4342–4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharma A, Qadri A. 2004. Vi polysaccharide of Salmonella typhi targets the prohibitin family of molecules in intestinal epithelial cells and suppresses early inflammatory responses. Proc. Natl. Acad. Sci. U. S. A. 101:17492–17497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Unkmeir A, Kammerer U, Stade A, Hubner C, Haller S, Kolb-Maurer A, Frosch M, Dietrich G. 2002. Lipooligosaccharide and polysaccharide capsule: virulence factors of Neisseria meningitidis that determine meningococcal interaction with human dendritic cells. Infect. Immun. 70:2454–2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kocabas C, Katsenelson N, Kanswai S, Kennedy MN, Cui X, Blake MS, Segal DM, Akkoyuniu M. 2007. Neisseria meningitidis type C capsular polysaccharide inhibits lipooliogosaccharide-induced cell activation by binding to CD14. Cell. Microbiol. 9:1297–1310 [DOI] [PubMed] [Google Scholar]
- 46.Weiser JN, Pan N, McGowan KL, Musher D, Martin A, Richards J. 1998. Phosphorylcholine on the lipooligosaccharide of Haemophilus influenzae contributes to persistence in the respiratory tract and sensitivity to serum killing mediated by C-reactive protein. J. Exp. Med. 187:631–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Andersen-Nissen E, Smith KD, Strobe KL, Barrett SLR, Cookson BT, Logan SM, Aderem A. 2005. Evasion of toll-like receptor 5 by flagellated bacteria. Proc. Natl. Acad. Sci. U. S. A. 102:9247–9252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galkin VE, Yu X, Bielnick J, Heuser J, Ewing CP, Guerry P, Egelman EH. 2008. Divergence of quaternary structures among bacterial flagellar filaments. Science 320:382–385 [DOI] [PubMed] [Google Scholar]
- 49.van Mourik A, Steeghs L, van Laar J, Meiring HD, Hamstra H-J, van Putten JPM, Wösten MMSM. 2010. Altered linkage of hydroxyacyl chains in lipid A of Campylobacter jejuni reduces TLR4 activation and antimicrobial resistance. J. Biol. Chem. 285:15828–15836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Sorge NM, Bleumink NM, van Vliet SJ, Saeland E, van der Pol WL, van Kooyk Y, van Putten JP. 2009. N-glycosylated proteins and distinct lipooligosaccharide glycoforms of Campylobacter jejuni target the human C-type lectin receptor MGL. Cell. Microbiol. 11:1768–1781 [DOI] [PubMed] [Google Scholar]
- 51.Blaser MJ, Glass RI, Huq MI, Stoll B, Kibriay GM, Alim AR. 1980. Isolation of Campylobacter fetus subspecies jejuni from Bangladeshi children. J. Clin. Microbiol. 12:744–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pazzaglia G, Bourgeois AL, El Diwany K, Nour N, Badran N, Hablas R. 1991. Campylobacter diarrhoea and an association of recent disease with asymptomatic shedding in Egyptian children. Epidemiol. Infect. 106:77–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rajan DP, Mathan VI. 1982. Prevalence of Campylobacter fetus subspecies jejuni in healthy populations in Southern India. J. Clin. Microbiol. 15:749–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baqar S, Tribble D, Carmolli M, Sadigh K, Poly F, Porter C, Larsson CJ, Pierce KK, Guerry P, Campylobacter Study Team, Darsley M, Kirkpatrick B. 2010. Recrudescent Campylobacter jejuni infection in an immunocompetent adult following experimental infection with a well-characterized organism. Clin. Vaccine Immunol. 17:80–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guerry P, Szymanski CM, Prendergast MM, Hickey TE, Ewing CP, Pattarini DL, Moran AP. 2002. Phase variation of Campylobacter jejuni 81-176 lipooligosaccharide affects ganglioside mimicry and invasiveness in vitro. Infect. Immun. 70:787–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hendrixson DR. 2006. A phase-variable mechanism controlling the Campylobacter jejuni FlgR response regulator influences commensalism. Mol. Microbiol. 61:1646–1659 [DOI] [PubMed] [Google Scholar]
- 57.Hendrixson DR. 2008. Restoration of flagellar biosynthesis by varied mutational events in Campylobacter jejuni. Mol. Microbiol. 70:519–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Linton D, Gilbert M, Hitchen PG, Dell A, Morris HR, Wakarchuk WW, Gregson NA, Wren BW. 2000. Phase variation of a β-1,3 galactosyltransferase involved in generation of the ganglioside GM1-like lipooligosaccharide of Campylobacter jejuni. Mol. Microbiol. 37:501–514 [DOI] [PubMed] [Google Scholar]