

Abstract
Secretion of proteins in Gram-negative bacteria is a high-energy-consuming process that requires translocation across two membranes and a periplasmic space composed of a mesh-like layer, the peptidoglycan. To achieve this, bacteria have evolved complex secretion systems that cross these barriers, and in many cases there are specific peptidoglycanases that degrade the peptidoglycan to allow the proper assembly of the secretion machinery. We describe here the identification and characterization of a muramidase in Brucella abortus that participates in the intracellular multiplication in professional and nonprofessional phagocytes. We demonstrated that this protein has peptidoglycanase activity, that a strain with a clean deletion of the gene displayed a defect in the early stages of the intracellular multiplication curve, and that this is dependent on the lytic activity. While neither the attachment nor the invasion of the strain was affected, we demonstrated that it had a defect in excluding the lysosomal marker LAMP-1 but not in acquiring the reticulum endoplasmic marker calnexin, indicating that the gene participates in the early stages of the intracellular trafficking but not in the establishment of the replicative niche. Analysis of the assembly status and functionality of the VirB secretion apparatus indicated that the mutant has affected the proper function of this central virulence factor.
INTRODUCTION
Pathogens adapted to an intracellular lifestyle have evolved sophisticated strategies to avoid or subvert the microbicidal activities of the host cells. These strategies are very diverse and involve a wide repertoire of virulence factors involved in the secretion and translocation, into the host cell, of proteins that highjack the cellular machinery in its own benefit. In many cases, the pathogen resides and multiplies in membrane-contained niches that avoid fusion with lysosomes. This is the case of Brucella spp., Gram-negative bacteria that belong to the alphaproteobacteria group and cause brucellosis, one of the most worldwide-spread zoonoses that affects livestock and humans (1, 2). Brucella is endemic in many developing countries, generating significant economic losses due to reproductive burden and, because of its zoonotic nature, important human health problems in areas with high incidence. The virulence of the bacterium is dependent on its ability to invade professional and nonprofessional phagocytes, avoid the fusion of the vacuole that contains it with the lysosomes, and redirect its traffic in order to generate a replicative niche with endoplasmic reticulum-derived membranes where it will exponentially multiply (3). Many of these activities are completely dependent on the virB system, a type IV secretion system that secretes and translocates into the host cell effector proteins that reprogram the fate of the Brucella-containing vacuole (BCV) and allow the establishment of the replicative niche (4). To date, only a few effector proteins of this secretion system have been identified, and the molecular mechanisms underlying their activities have not been elucidated yet (5–7). Although most of the work performed so far in understanding how this bacterium survives intracellularly has focused on the virB system, it does not mean that this is the only virulence factor that participates in this stage of the life cycle, as the existence of translocated proteins in a virB-independent manner has been described (7), thus suggesting that other secretion systems might be participating in the intracellular life cycle.
Secretion in Gram-negative bacteria is a high-energy-consuming process, as it implicates the passage of the proteins through the inner membrane, the periplasm, the peptidoglycan, and the outer membrane. Because of this, bacteria have evolved a wide range of secretion systems that operate with diverse mechanisms depending on the final localization of the secreted protein (8–11). In many cases, when the protein is secreted to the extracellular space, translocated into the host cell, or anchored in the outer membrane, it must transit through the peptidoglycan, a polymer of sugars and amino acids that form a mesh-like layer outside the plasma membrane. The peptidoglycan is composed of linear chains of N-acetylglucosamine and N-acetylmuramic acid (MurNAc), and the alternating sugars are connected by β-1,4-glycosidic bonds. Each MurNAc is attached to a short (4- to 5-residue) amino acid chain containing, in the majority of Gram-negative organisms, d-alanine and d-glutamic acid (12). Due to its structure, the peptidoglycan imposes an important physical barrier for the assembly of macromolecular complexes that connect the cytoplasm with the outer space or for the trans-envelope transport of proteins or DNA. For this reason, all bacteria encode muramidases that degrade the peptidoglycan in order to allow, for example, the assembly of type III or type IV secretion systems essential in virulence, motility, or conjugation (12). Muramidases are specialized enzymes of the lysozyme superfamily that generate, locally, gaps in the peptidoglycan meshwork necessary for the assembly of these macromolecular systems.
In the present work, we analyzed the function of a gene in Brucella abortus with homology to muramidases of the lysozyme family. We show that this gene encodes an active peptidoglycanase and that the canonical catalytic active site is conserved. We demonstrate that this gene plays an important role in the early stages of intracellular replication in professional and nonprofessional phagocytes but is not required for attachment or invasion. Moreover, we show that the mutant is less effective in excluding the lysosomal marker LAMP-1 from the phagosomes but not in the late stages of the intracellular replication process and that this defect is, most probably, the consequence of an altered assembly and lack of proper function of the VirB secretion system.
MATERIALS AND METHODS
Media and culture conditions.
Brucella strains were grown at 37°C in tryptic soy broth (TSB). E. coli strains were grown at 37°C in Luria-Bertani broth. If necessary, media were supplemented with the appropriate antibiotics at the indicated final concentrations: ampicillin, 100 μg/ml; kanamycin, 50 μg/ml; and nalidixic acid, 5 μg/ml.
Growth assay.
Brucella growth curves were done in tryptic soy broth supplemented with 5 μg/ml of nalidixic acid. Overnight cultures were diluted to an optical density at 600 nm (OD600) of 0.1 and grown at 37°C. At the indicated time, aliquots were taken, and the OD600 was determined.
Recombinant DNA techniques. (i) Construction of plasmid pDK51/sagA.
A DNA fragment of 1,420 bp containing the sagA gene was amplified from B. abortus genomic DNA using primers CC8 (5′-CGCGGATCCTTCGCATCCCAAGTTTCGTCCAC-3′) and CC11 (5′-CCC AAGCTTCGCTTTCCCGAATGCATTATG-3′). This fragment was digested with BamHI and HindIII and ligated to pDK51 plasmid (13) digested with the same enzymes.
Construction of plasmid pDK51/sagA-tr.
In order to clone the sagA gene without the sequence encoding the transmembrane region (sagA-tr), a DNA fragment of 990 bp was amplified from B. abortus genomic DNA using primers CC8 and MDG14 (5′-CCC AAGCTT CGCCCATTGCATCGGGCCG-3′). The resulting fragment was digested with BamHI and HindIII and ligated into pDK51 plasmid digested with the same enzymes.
Site-directed mutagenesis and construction of expression plasmids.
Mutagenesis of the sagA gene was carried out by the PCR-based site-directed mutagenesis. Two point mutations were constructed: E17A (GAA→GCA) and D26A (GAC→GCC). The PCRs were carried out using as the template the pDK51/sagA-tr plasmid with two pairs of mutagenic primers: MDG08 (5′-TGTCTTCTCGGATGCAGGCGGGTATGTC-3′) and MDG09 (5′-CGACATACCCGCCTGCATCCGAGAAGACA-3′) for the GAA→GCA mutation and MDG10 (5′-CGATCATCCGAAAGCCCCCGGCGGCGCAAC-3′) and MDG11 (5′-GTTGCGCCGCCGGGGGCTTTCGGATGATCG-3′) for the GAC→GCC mutation. Both mutations were confirmed by DNA sequencing.
Using primers MDG16 (5′-CGCGGATCC GCCGAAAGGAAATCACCAATGG-3′) and MDG14 (5′-CCC AAGCTT CGCCCATTGCATCGGGCCG-3′), the sagA-tr wild type and point mutants were amplified from the corresponding pDK51/sagA plasmids. The PCR products were digested with BamHI and HindIII and ligated to the expression vector pQE30 (Qiagen; the QIAexpress system) digested with the same enzymes.
Construction of ΔsagA, ΔsagB, and ΔsagA ΔsagB mutant strains.
To construct a B. abortus 2308 ΔsagA mutant strain, the regions flanking the sagA gene were amplified and ligated using the recombinant PCR technique (14). The resulting fragment was digested with BamHI and HindIII and ligated to the pK18mobSacB plasmid digested with the same enzymes. The primers used for PCR amplification were CC8 (5′-CGCGGATCCTTCGCATCCCAAGTTTCGTCCAC-3′) and CC9 (5′-ATTCGTTGCGCCGCCGGGGTCTTT-3′) to amplify a 300-bp upstream region and CC10 (5′-GGCGGCGCAACGAATTCTGGGCTCTCATCCCAAACT-3′) and CC11 (5′-CCC AAGCTTCGCTTTCCCGAATGCATTATG-3′) to amplify a 400-bp downstream region; CC8 and CC11 were used in overlapping PCR.
To construct a B. abortus 2308 ΔsagB mutant strain, the regions flanking the sagB gene were amplified and ligated using the recombinant PCR technique (14). The resulting fragment was digested with BamHI and PstI and ligated to the pK18mobSacB plasmid digested with the same enzymes. The primers used for PCR amplification were MDG1 (5′-CGCGGATCCAAGGAAGTTCGGCTCCACCTC-3′) and MDG2 (5′-AATTTGCCCCTACTTTAGATTTTG-3′) to amplify a 500-bp upstream region and MDG3 (5′-AAGTAGGGGCAAATTATAACTCTCTTTTGAAAACAT-3′) and MDG4 (5′-CCC AACTGCAGAACCGCCGTATTACGCGATCAGCC-3′) to amplify a 500-bp downstream region; MDG1 and MDG4 were used in the overlapping PCR.
In both cases, the resulting plasmids were introduced into B. abortus 2308 by biparental mating using the Escherichia coli S17-λpir strain. Double recombination events (Kms Sacr) were selected, and the gene knockout was confirmed by genomic PCR.
To construct the ΔsagA ΔsagB double mutant strain, the sagB gene was deleted in the ΔsagA mutant.
Construction of complemented strains.
Plasmids pDK51 containing sagA wild-type and mutant genes were introduced into B. abortus 2308 and ΔsagA strains by biparental mating.
Construction of sagA-3×Flag and bpe123-3×Flag merodiploid strains.
To construct a B. abortus 2308 sagA merodiploid strain, the 3× Flag-tagged sagA gene was amplified from the pBBR1-MCS4/sagA-3×Flag plasmid using the primers CC7-Flag (CGCGGATCCACT CTAGAGATCGTCATCCTTGTAA) and MDG16. The resulting fragment was digested with BamHI and ligated to the pK18mobSacB plasmid digested with the same enzyme. The resulting plasmid was introduced into B. abortus 2308 by biparental mating using the E. coli S17-λpir strain. Simple recombination events (Kmr Sacs) were selected, and the presence of the integrated plasmid was confirmed by genomic PCR.
B. abortus 2308 and ΔsagA bpe123-3×Flag merodiploid strains were constructed as indicated for sagA merodiploid construction and by using the plasmid described in reference 7.
Protein expression and purification.
The recombinant poly-histidine-tagged SagA wild type or mutants were expressed in E. coli and purified using nickel affinity chromatography under native conditions. Briefly, E. coli was grown at 37°C at 250 rpm, and the expression was induced with IPTG (isopropyl-β-d-thiogalactopyranoside) at an OD600 of 0.6. Three hours postinduction, cells were harvested and lysed by sonication. Supernatants were recovered and applied to a HisTrap HP column (GE Healthcare). The protein was eluted with 200 mM imidazole. For the peptidoglycanase assays, imidazole was eliminated by dialysis.
Peptidoglycanase activity assay.
Peptidoglycanase activity was determined according to the turbidimetric method (15), based on the decrease in turbidity of a Micrococcus lysodeikticus cell suspension in 50 mM potassium phosphate buffer (pH 6.2). Briefly, 20 μg of purified recombinant SagA was added to 1.9 ml of lyophilized M. lysodeikticus (Sigma) cell suspension (OD450 = 0.75 to 0.8). The decrease in absorbance at 450 nm was monitored during 2 min using a Beckman DU650 spectrophotometer. Each assay was the result of triplicate measurements. The activity was expressed as the rate of decrease in absorbance per minute.
Intracellular replication assays.
A standard antibiotic protection assay was performed in human epithelial cell line HeLa and murine macrophage-like J774 A.1 cells (16). Cells were seeded in 24-well plates in suitable culture medium at 105 cells per ml and incubated overnight at 37°C. Brucella strains were grown in TSB with the appropriate antibiotics for 24 h and diluted in culture medium prior to infection. The suspension was added at the indicated multiplicity of infection (50:1 for J774 A.1 cells and 500:1 for HeLa cells) and centrifuged at 300 × g for 10 min. After 30 min or 1 h (depending on the assay) of incubation at 37°C, cells were washed and fresh medium containing 100 μg per ml of streptomycin and 50 μg per ml of gentamicin was added. At 1, 2, 4, 24, or 48 h postinfection (depending on the assay), cells were washed and lysed with 0.1% Triton X-100. The intracellular number of CFU was determined by direct plating on TSB agar plates.
Immunofluorescence microscopy.
Cells were seeded on glass coverslips and infected as described above. At different times postinfection, cells were washed three times with phosphate-buffered saline (PBS) and fixed for 15 min in 4% paraformaldehyde and then processed for immunofluorescence labeling. Briefly, coverslips were washed three times with PBS and incubated for 15 min with PBS added with 50 mM NH4Cl in order to quench free aldehyde groups. Coverslips were then incubated with the primary antibodies in a PBS-10% horse serum-5% bovine serum albumin-0.1% saponin solution for 1 h at room temperature, washed in PBS, and then incubated with the secondary antibodies in a PBS-10% horse serum-5% bovine serum albumin-0.1% saponin solution under the same conditions. The coverslips were mounted onto glass slides using FluorSave reagent (Calbiochem). Cells were observed in an immunofluorescence microscope (Nikon-Eclipse T2000) using a 100× oil immersion objective. Projections were saved in TIFF format and imported to Adobe Photoshop CS, where images were merged using RGB format. To determine the percentages of bacteria that colocalized with the lysosomal marker LAMP-1, a minimum of 100 intracellular bacteria (visualized by indirect immunofluorescence) were counted. The assays were performed in triplicate. The primary antibodies used were mouse anti-human LAMP-1 H4A3 (Developmental Studies Hybridoma Bank, Department of Biological Sciences, University of Iowa; dilution, 1:200), rabbit anti-human calnexin (Sigma; dilution, 1:1,000), rabbit anti-Brucella polyclonal antibody (dilution, 1:1,500), and mouse anti-O-polysaccharide monoclonal antibody (M84; dilution, 1:1,000) (17).
The secondary antibodies used were goat anti-mouse or goat anti-rabbit Alexa Fluor 568 or 488 antibodies (Molecular Probes, Invitrogen Co.) at a 1:4,000 dilution. For DNA staining, DAPI (4′,6-diamidino-2-phenylindole) dye at 0.5 mg/ml (final concentration) was used.
Adherence and internalization assays.
To determine adhesion and invasion, infected HeLa cells were fixed for 15 min in 4% paraformaldehyde (pH 7.4) at room temperature at 4 h postinfection. Coverslips were washed three times with PBS, incubated for 10 min with PBS added with 50 mM NH4Cl, and, before permeabilization, incubated with the primary antibody rabbit anti-Brucella polyclonal antibody (dilution 1:1,500) in a PBS-10% horse serum solution. Then cells were washed and incubated with the other primary antibody mouse anti-M84 (anti-O-antigen) monoclonal antibody (dilution, 1:1,000) in a PBS-10% horse serum-0.1% saponin solution followed by incubation with the secondary antibodies (Alexa Fluor 568 or 488; Molecular Probes, Invitrogen Co.) in a PBS-10% horse serum-0.1% saponin solution. The coverslips were mounted as described before. Invasion was determined as the number of bacteria positive for both labels versus the ones positive for the anti-mouse antibody labeling. Adhesion was determined counting the number of bacteria associated per 100 HeLa cells.
Western blot analysis. (i) Outer membrane protein analysis.
Cells were harvested at 8,000 × g, resuspended in buffer A (15 mM Tris-HCl [pH 8], 0.45 mM sucrose, 8 mM EDTA [pH 8], 0.4 mg/ml) and incubated for 15 min at 4°C. Then, cells were centrifuged (8,000 × g, 15 min) and sonicated in buffer B (50 mM Tris-HCl [pH 7.6], 5 mM MgCl2, 2 mM phenylmethylsulfonyl fluoride [PMSF], deoxyribonuclease I [DNAse]). The sonicated cells were centrifuged, and supernatant was recovered. Insoluble membrane fractions were recovered by ultracentrifugation and homogenized in 50 mM Tris-HCl, pH 7.6.
Brucella membrane extracts were submitted to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; 10%) and transferred to nitrocellulose membranes. The presence of outer membrane proteins (OMPs) was assessed by immunoblot analysis with monoclonal antibodies (dilution, 1:2,000) specific for OMPs 10, 16, 19, and 25 (18, 19).
(ii) VirB proteins.
Brucella whole-cell extracts and cell membranes were resuspended in Laemmli sample buffer and heated to 100°C for 5 min. For cell membranes, nonreducing conditions were preserved. Samples were submitted to SDS-PAGE (12.5%) and transferred to nitrocellulose membranes. The presence of VirB5 and VirB8 proteins was carried out by immunoblot analysis using rabbit polyclonal antibodies specific for VirB5 and VirB8 (dilution, 1:2,000) (20) and IRDye secondary anti-rabbit antibody (LI-COR, Inc.). Detection was performed using the Odyssey Imaging System (LI-COR, Inc.).
Secretion of Bpe123.
J774 A.1 cells were infected with the bpe123 merodiploid strains expressing bpe123-3×Flag (multiplicity of infection [MOI], 50:1). At 4 h postinfection, cells were washed three times with PBS and fixed for 15 min in 4% paraformaldehyde and then processed for immunofluorescence labeling using rabbit anti-Brucella polyclonal antibody (dilution, 1:1,500) and anti-Flag monoclonal antibody (dilution, 1:4,000). The secondary antibodies used were goat anti-mouse Alexa Fluor 488 and goat anti-rabbit Alexa Fluor 568 (Molecular Probes, Invitrogen Co.) at a 1:4,000 dilution. For DNA staining, DAPI dye at 0.5 mg/ml (final concentration) was used. Colocalization was determined by counting the number of bacteria positive for both labels and expressed as percentages of Flag-positive BCVs.
In vitro susceptibility to low pH.
PBS was adjusted to pH 3.5, 4, 5, or 7 by adding 1 N HCl. PBS solutions with different pHs were inoculated with 1 × 107 CFU/ml of wild-type or mutant strains and incubated at 37°C for 1 h, after which serial dilutions of each culture were plated on TSB agar to determine viable bacteria.
RESULTS
Bab1_1002 of Brucella abortus encodes an active muramidase.
With the purpose of identifying novel virulence factors in B. abortus, we performed a bioinformatic search seeking for potential horizontally transmitted regions. For this, we identified transposases or recombinases close to transfer RNAs and analyzed the genes present in those regions to determine homology to known virulence factors or eukaryotic proteins. Using this approach, we identified several potentially horizontally transmitted regions, among which was a previously identified pathogenicity island named genomic island II (21) and an island coding for a novel adhesin (14). The genomic island II is absent in the nonpathogenic species Brucella ovis and encodes 20 genes, many of which have no function assigned. One of these genes is Bab1_1002, which encodes a potential membrane-bound 252-amino-acid protein with homology to lysozymes, enzymes that degrade the peptidoglycan. A more detailed examination of this homology revealed that this gene is similar to proteins identified as secretion activators, as they stimulate the secretion of levansucrase and invertase in Zymomonas mobilis (22) and participate in the secretion of the typhoid toxin (23) in Salmonella enterica serovar Typhi (J. E. Ugalde, S. Spano, and J. E. Galán, unpublished data) (Fig. 1A). For these reasons, we renamed Bab1_1002 sagA, for secretion activator gene A.
Fig 1.
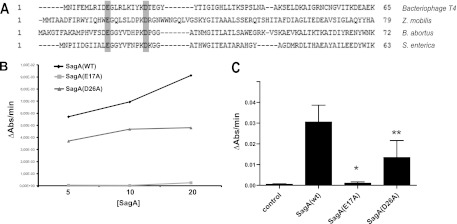
SagA is an active muramidase. (A) Protein alignment of SagA of Brucella abortus with enzymes from the bacteriophage T4, Zymomonas mobilis and Salmonella enterica. In gray are highlighted two conserved catalytic amino acids. (B) Enzymatic activity determined with different concentrations of recombinant SagA or the enzyme with the point mutations E17A and D26A. Activity determined at 10 min of reaction. (C) Activity of the site-directed mutants. *, P < 0.0001; **, P < 0.05.
To determine if the protein product of sagA has muramidase activity, we determined the enzymatic activity of a recombinant protein lacking the transmembrane region (amino acids 223 to 245) by measuring the decrease in absorbance at 480 nm of a Micrococcus lysodeikticus suspension, a substrate of lysozymes. As negative controls, we included the recombinant proteins with point mutations in two residues involved in the catalytic activity (Fig. 1A). As can be observed in Fig. 1B and C, recombinant SagA showed muramidase activity that was completely abolished when amino acid glutamic 17 was replaced by alanine and significantly reduced when the amino acid aspartic 26 was replaced by alanine. These results indicate that sagA codes for an active muramidase.
SagA is required for the early stages of intracellular replication.
To determine if sagA plays a role in the pathogenicity of B. abortus, a deletion mutant was constructed as indicated in Materials and Methods, and the intracellular multiplication in HeLa and J774 A.1 cells was determined in comparison to the parental wild-type strain. Figure 2A and B show that deletion of sagA resulted in a significant reduction (over 10-fold) in the intracellular bacterial load at 4 h postinfection in both HeLa and J774 A.1 cells. Despite this early-stage reduction in the intracellular bacterial load, the mutant was able to replicate with similar kinetics to that of the wild-type parental strain, indicating that the defect was restricted to the initial phases of the infection. To determine if this difference was dependent on the multiplicity of infection (MOI), an antibiotic protection assay was performed with dilutions of 1:50, 1:100, and 1:500 in HeLa cells, and the intracellular bacterial load was determined at 4 h postinfection. As shown in Fig. 2C, at all tested MOI, the mutant displayed a significant reduction in the intracellular survival. This phenotype was not the result of a general growth deficiency, as the growth curve of the mutant was undistinguishable from that of the parental wild-type strain (Fig. 2D). Moreover, the mutant did not show any defects in the membrane expression of several major outer membrane proteins (Fig. 2E), indicating that the mutation did not result in a generalized outer membrane alteration.
Fig 2.
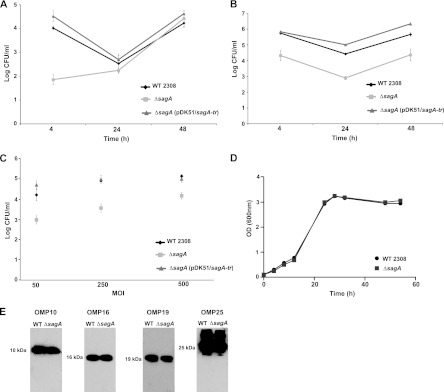
sagA is involved in the early stages of the intracellular replication process. (A) Intracellular replication in HeLa cells; (B) intracellular replication in J774 A1 cells; (C) intracellular CFU 4 h postinfection determined with different multiplicity of infection (MOI) in HeLa cells; (D) growth curve of the wild type and the ΔsagA deletion strain in TSB; (E) abundance of OMPs 10, 16, 19, and 25 in the membranes of wild-type and ΔsagA strains.
The reduction in bacterial survival at 4 h postinfection could be the consequence of a reduced attachment capacity, less invasiveness, or a defect in the ability to resist the initial bactericidal attack of the cell. To distinguish between these possibilities, we first evaluated the attachment and the invasion capacity of the mutant strain determining the number of cell-associated bacteria (attachment) and the percentage of internalized bacteria by an in-out differential staining (invasion) (14). As can be observed in Fig. 3A, the ΔsagA strain showed no significant differences in the attachment or in the invasion of HeLa cells compared to those of the wild-type strain. These results prompted us evaluate if the differences observed in the intracellular viable counts were the consequence of an increased degradation or inactivation of the mutant. For this, we determined the number of LAMP-1-positive BCVs at 4 and 24 h postinfection in HeLa cells for the ΔsagA mutant and the parental wild-type strains. As shown in Fig. 3B, the mutant showed a reduced capacity to exclude the lysosomal marker LAMP-1 at 24 h postinfection in comparison with the wild-type strain, indicating that the absence of sagA decreases the ability of the bacteria to avoid the fusion of the BCV with lysosomes. Albeit this, the bacteria that were able to avoid its degradation replicated in a compartment calnexin positive as the wild-type strain (Fig. 3D). All together, these results indicate that sagA is required during the initial phases of the intracellular life cycle participating in the process in which the BCV avoids maturation into an active lysosome. To determine if this early intracellular deficiency was the consequence of an increased acid sensitivity of the mutant, we performed an acid resistance assay as described in Materials and Methods. The results shown in Fig. S1 in the supplemental material indicate that the mutant did not display any increased sensitivity to low pH.
Fig 3.

sagA is involved in the early stages of the intracellular trafficking of the BCV. (A) Adherence and internalization of the wild-type 2308 and the ΔsagA strains to HeLa cells at 4 h postinfection. The percentage of internalized bacteria and the number of bacteria adhered per 100 HeLa cells were calculated as indicated in Materials and Methods. (B) Determination of LAMP-1-positive BCVs at 4 and 24 h postinfection of the wild-type and the ΔsagA mutant in HeLa cells. (C) Representative images of the LAMP-1-positive and -negative BCVs. *, P < 0.05. (D) Representative image of calnexin-positive BCVs of the ΔsagA mutant at 48 h postinfection.
To determine if the peptidoglycanase activity of SagA is required in the early stages of the intracellular life cycle, the ΔsagA mutant was complemented with a plasmid coding for a version of the gene with the point mutation E17A, and the intracellular replication capacity was evaluated in HeLa cells. As shown in Fig. 4, complementation with this mutant did not restore the wild-type levels of intracellular CFU at 4 h postinfection, indicating that the peptidoglycanase activity is required for the activity of sagA during the infectious process.
Fig 4.

The muramidase activity of sagA is required for the early stages of the intracellular replication process. Intracellular replication curve in HeLa cells with the ΔsagA strain complemented with the wild type and the site-directed E17A mutant.
To determine if the early intracellular survival defect at 4 h postinfection was also observed at earlier time points, we performed an intracellular survival assay in J774 A.1 cells and determined the bacterial loads at 1 and 2 h postinfection. We also included in this assay the B. abortus virB10 mutant (24). As shown in Fig. S2 in the supplemental material, the mutant displayed a defect at very early time points postinfection, indicative that the gene plays a role in the initial biogenesis of the BCV. Surprisingly, the virB mutant also displayed a similar intracellular defect at these very early time points, even though it has been revealed that until 4 h postinfection, a virB9 mutant had no phenotype in bone marrow-derived macrophages (25).
SagA participates in the assembly and proper function of the VirB secretion system.
The defect in the early stages of the intracellular replication process observed with the mutant and also with the virB10 mutant suggested that one of the systems potentially affected in this strain could be the VirB secretion apparatus. To test this, we analyzed the assembly status of two proteins of the system in the membrane of the bacteria (VirB5 and VirB8) and the functionality of the translocation apparatus determining the secretion of a known substrate, the product of Bab2_0123 (7). As can be observed in Fig. 5A, while both proteins were expressed at similar levels in the wild-type and mutant strains (whole bacteria), when total membranes submitted to SDS-PAGE without the addition of a reducing agent were used, the wild-type and mutant strains showed different band patterns for both VirB5 and VirB8 proteins. More specifically, while in the wild-type parental strain both proteins showed a band corresponding to the monomer and a higher band most probably due to an association with another protein of the complex, the mutant strain exhibited only the monomer form in VirB8 and a significantly less abundant complex band in the case of VirB5. As a control for a membrane protein, an OMP19 Western blot was included. This result demonstrates that, even though we are not able to explain the associations of these proteins observed in the wild-type strain because, to date, there is no thorough physical map of the VirB system in Brucella, whatever VirB5 and VirB8 complexes are formed in the membrane of the bacteria, these are not present in the mutant, indicating that the VirB complex is altered. Because an altered assembly of the VirB system does not imply a defect in the secretion, we analyzed functionality determining, at 4 h postinfection, the secretion of an effector protein (Bpe123) recently identified as a type IV substrate in B. abortus (7). To do this, we constructed two merodiploid strains carrying a chromosomal 3× Flag of Bpe123 in the wild-type and ΔsagA mutant strains and analyzed, with them, secretion of the protein during the infection of J774 A.1 cells by immunofluorescence with a monoclonal anti-3× Flag antibody. As can be observed in Fig. 5B, C, and D, the mutant strain showed significantly less surface-exposed Bpe123 than the wild-type strain, even though both strains expressed the protein at similar levels. All together, these results demonstrate that SagA is involved in the assembly of a functional VirB system and suggest that the phenotype observed during the early stages of the intracellular replication stage is probably the consequence of this defective assembly.
Fig 5.

sagA is involved in the assembly of a functional VirB secretion system. (A) Western blot analysis of VirB5 and VirB8 components of the VirB macromolecular complex in whole bacteria and membranes of the wild-type and ΔsagA mutant strains. Total membranes of the wild-type and mutant strains were subjected to SDS-PAGE in the absence of any reducing agent and probed with rabbit anti-VirB5 and -VirB8 antibodies. (B) Percent of bacteria positive for surface-exposed Bpe123 in J774 A.1-infected cells with the merodiploid strains (wild type and ΔsagA) carrying a copy of the gene fused to a 3× Flag tag. Cells were infected for 4 h, fixed, and stained with a monoclonal anti-Flag and a rabbit polyclonal anti-Brucella antibody. The inset shows the expression levels of the tagged Bpe123 in both strains. **, P < 0.005. (C and D) Representative pictures of the quantification shown in panel B.
sagB, a homologue of sagA, also participates in the early stages of intracellular replication.
Analysis of the chromosome of B. abortus 2308 revealed the presence of an open reading frame with high similarity to sagA, Bab1_0875 (Fig. 6A), that we named sagB. The protein encoded by this gene, with a 54% identity and a 72% similarity to SagA, has the conserved catalytic residues and a transmembrane domain in the carboxy terminus. To determine if this gene plays a role similar to the one of sagA, we generated a deletion mutant strain and a double mutant (see Materials and Methods) and analyzed the intracellular replication capacity in HeLa cells. As can be observed in Fig. 6B, deletion of sagB resulted in a strain with a phenotype similar to that of the ΔsagA strain, with reduced bacterial load at 4 h postinfection but with the capacity to replicate with kinetics similar to the wild type. Interestingly, the double mutant did not show an enhanced defect but had, at 4 h postinfection, a bacterial load similar to the single mutant. One interesting observation was that complementation of the ΔsagB mutant with plasmid pDK51/sagA-tr coding for sagA did not complement the phenotype (not shown). This strongly suggests that, even though there is a high conservation between genes, they do not play redundant roles, a hypothesis supported by the fact that each single mutant per se has a phenotype.
Fig 6.
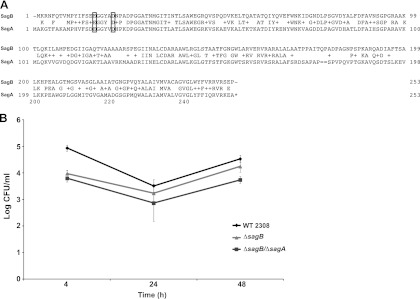
sagB is highly similar to sagA and is also required during the early stages of intracellular replication. (A) Comparison of SagB and SagA. The conserved catalytic residues are marked with a box. (B) Intracellular replication in HeLa cells of the 2308 wild-type, ΔsagB, and ΔsagA ΔsagB double mutant strains.
DISCUSSION
Secretion of proteins is central for the virulence of all pathogenic bacteria known to date and, in the case of Gram-negative organisms, this process requires the translocation of the cognate proteins across the peptidoglycan. To achieve this, the proteins must cross this mesh-like polymer for which there are specialized enzymes that degrade it in a precise temporal and spatial organization. These proteins belong to the family of the lysozyme-like enzymes that are widely distributed in bacteria (26) and have the capacity to cleave the β-1,4 glycosidic bond between the MurNAc and GlcNAc in the peptidoglycan. Even though there are several genes with homology to lysozyme-like enzymes in the Brucella genomes, none have been characterized to date. We report here the identification and characterization of a lysozyme-like enzyme of Brucella abortus, which we have named SagA. We have demonstrated that this protein has peptidoglycanase activity that is dependent on conserved catalytic amino acids of this family of enzymes. The deletion of sagA rendered a strain with a significant defect in the early stages of the intracellular cycle in professional and nonprofessional phagocytes determined by standard intracellular multiplication assays. Moreover, this defect is the consequence of the absence of the enzymatic activity, because an inactive protein was unable to complement the mutant. Even though the mutant showed a significant decrease in the viable counts at 4 h postinfection, the strain was able to replicate with kinetics similar to that of the wild type, indicating that the defects were in the attachment, invasion, or early intracellular trafficking. Our results indicate that neither the attachment nor the invasion was affected but rather the capacity to avoid the fusion of the BCV with the lysosomes as evidenced by a defect in excluding the lysosomal marker LAMP-1, indicating that the gene plays a role in modulating the intracellular trafficking. To explore if this early intracellular trafficking defect was the consequence of an altered VirB system, we analyzed the assembly status and functionality of the complex, determining the migratory properties of two proteins of the macromolecular complex in the bacterial membrane and the secretion of a known substrate of the system. Our results indicate that SagA participates in the assembly of the VirB system and that the defective early intracellular survival is probably the consequence of this defect. It is important to highlight the fact that the first gene in the virB operon (virB1) encodes a lysozyme-like enzyme, and it has been shown to be important for the intracellular replication but not in the animal model (27). The fact that the virB1 mutant is still able to replicate in cells but with difficulties indicates that the trans-glycosidase activity is redundant, a hypothesis strengthened by our results that indicate that sagA also plays a role in the assembly of the VirB complex.
Examination of the genome of B. abortus resulted in the identification of a gene, sagB, highly similar to sagA. A deletion mutant in this gene also resulted in a strain with a defect in the early stages of the intracellular replication process, but sagA was not capable of cross-complementation, indicating, a priori, that they do not have redundant functions. Because bacteria have several macromolecular membrane complexes devoted to virulence, it could be possible that sagA and sagB play a similar function in the assembly of different secretion complexes involved in the intracellular life cycle.
Supplementary Material
ACKNOWLEDGMENTS
We thank María Inés Marchesini for kindly providing the plasmid coding for the Bab2_0123-3×Flag fusion and Christian Baron for the anti-Vir5 and -VirB8 antibodies.
This work was supported by grants PICT-PRH08-160 and PICT-PRH08-230 to C.C. and J.E.U., respectively, NIAID grant AI078891 to J.E.U., and grants from the University of San Martín to C.C. and J.E.U.
C.C. and J.E.U. are members of the National Research Council of Argentina (CONICET).
Footnotes
Published ahead of print 14 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01158-12.
REFERENCES
- 1. Corbel MJ. 1997. Brucellosis: an overview. Emerg. Infect. Dis. 3:213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ugalde RA. 1999. Intracellular lifestyle of Brucella spp. common genes with other animal pathogens, plant pathogens, and endosymbionts. Microbes Infect. 1:1211–1219 [DOI] [PubMed] [Google Scholar]
- 3. Gorvel JP, Moreno E. 2002. Brucella intracellular life: from invasion to intracellular replication. Vet. Microbiol. 90:281–297 [DOI] [PubMed] [Google Scholar]
- 4. de Jong MF, Tsolis RM. 2012. Brucellosis and type IV secretion. Future Microbiol. 7:47–58 [DOI] [PubMed] [Google Scholar]
- 5. de Barsy M, Jamet A, Filopon D, Nicolas C, Laloux G, Rual JF, Muller A, Twizere JC, Nkengfac B, Vandenhaute J, Hill DE, Salcedo SP, Gorvel JP, Letesson JJ, De Bolle X. 2011. Identification of a Brucella spp. secreted effector specifically interacting with human small GTPase Rab2. Cell. Microbiol. 13:1044–1058 [DOI] [PubMed] [Google Scholar]
- 6. de Jong MF, Sun YH, den Hartigh AB, van Dijl JM, Tsolis RM. 2008. Identification of VceA and VceC, two members of the VjbR regulon that are translocated into macrophages by the Brucella type IV secretion system. Mol. Microbiol. 70:1378–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marchesini MI, Herrmann CK, Salcedo SP, Gorvel JP, Comerci DJ. 2011. In search of Brucella abortus type IV secretion substrates: screening and identification of four proteins translocated into host cells through VirB system. Cell. Microbiol. 13:1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hueck CJ. 1998. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol. Mol. Biol. Rev. 62:379–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Korotkov KV, Sandkvist M, Hol WG. 2012. The type II secretion system: biogenesis, molecular architecture and mechanism. Nat. Rev. Microbiol. 10:336–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pukatzki S, McAuley SB, Miyata ST. 2009. The type VI secretion system: translocation of effectors and effector-domains. Curr. Opin. Microbiol. 12:11–17 [DOI] [PubMed] [Google Scholar]
- 11. Wallden K, Rivera-Calzada A, Waksman G. 2010. Type IV secretion systems: versatility and diversity in function. Cell. Microbiol. 12:1203–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Koraimann G. 2003. Lytic transglycosylases in macromolecular transport systems of Gram-negative bacteria. Cell. Mol. Life Sci. 60:2371–2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marchesini MI, Ugalde JE, Czibener C, Comerci DJ, Ugalde RA. 2004. N-terminal-capturing screening system for the isolation of Brucella abortus genes encoding surface exposed and secreted proteins. Microb. Pathog. 37:95–105 [DOI] [PubMed] [Google Scholar]
- 14. Czibener C, Ugalde JE. 2012. Identification of a unique gene cluster of Brucella spp. that mediates adhesion to host cells. Microbes Infect. 14:79–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ibrahim HR, Matsuzaki T, Aoki T. 2001. Genetic evidence that antibacterial activity of lysozyme is independent of its catalytic function. FEBS Lett. 506:27–32 [DOI] [PubMed] [Google Scholar]
- 16. Sieira R, Comerci DJ, Sanchez DO, Ugalde RA. 2000. A homologue of an operon required for DNA transfer in Agrobacterium is required in Brucella abortus for virulence and intracellular multiplication. J. Bacteriol. 182:4849–4855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nielsen KH, Kelly L, Gall D, Nicoletti P, Kelly W. 1995. Improved competitive enzyme immunoassay for the diagnosis of bovine brucellosis. Vet. Immunol. Immunopathol. 46:285–291 [DOI] [PubMed] [Google Scholar]
- 18. Cloeckaert A, de Wergifosse P, Dubray G, Limet JN. 1990. Identification of seven surface-exposed Brucella outer membrane proteins by use of monoclonal antibodies: immunogold labeling for electron microscopy and enzyme-linked immunosorbent assay. Infect. Immun. 58:3980–3987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cloeckaert A, Jacques I, Bosseray N, Limet JN, Bowden R, Dubray G, Plommet M. 1991. Protection conferred on mice by monoclonal antibodies directed against outer-membrane-protein antigens of Brucella. J. Med. Microbiol. 34:175–180 [DOI] [PubMed] [Google Scholar]
- 20. Paschos A, den Hartigh A, Smith MA, Atluri VL, Sivanesan D, Tsolis RM, Baron C. 2011. An in vivo high-throughput screening approach targeting the type IV secretion system component VirB8 identified inhibitors of Brucella abortus 2308 proliferation. Infect. Immun. 79:1033–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rajashekara G, Covert J, Petersen E, Eskra L, Splitter G. 2008. Genomic island 2 of Brucella melitensis is a major virulence determinant: functional analyses of genomic islands. J. Bacteriol. 190:6243–6252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kondo Y, Toyoda A, Fukushi H, Yanase H, Tonomura K, Kawasaki H, Sakai T. 1994. Cloning and characterization of a pair of genes that stimulate the production and secretion of Zymomonas mobilis extracellular levansucrase and invertase. Biosci. Biotechnol. Biochem. 58:526–530 [DOI] [PubMed] [Google Scholar]
- 23. Spano S, Ugalde JE, Galan JE. 2008. Delivery of a Salmonella Typhi exotoxin from a host intracellular compartment. Cell Host Microbe 3:30–38 [DOI] [PubMed] [Google Scholar]
- 24. Comerci DJ, Martinez-Lorenzo MJ, Sieira R, Gorvel JP, Ugalde RA. 2001. Essential role of the VirB machinery in the maturation of the Brucella abortus-containing vacuole. Cell. Microbiol. 3:159–168 [DOI] [PubMed] [Google Scholar]
- 25. Celli J, de Chastellier C, Franchini DM, Pizarro-Cerda J, Moreno E, Gorvel JP. 2003. Brucella evades macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J. Exp. Med. 198:545–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pei J, Grishin NV. 2005. COG3926 and COG5526: a tale of two new lysozyme-like protein families. Protein Sci. 14:2574–2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. den Hartigh AB, Sun YH, Sondervan D, Heuvelmans N, Reinders MO, Ficht TA, Tsolis RM. 2004. Differential requirements for VirB1 and VirB2 during Brucella abortus infection. Infect. Immun. 72:5143–5149 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.