

Abstract
Fatty acid metabolism impacts multiple intracellular signaling pathways in many cell types, but its role in prostate cancer cells is still unclear. Our previous studies have shown that the n-3 polyunsaturated fatty acid docosahexaenoic acid (DHA) induces apoptosis in human prostate cancer cells by a syndecan-1 (SDC-1)-dependent mechanism. Here, we examined the contribution of lipoxygenase (LOX)- and cyclooxygenase (COX)-mediated DHA metabolism to this effect. Pan-LOX inhibitor (nordihydroguaiaretic acid), 15-LOX inhibitor (luteolin) or 15/12-LOX inhibitor (baicalein) blocked the induced effect of DHA on SDC-1 expression and apoptosis in human prostate cancer cells, whereas 5-LOX inhibitor, AA861, was ineffective. Human prostate cancer cells lines (PC3, LNCaP and DU145 cells) expressed two 15-LOX isoforms, 15-LOX-1 and 15-LOX-2, with higher 15-LOX-1 and lower 15-LOX-2 expressions compared with human epithelial prostate cells. Knockdown of 15-LOX-1 blocked the effect of DHA on SDC-1 expression and caspase-3 activity, whereas silencing 15-LOX-2, 5-LOX, COX-1, COX-2 or 12-LOX had no effect. Moreover, the ability of DHA to inhibit the activity of the PDK/Akt (T308) signaling pathway was abrogated by silencing 15-LOX-1. These findings demonstrate that 15-LOX-1-mediated metabolism of DHA is required for it to upregulate SDC-1 and trigger the signaling pathway that elicits apoptosis in prostate cancer cells.
Introduction
Prostate cancer, the most frequently diagnosed cancer in males and a leading cause of male cancer-related death in the USA, can be modified by dietary fatty acids. Higher intake of n-3 polyunsaturated fatty acids (PUFA) is associated with lower prostate cancer risk (1,2) and in prolonging survival of men diagnosed with advanced prostate cancer (3). Docosahexaenoic acid (22:6 n-3, DHA) is a long chain n-3 PUFA that is mainly acquired in humans through consumption of coldwater fish products. Epidemiological studies showed that DHA inhibits the incidence and development of several types of cancer (2,4–7) and increases the efficacy of anticancer radiation and chemotherapies (8,9). An accumulating body of evidence including our data shows that DHA inhibits proliferation and promotes apoptosis of a variety of cancer cells in vitro and in vivo (10–16). These biological effects may directly or indirectly link to induction of intracellular oxidative stress (17,18) and lead to disruption of the tumor microenvironment by anti-inflammatory, antiangiogenic and immunomodulatory processes (19–21). However, the molecular mechanisms remain unclear.
DHA accumulates primarily in the phosphatidylethanolamine and phosphatidylserine of cellular membranes, but it can also be oxygenated to produce bioactive lipids. This oxygenation is thought to principally occur through lipoxygenase (LOX)- and the cyclooxygenase (COX)-mediated pathways, which form a wide range of lipid mediators causing a plethora of physiological or pathological effects on the organism (22,23). One proposed explanation for the tumor-inhibitory effect of n-3 PUFA is that n-3 PUFA compete for the same metabolic enzymes (LOX and COX) with n-6 PUFA (24). However, until now, no specific enzyme has been confirmed relevant to this antitumorigenic process. Although the potential use of DHA as a chemopreventive agent has been reported, the knowledge of lipid-processing pathways and bioactive lipid metabolites, which regulate the inhibitory effects, in cancer remains relatively limited. One study from neuroblastoma cells showed that metabolites of DHA, 17-hydroxydocosahexaenoic acid (17-HDHA) via 17-hydroperoxydocosahexaenoic acid (17-HpDHA) through 15-LOX and auto-oxidation, contributed to direct cytotoxicity by inducing apoptosis and decreasing proliferation (25). This implied a tumor-suppressing role for DHA metabolites.
Our previous work has demonstrated that DHA induces apoptosis in human prostate cancer cells through a pathway that involves activation of the nuclear receptor peroxisome proliferator-activating receptor-γ, to upregulate a peroxisome proliferator-activating receptor-γ target gene, syndecan-1 (SDC-1). SDC-1 is a transmembrane heparan sulfate proteoglycan that, when upregulated by DHA, induces apoptosis through inhibition of the PDK1/Akt/Bad survival pathway (12,14). We, therefore, sought to determine if specific LOX and COX enzymatic pathways of DHA are involved in the SDC-1-associated antitumorigenic effect. In this study, we have used human prostate cancer cells to explore the potential of DHA metabolism by 15-LOX-1, 15-LOX-2, 12-LOX, 5-LOX, COX-1 and COX-2 on SDC-1, its signaling pathway and subsequent apoptosis. We identified 15-LOX-1 as a critical enzyme in DHA upregulation of SDC-1 and induction of apoptosis.
Materials and methods
Materials
Anti-SDC-1 (H-174) antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-Akt, antiphospho-Akt (S473), antiphospho-Akt (T308), antiprotein serine/threonine kinase 3′-phosphoinositide-dependent kinase 1 (PDK1), antiphospho-PDK1 (S243), antipoly(ADP-ribose) polymerase (PARP) and antihorseradish peroxidase-conjugated secondary antibody against rabbit were purchased from Cell Signaling Technology (Danvers, MA). Caspase-Glo® 3/7 assay was from Promega (Madison, WI). Anticaspase 3 was from Santa Cruz Biotechnology. DHA was from Sigma Chemical Co (St. Louis, MO) and prepared as 600 µmol/l stocks complexed to bovine serum albumin (BSA) as described (26). Nordihydroguaiaretic acid (NDGA), luteolin, baicalein and AA861 were purchased from Cayman Chemical Company, Ann Arbor, MI. Hoechst 33342 was from Invitrogen (Eugene, OR). COX-1 small interfering RNA (siRNA) (Cat. No. SI00301511), COX-2 siRNA (Cat. No. SI00301525), 15-LOX-1-siRNA (Cat. No. SI00022449), 15-LOX-2 siRNA (Cat. No. SI00022498), 12-LOX-siRNA (Cat. No. SI00294679), 5-LOX-siRNA (Cat. No. SI00294714) and control siRNA with no known human target (Cat. No. 1027280) were purchased from Qiagen (Austin, TX).
Cell lines and cell culture
PC3, LNCaP and DU145 cells were purchased from the American Type Culture Collection (Manassas, VA) and maintained in advanced Dulbecco’s modified Eagle’s medium (Invitrogen, Grand Island, NY) containing 1% fetal bovine serum (FBS) (PC3 cells), RPMI 1640 medium (Invitrogen) with 10% FBS (LNCaP cells) or Eagle’s minimum essential medium (Invitrogen) containing 10% FBS (DU145 cells). Normal human prostate epithelial cells (PrEC) were purchased from Clonetics (San Diego, CA) and maintained in Clonetics Prostate Epithelial Cell medium.
Western blot assay
Human prostate cancer cells were lysed in ice-cold lysis buffer (25mM Tris–HCl, 150mM NaCl, 1% Triton X-100, 0.1mg/ml phenylmethylsulfonyl fluoride) with 1× proteinase and 1× phosphatase inhibitors (Roche Applied Science). For the analysis of SDC-1 protein, lysates were dialyzed against 100mM Tris, 30mM sodium acetate, pH 8.0 for 24h at 4°C and then digested by chondroitinase ABC (Seikagaku) and heparinase III (Sigma–Aldrich Company, Allentown, PA) at 37°C overnight. Protein extracts were electrophoresed by 12.5% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. After blocking with 5% non-fat dry milk, the membrane was washed three times with Tris-buffered saline/Tween-20 and incubated with the primary antibody diluted in 3% BSA at 4°C overnight. The blot was washed in Tris-buffered saline/Tween-20 and incubated for 1h with a horseradish peroxidase-conjugated secondary antibody diluted 1:2000 for goat antimouse and goat antirabbit. The signal was detected using the chemiluminescent detection system (Pierce). Band densities on photographic films were analyzed using Image J 1.37v (NIH).
Gene silencing by siRNA
Cells were plated in six-well plates at a density of 2.0×105 cells per well and transfected with siRNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Control cells received a negative control siRNA with no known target. At 6h after transfection, wells were supplemented with medium containing 1% FBS overnight. Cells were harvested after transfection 24, 48 and 72h to measure gene expressions.
Quantitative real-time PCR
Quantitative real-time PCR (RT-PCR) was performed as described (13,26). Briefly, total cell RNA was prepared. Quantitative analysis of gene expression was generated with Applied Biosystems 7500 Real-Time PCR System and the SYBR Green Master Mix kit. The primers used for human SDC-1 were 5′-ggagcaggacttcacctttg (forward) and 5′-ctcccagcacctctttcct (reverse). Primers for human COX-1 were 5′-tgcccagctcctggcccgccgctt-3′ (forward) and 5′-gtgcatcaacacaggcgcctcttc-3′ (reverse). Primers for human COX-2 were 5′-gtttgcattctttgcccagc-3′ (forward) and 5′-caggcaccagaccaaagacc-3′ (reverse). Primers for human 15-LOX-1 were 5′-gctgcggctctgggaaatcatct-3′ (forward) and 5′-gggcccgaaaaatactcctcctca-3′ (reverse). Primers for human 15-LOX-2 were 5′-tgatgacaagtgggactggttgct-3′ (forward) and 5′-actcagagaagccttcaatgccga-3′ (reverse). Primers for human 12-LOX were 5′-agaatggttccctgtttgaagct-3′ (forward) and 5′-ccattgggctccatcttcag-3′ (reverse). Primers for human 5-LOX were 5′-ccaaagcgatggagaacctg-3′ (forward) and 5′-gcagaaggtgggtgatggtc-3′ (reverse). Primers for human peptidylprolylisomerase B housekeeping gene were 5′-gcccaaagtcaccgtcaa (forward) and 5′-tccgaagagaccaaagatcac (reverse). Data were normalized to the housekeeping control peptidylprolylisomerase B and are presented relative to control.
Detection of apoptosis
PC3, DU145 and/or LNCaP cells were treated with 30 µM DHA and/or luteolin (1 µM) or transfection with specific oxygenase or control siRNA. Apoptosis was measured by caspase-3 activity using Caspase-Glo® 3/7 assay following the manufacturer’s protocol and/or Hoechst 33342 staining or cleaved PARP using western blot assay.
Statistical analysis
Data are expressed as mean and standard deviation or standard error. Statistical analysis was performed using one-way analysis of variance. P values <0.05 were considered statistically significant.
Results
DHA-induced apoptosis and SDC-1 are blocked by pan-LOX or 15-LOX inhibitor
Because our previous studies had shown that DHA upregulated SDC-1 expression and induced apoptosis in prostate cancer cells (12,14), we sought to clarify the requirement for DHA metabolism in this process. The human LOX enzymes that may be involved in DHA signaling are 5-LOX, 12-LOX, 15-LOX-1 and 15-LOX-2. To determine whether DHA itself or an oxygenated metabolite by LOX caused those effects, we treated cells with pan-oxygenase inhibitor, NDGA, 15-LOX inhibitor, luteolin, 15-LOX-1/2 and 12-LOX inhibitor, baicalein or 5-LOX inhibitor, AA861 and measured SDC-1 gene expression and caspase 3 activity. At doses below those required to inhibit cell proliferation, NDGA prevented DHA stimulation of both caspase-3 activity (Figure 1A) and SDC-1 (Figure 1B). Luteolin likewise blocked the effect of DHA on caspase-3 activity (Figure 1C) and SDC-1 gene expression (Figure 1D). Hoechst staining further confirmed that luteolin suppressed DHA induced-chromatin condensation and/or DNA fragmentation, which are typical apoptotic nuclear morphology (Figure 1E). Baicalein also abrogated the ability of DHA to upregulate SDC-1 (Supplementary Figure 1A), whereas AA861 was not effective (Supplementary Figure 1B). Although these drugs have other effects, these data suggest that DHA-induced apoptosis and SDC-1 are driven by 15-LOX or 12-LOX, but not by 5-LOX.
Fig. 1.

Inhibition of DHA-induced apoptosis and SDC-1 by pan-oxgenase and 15-LOX inhibitors. A and C. PC3 cells was treated with DHA-BSA (30 µM) and indicated concentrations of NDGA (A) or luteolin (C) for 48h. Caspase-3 activity was measured by Caspase-Glo 3/7 assay. Values representing the mean ± SD (n = 3) with different letters are significantly different (P < 0.05). B and D. PC3 cells were treated with DHA-BSA (30 µM) and indicated concentration of NDGA (B) or luteolin (D) for 8h. Total RNA was isolated and SDC-1 mRNA was determined by RT-PCR. Values, expressed relative to controls (mean ± SD, n = 3), with different letters are significantly different (P < 0.05). E. PC3, DU145 and LNCaP cells were treated with DHA-BSA (30 µM) and/or luteolin (1 µM) for 48h. Cell morphologic changes were examined by Hoechst 33342 staining and observed by fluorescence microscopy. Representative photographs were obtained from three separate experiments. Apoptotic cells with condensed nuclei and/or fragmented chromatin are indicated by arrows. The percentages of apoptotic cells are expressed in the graph as means ± SE of three individual experiments. Values with different letters are significantly different (P < 0.05).
15-LOX are expressed in human prostate cancer cells
15-LOX belongs to the structurally and functionally related non-heme iron dioxygenase family. It has two isoforms, 15-LOX-1 and 15-LOX-2. There is evidence that 15-LOX converts DHA to new families of lipid mediators that have potent anti-inflammatory and immunoregulatory actions (27). To investigate the role of 15-LOX isoforms in DHA-induced apoptosis and SDC-1 in prostate cancer, we confirmed that human prostate cancer cells express 15-LOX-1 and 15-LOX-2 and compared their expression with that of normal PrEC. As shown in Figure 2, messenger RNA (mRNA) for 15-LOX-1 was barely detectable in PrEC cells, but it was markedly higher in prostate cancer cell lines (Figure 2A). The level of 15-LOX-2 mRNA was >20-fold lower in cancer cells than in PrEC cells (Figure 2B). Western blots confirmed the presence of both 15-LOX-1 and 15-LOX-2 proteins in the cancer cells with relative protein levels consistent with corresponding mRNA levels (Figure 2C).
Fig. 2.

Expressions of 15-LOX-1 and 15-LOX-2 in human prostate cancer cells. A and B. Human prostate cancer cells (PC3, DU145 and LNCaP cells) and human PrEC were cultured and harvested at the same time. Total RNA was isolated, and 15-LOX-1 and 15-LOX-2 mRNA were measured by RT-PCR. Values expressed relative to PrEC cells (mean ± SD, n = 3) with different letters are significantly different (P < 0.05). C. Protein extracts from PrEC, PC3, DU145 and LNCaP cells were used for western blot analysis of 15-LOX-1, 15-LOX-2 and β-actin.
DHA-induced apoptosis in human prostate cancer cells is mediated through 15-LOX-1 pathway
15-LOX attacks DHA to form a series of 17-hydroperoxy products (28) and also can act in concert with COX-2 and 5-LOX to form other products (29); 12-LOX attacks DHA to form monohydroxy products (30,31). To test the contribution of these individual LOX and COX to DHA-induced apoptosis, PC3 and LNCaP cells were transfected with 15-LOX-1, 15-LOX-2, 5-LOX, 12-LOX, COX-1 and COX-2 siRNAs. As shown in Figure 3, knockdown of 15-LOX-1 was specific for 15-LOX-1 expression and knockdown of 15-LOX-2 was specific for 15-LOX-2 expression in both cell lines (Figure 3A and 3B). Neither 15-LOX-1 nor 15-LOX-2 siRNA inhibited the expression of 5-LOX, 12-LOX, COX-1 and COX-2 (Supplementary Figure 2). Knockdown of 15-LOX-1 blocked DHA-induced apoptosis as measured by caspase-3 activity (Figure 3C) and caspase-cleaved PARP expression (Figure 3E), whereas 15-LOX-2 knockdown had no effect (Figure 3D and 3E). 5-LOX and 12-LOX, which were likewise silenced by their specific siRNAs (Supplementary Figure 3A and B), failed to block the ability of DHA to induce apoptosis (Figure 4A and 4B). Similar data were obtained with COX showing that knockdown of COX-1 and COX-2 (Supplementary Figure 3C and D) did not impede the ability of DHA to induce caspase activity (Figure 4C and 4D). These data indicate that DHA-induced apoptosis is mainly mediated by 15-LOX-1, but not by the other oxygenase enzymes.
Fig. 3.
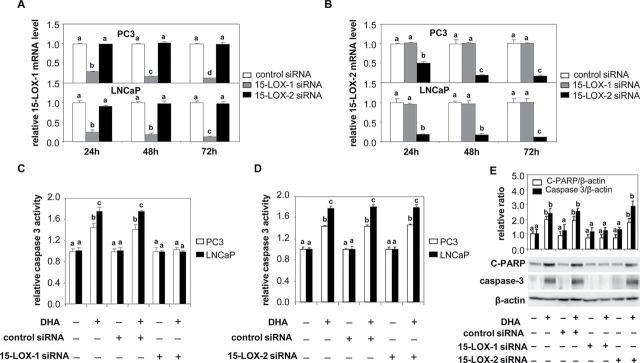
Effects of 15-LOX-1 and 15-LOX-2 knockdown on DHA-induced apoptosis in PC3 and LNCaP cells. A and B. PC3 and LNCaP cells were transfected with a control siRNA, 15-LOX-1 siRNA or 15-LOX-2 siRNA. At indicated times after transfection, total RNA was isolated and 15-LOX-1 or 15-LOX-2 mRNA was determined by RT-PCR. Data shown are the levels of 15-LOX-1 (A) or 15-LOX-2 (B) gene expression relative to control (n = 3). Values representing the mean ± SD (n = 3) with different letters are significantly different (P < 0.05). C and D. Cells were transfected with control siRNA, 15-LOX-1 siRNA (C) or 15-LOX-2 siRNA (D) for 6h and then supplemented with growth medium containing 1% FBS overnight. Cells were treated with DHA-BSA (30 µM) for 48h. Caspase-3 activity was measured with the Caspase-Glo® 3/7 assay. Values expressed relative to controls representing the mean ± SD (n = 3) with different letters are significantly different (P < 0.05). E. LNCaP cells were transfected with control siRNA, 15-LOX-1 siRNA or 15-LOX-2 siRNA for 6h and then supplemented with growth medium containing 1% FBS overnight. Cells were treated with DHA-BSA (30 µM) for 48h. Protein extracts were used for western blot analysis of cleaved PARP, caspase-3 and β-actin. Data shown are representative of three independent experiments. Bar graphs are scans of cleaved PARP and caspase 3 bands corrected for β-actin and shown relative to control. Relative ratios are shown in graphs, and values representing the mean ± SE (n = 3) with different letters are significantly different (P < 0.05).
Fig. 4.

Effects of 5-LOX, 12-LOX, COX-1 and COX-2 knockdown on DHA-induced apoptosis in PC3 cells. PC3 cells were transfected with control siRNA, 5-LOX siRNA (A), 12-LOX siRNA(B), COX-1 siRNA (C) or COX-2 siRNA (D) for 6h and then supplemented with growth medium containing 1% FBS overnight. Cells were treated with DHA-BSA (30 µM) for 48h. Caspase-3 activity was measured with the Caspase-Glo® 3/7 assay. Values expressed relative to controls representing the mean ± SD (n = 3) with different letters are significantly different (P < 0.05).
SDC-1 upregulation by DHA is 15-LOX-1 dependent
Our previous studies found that upregulation of SDC-1 is an essential requirement for DHA-induced apoptosis in prostate cancer cells (12), and as shown in Figure 1, inhibition of LOX activity with relatively non-selective pharmacological agents inhibited DHA upregulation of SDC-1. To identify the involvement of a specific oxygenase on the DHA effect on SDC-1, we examined SDC-1 expression in PC3 cells transfected with 15-LOX-1, 15-LOX-2, 5-LOX, 12-LOX, COX-1 and COX-2 siRNAs. SDC-1 message and/or protein levels were measured by RT-PCR and western blot assay, respectively. Specific knockdown of 15-LOX-1 blocked DHA-mediated SDC-1 upregulation (Figure 5A and 5C), whereas silencing 15-LOX-2 (Figure 5B and 5D), 5-LOX (Supplementary Figure 4A), 12-LOX (Supplementary Figure 4B), COX-1 (Supplementary Figure 4C) and COX-2 (Supplementary Figure 4D) had no effect. Thus, DHA upregulation of SDC-1 is a 15-LOX-1-dependent process.
Fig. 5.

Effects of 15-LOX-1 and 15-LOX-2 knockdown on SDC-1 upregulation by DHA in PC3 cells. A and B. PC3 were transfected with control siRNA, 15-LOX-1 siRNA (A) or 15-LOX-2 siRNA (B) for 6h and then supplemented with growth medium containing 1% FBS overnight. Cells were treated with DHA-BSA (30 µM) for 8h. Total RNA from cells was isolated, and SDC-1 mRNA was measured by RT-PCR. Values compared with relevant control with different letters are significantly different (P < 0.05). C and D. PC3 were transfected with control siRNA, 15-LOX-1 siRNA (C) or 15-LOX-2 siRNA (D) for 6h and then supplemented with growth medium containing 1% FBS overnight. Cells were treated with DHA-BSA (30 µM) for 48h. Protein extracts were used for western blot analysis of SDC-1 and β-actin. Bar graphs are scans of SDC-1 corrected for β-actin and shown relative to control. Data shown are representative of three independent experiments. Relative ratios are shown in graphs, and values representing the mean ± SE (n = 3) with different letters are significantly different (P < 0.05).
Suppression of PDK1/Akt activation by DHA is 15-LOX-1 dependent
Our previous studies have shown that DHA upregulation of SDC-1 results in decreased phosphorylation of PDK1 and Akt (T308) that leads to induction of apoptosis (12). To examine the requirement of 15-LOX-1 in this signaling pathway, PC3 cells were transfected with either 15-LOX-1 or 15-LOX-2 siRNA prior to treatment with DHA, and protein phosphorylation was measured by western blot. As shown in Figure 6, following 15-LOX-1 knockdown, DHA was unable to inhibit the phosphorylation of either PDK1 or Akt (T308). As shown previously, pAkt (S473) was not affected by DHA and was not impacted by 15-LOX-1 knockdown in these experiments. These results indicate that 15-LOX-1 metabolism of DHA is critical for its ability to suppress PDK1/Akt activation.
Fig. 6.

15-LOX-1 knockdown blocks the effect of DHA on phosphorylations of PDK1 and Akt. PC3 cells were transfected with control siRNA, 15-LOX-1 siRNA or 15-LOX-2 siRNA for 6h and then supplemented with growth medium containing 1% FBS overnight. Cells were treated with DHA (30 µM) for 48h before harvest. PDK1, p-PDK1 (S243), total Akt and pAkt (T308 and S473) were measured by western blot assay. Data shown are representative of three independent experiments. Relative ratios are shown in the graph, and values representing the mean ± SE (n = 3) with different letters are significantly different (P < 0.05)
Discussion
Metabolites of PUFA serve as important messengers in many physiological and pathological events. Although beneficial effects of DHA on human health have been reported, the role of its metabolism in diseases especially cancer remains to be understood. Our previous studies showed that an n-3 PUFA-enriched diet inhibits tumor progression in a mouse model of prostate cancer and that DHA specifically induces apoptosis in cultured human prostate cancer cells by upregulation of SDC-1 (12,14,32). DHA metabolism is catalyzed by two major groups of enzymes, LOX and COX, and in this study, we explored the contribution of these enzymes to DHA’s ability to induce apoptosis in prostate cancer cells. We have demonstrated that 15-LOX-1 is required for the SDC-1 signaling involved in DHA-induced apoptosis. These findings offer clues to systematically explore the chemoprevention activity of DHA in prostate cancer.
The oxygenation of DHA by LOX produces diverse lipid mediators. 15-LOX converts DHA to its 17S-hydroxy and a series of other products (33); aspirin-acetylated COX-2 generates its 17R-hydroxy product, which then can be converted by 5-LOX to new families of lipid mediators such as resolvin D series and neuroprotectins D formed through epoxidation and enzymatic hydrolysis (23). DHA can also be converted by human platelet 12-LOX to 14- and 11-HDHA (31,34) and by human neutrophil 5-LOX to 7-HpDHA and its 7-hydroxy derivative 7-HDHA (35). A recent study showed that a DHA oxygenation product, 17-HpDHA, induces cytotoxicity in neuroblastoma cells (10), which suggests that DHA metabolism may impact cellular activities related to cancer.
It is reported that reduced prostate cancer risk was associated with high erythrocyte DHA (36), and in prostate tissue, reduced DHA level was associated with cancer compared with benign prostatic hyperplasia and advanced stage disease (37). Therefore, dietary supplementation with DHA may prevent or delay metastases in prostate cancer (38). In this study, we have demonstrated that the major metabolic enzymes of DHA including 15-LOX-1, 15-LOX-2, 5-LOX, 12-LOX, COX-1 and COX-2 were all expressed in prostate cancer cells. Exogenous DHA induced apoptosis in these cells. Moreover, specific knockdown of 15-LOX-1 prevented this effect. Individual silence of 15-LOX-2, 12-LOX, 5-LOX, COX-1 or COX-2 did not change 15-LOX-1 expression and also did not block DHA-induced apoptosis. In addition, 15-LOX-1-mediated DHA metabolites including 17-HpDHA, 17-HDHA, 10,17-dihydroxy-DHA and 7,17-dihydroxy-DHA induced SDC-1 expression and activated caspase-3 activity in PC3 cells (39). These data support a potential role of 15-LOX-1-guided DHA metabolism in the induction of apoptosis of prostate cancer.
Apoptosis is controlled by a diverse range of cell signals, which may originate either extracellularly (extrinsic inducers) or intracellularly (intrinsic inducers). Extracellular signals must either cross the plasma membrane or be transduced to impact a response. In this study, we explored the role of enzyme-dependent oxygenation of DHA supplement in the membrane-mediated processes of prostate cells. It is known that SDC-1, as a prototypical cell-surface proteoglycan with a type I transmembrane core protein, regulates cell–cell and cell–matrix interactions, cell migration, adhesion, differentiation and tumorigenesis (40–44). In human prostate cancer, recent studies show that SDC-1 plays an important role in support of homeostasis of the epithelial compartment of normal prostate and non-malignant hormone-responsive and well-differentiated prostate tumors (45,46). Our previous studies have shown SDC-1 to be effective in inducing apoptosis and concomitantly inhibiting the growth of prostate cancer cells (12). By demonstrating that DHA is unable to upregulate SDC-1 in the absence of 15-LOX-1 or in the presence of 15-LOX-1 pharmaceutical inhibitors, this study confirmed a role for SDC-1 in mediating the apoptotic properties of 15-LOX-1-guided DHA oxygenated products in prostate cancer cells. Moreover, this study also shows that SDC-1-dependent suppression of phosphorylation of PDK1/Akt by DHA was blocked by 15-LOX-1 siRNA, implying that 15-LOX-1-mediated metabolism of DHA initiates SDC-1 signaling, which leads to apoptosis. The PDK1/Akt pathway appears to be very sensitive to SDC-1 with a modest DHA-induced increase in SDC-1 resulting in a marked inhibition of PDK1 and Akt phosphorylation (Figures 5 and 6) (12).
The standard therapies of prostate cancer include androgen ablation that initially causes tumor regression. However, the tumor often eventually relapses and develops into hormone refractory prostate cancer (47). A failure to undergo apoptosis maybe a major cause of resistance of prostate cancer cells to this therapy because the majority of tumors following androgen ablation have no increase in apoptotic indices (48,49) and promotion of apoptosis in prostate cancer cells leads to the regression of cancer cells and improved prognosis of refractory disease (50). Thus, an agent that induces apoptosis may be useful for chemotherapy against prostate cancer. This study assigned a central role for 15-LOX-1-mediated metabolism of DHA in inducing apoptosis in cultured human prostate cancer cells. We also demonstrated that DHA-induced apoptosis persists in COX-1, COX-2, 5-LOX, 12-LOX or 15-LOX-2 knockout prostate cancer cells, suggesting the effect of DHA is independent of these enzymes. This specificity of the 15-LOX-1 involvement in DHA metabolism could be beneficial in enhancing the efficacy of conventional chemotherapy of prostate cancer.
A recent study from Sapieha and colleagues (51) disclosed a key role of a 5-LOX-product, 4-HDHA, in antiangiogenic effect of dietary DHA in mouse retinopathy, whereas our data showed that in prostate cancer cells, only 15-LOX-1-mediated DHA metabolism promotes apoptosis. These results imply that a specific DHA metabolic pathway may mediate a unique biological effect and may depend on the cell of origin. Many lines of evidence support important roles for inflammation and angiogenesis in the pathogenesis of prostate cancer (52–55). Therefore, it will be important to investigate the possible role of COX and LOX metabolism of DHA in anti-inflammatory and antiangiogenic effects in prostate cancer.
In summary, this study describes an obligatory step in DHA metabolism required for its upregulation of SDC-1 and induction of apoptosis in prostate cancer cells. We identified 15-LOX-1 as the critical oxygenase for DHA to induce SDC-1 signaling and subsequent apoptosis in prostate tumor cells. The findings provide insight into how DHA modulates tumor progression.
Supplementary material
Supplementary Figures 1–4 can be found at http://carcin.oxfordjournals.org/
Funding
The National Institutes of Health, National Cancer Institute P01CA106742 (I.J.E. and J.O.) and in part by R01CA115958 (I.J.E.).
Conflict of Interest Statement: None declared.
Supplementary Material
Glossary
Abbreviations:
- BSA
bovine serum albumin
- COX
cyclooxygenase
- DHA
docosahexaenoic acid
- FBS
fetal bovine serum
- HDHA
hydroxydocosahexaenoic acid
- HpDHA
hydroperoxydocosahexaenoic acid
- LOX
lipoxygenases
- mRNA
messenger RNA
- NDGA
nordihydroguaiaretic acid
- PARP
poly(ADP-ribose) polymerase
- PDK1
3′-phosphoinositide-dependent kinase 1
- PrEC
prostate epithelial cells
- PUFA
polyunsaturated fatty acids
- RT-PCR
real-time PCR
- SDC-1
syndecan-1
- siRNA
small interfering RNA.
References
- 1. Augustsson K., et al. (2003). A prospective study of intake of fish and marine fatty acids and prostate cancer. Cancer Epidemiol. Biomarkers Prev. 12 64–67 [PubMed] [Google Scholar]
- 2. Leitzmann M.F., et al. (2004). Dietary intake of n-3 and n-6 fatty acids and the risk of prostate cancer. Am. J. Clin. Nutr. 80 204–216 [DOI] [PubMed] [Google Scholar]
- 3. McEntee M.F., et al. (2008). Dietary n-3 polyunsaturated fatty acids enhance hormone ablation therapy in androgen-dependent prostate cancer. Am. J. Pathol. 173 229–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kuriki K., et al. (2007). Breast cancer risk and erythrocyte compositions of n-3 highly unsaturated fatty acids in Japanese. Int. J. Cancer 121 377–385 [DOI] [PubMed] [Google Scholar]
- 5. Patterson R.E., et al. (2011). Marine fatty acid intake is associated with breast cancer prognosis. J. Nutr. 141 201–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pot G.K., et al. (2008). Opposing associations of serum n-3 and n-6 polyunsaturated fatty acids with colorectal adenoma risk: an endoscopy-based case-control study. Int. J. Cancer 123 1974–1977 [DOI] [PubMed] [Google Scholar]
- 7. Berg J.P., et al. (1994). Longchain serum fatty acids and risk of thyroid cancer: a population-based case-control study in Norway. Cancer Causes Control 5 433–439 [DOI] [PubMed] [Google Scholar]
- 8. Bougnoux P., et al. (2009). Improving outcome of chemotherapy of metastatic breast cancer by docosahexaenoic acid: a phase II trial. Br. J. Cancer 101 1978–1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bougnoux P., et al. (2010). Fatty acids and breast cancer: sensitization to treatments and prevention of metastatic re-growth. Prog. Lipid Res. 49 76–86 [DOI] [PubMed] [Google Scholar]
- 10. Gleissman H., et al. (2011). Omega-3 fatty acid supplementation delays the progression of neuroblastoma in vivo. Int. J. Cancer 128 1703–1711 [DOI] [PubMed] [Google Scholar]
- 11. Wu M., et al. (2005). Omega-3 polyunsaturated fatty acids attenuate breast cancer growth through activation of a neutral sphingomyelinase-mediated pathway. Int. J. Cancer 117 340–348 [DOI] [PubMed] [Google Scholar]
- 12. Hu Y., et al. (2010). Syndecan-1-dependent suppression of PDK1/Akt/bad signaling by docosahexaenoic acid induces apoptosis in prostate cancer. Neoplasia 12 826–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sun H., et al. (2008). Peroxisome proliferator-activated receptor gamma-mediated up-regulation of syndecan-1 by n-3 fatty acids promotes apoptosis of human breast cancer cells. Cancer Res. 68 2912–2919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Edwards I.J., et al. (2008). In vivo and in vitro regulation of syndecan 1 in prostate cells by n-3 polyunsaturated fatty acids. J. Biol. Chem. 283 18441–18449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yamagami T., et al. (2009). Docosahexaenoic acid induces dose dependent cell death in an early undifferentiated subtype of acute myeloid leukemia cell line. Cancer Biol. Ther. 8 331–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blanckaert V., et al. (2010). Docosahexaenoic acid intake decreases proliferation, increases apoptosis and decreases the invasive potential of the human breast carcinoma cell line MDA-MB-231. Int. J. Oncol. 36 737–742 [DOI] [PubMed] [Google Scholar]
- 17.Kang K.S., et al. Docosahexaenoic acid induces apoptosis in MCF-7 cells in vitro and in vivo via reactive oxygen species formation and caspase 8 activation. PLoS ONE. (2010);5:e10296. doi: 10.1371/journal.pone.0010296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ding W.Q., et al. (2007). Phospholipid hydroperoxide glutathione peroxidase plays a role in protecting cancer cells from docosahexaenoic acid-induced cytotoxicity. Mol. Cancer Ther. 6 1467–1474 [DOI] [PubMed] [Google Scholar]
- 19. Wall R., et al. (2010). Fatty acids from fish: the anti-inflammatory potential of long-chain omega-3 fatty acids. Nutr. Rev. 68 280–289 [DOI] [PubMed] [Google Scholar]
- 20. Spencer L., et al. (2009). The effect of omega-3 FAs on tumour angiogenesis and their therapeutic potential. Eur. J. Cancer 45 2077–2086 [DOI] [PubMed] [Google Scholar]
- 21. Calder P.C. (2007). Immunomodulation by omega-3 fatty acids. Prostaglandins Leukot. Essent. Fatty Acids 77 327–335 [DOI] [PubMed] [Google Scholar]
- 22. Serhan C.N. (2005). Novel eicosanoid and docosanoid mediators: resolvins, docosatrienes, and neuroprotectins. Curr. Opin. Clin. Nutr. Metab. Care 8 115–121 [DOI] [PubMed] [Google Scholar]
- 23. Sun Y.P., et al. (2007). Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J. Biol. Chem. 282 9323–9334 [DOI] [PubMed] [Google Scholar]
- 24. Larsson S.C., et al. (2004). Dietary long-chain n-3 fatty acids for the prevention of cancer: a review of potential mechanisms. Am. J. Clin. Nutr. 79 935–945 [DOI] [PubMed] [Google Scholar]
- 25. Gleissman H., et al. (2010). Docosahexaenoic acid metabolome in neural tumors: identification of cytotoxic intermediates. FASEB J. 24 906–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sun H., et al. (2005). Omega-3 polyunsaturated fatty acids regulate syndecan-1 expression in human breast cancer cells. Cancer Res. 65 4442–4447 [DOI] [PubMed] [Google Scholar]
- 27. Serhan C.N., et al. (2004). Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their aspirin-triggered endogenous epimers: an overview of their protective roles in catabasis. Prostaglandins Other Lipid Mediat. 73 155–172 [DOI] [PubMed] [Google Scholar]
- 28. Stables M.J., et al. (2011). Old and new generation lipid mediators in acute inflammation and resolution. Prog. Lipid Res. 50 35–51 [DOI] [PubMed] [Google Scholar]
- 29. Calder P.C. (2009). Polyunsaturated fatty acids and inflammatory processes: New twists in an old tale. Biochimie 91 791–795 [DOI] [PubMed] [Google Scholar]
- 30. Lu Y., et al. (2010). Novel 14,21-dihydroxy-docosahexaenoic acids: structures, formation pathways, and enhancement of wound healing. J. Lipid Res. 51 923–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morgan L.T., et al. (2010). Thrombin-activated human platelets acutely generate oxidized docosahexaenoic-acid-containing phospholipids via 12-lipoxygenase. Biochem. J. 431 141–148 [DOI] [PubMed] [Google Scholar]
- 32. Berquin I.M., et al. (2007). Modulation of prostate cancer genetic risk by omega-3 and omega-6 fatty acids. J. Clin. Invest. 117 1866–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Serhan C.N., et al. (2005). Resolution of inflammation: the beginning programs the end. Nat. Immunol. 6 1191–1197 [DOI] [PubMed] [Google Scholar]
- 34. Aveldaño M.I., et al. (1983). Synthesis of hydroxy fatty acids from 4, 7, 10, 13, 16, 19-[1-14C] docosahexaenoic acid by human platelets. J. Biol. Chem. 258 9339–9343 [PubMed] [Google Scholar]
- 35. Sperling R.I. (1998). The effects of dietary n-3 polyunsaturated fatty acids on neutrophils. Proc. Nutr. Soc. 57 527–534 [DOI] [PubMed] [Google Scholar]
- 36. Yang Y.J., et al. (1999). Comparison of fatty acid profiles in the serum of patients with prostate cancer and benign prostatic hyperplasia. Clin. Biochem. 32 405–409 [DOI] [PubMed] [Google Scholar]
- 37. Mamalakis G., et al. (2002). Prostate cancer vs hyperplasia: relationships with prostatic and adipose tissue fatty acid composition. Prostaglandins Leukot. Essent. Fatty Acids 66 467–477 [DOI] [PubMed] [Google Scholar]
- 38. Brown M.D., et al. (2006). Promotion of prostatic metastatic migration towards human bone marrow stoma by Omega 6 and its inhibition by Omega 3 PUFAs. Br. J. Cancer 94 842–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Flaherty J.T., et al. 15-lipoxygenase metabolites of docosahexaenoic Acid inhibit prostate cancer cell proliferation and survival. PLoS ONE. (2012);7:e45480. doi: 10.1371/journal.pone.0045480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liebersbach B.F., et al. (1994). Expression of syndecan-1 inhibits cell invasion into type I collagen. J. Biol. Chem. 269 20013–20019 [PubMed] [Google Scholar]
- 41. Liu W., et al. (1998). Heparan sulfate proteoglycans as adhesive and anti-invasive molecules. Syndecans and glypican have distinct functions. J. Biol. Chem. 273 22825–22832 [DOI] [PubMed] [Google Scholar]
- 42. Couchman J.R. (2003). Syndecans: proteoglycan regulators of cell-surface microdomains? Nat. Rev. Mol. Cell Biol. 4 926–937 [DOI] [PubMed] [Google Scholar]
- 43. Inki P., et al. (1996). The role of syndecan-1 in malignancies. Ann. Med. 28 63–67 [DOI] [PubMed] [Google Scholar]
- 44. Rapraeger A.C. (2000). Syndecan-regulated receptor signaling. J. Cell Biol. 149 995–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Contreras H.R., et al. (2010). The expression of syndecan-1 and -2 is associated with Gleason score and epithelial-mesenchymal transition markers, E-cadherin and beta-catenin, in prostate cancer. Urol. Oncol. 28 534–540 [DOI] [PubMed] [Google Scholar]
- 46. Kiviniemi J., et al. (2004). Altered expression of syndecan-1 in prostate cancer. APMIS 112 89–97 [DOI] [PubMed] [Google Scholar]
- 47. Mimeault M., et al. (2006). Recent advances on multiple tumorigenic cascades involved in prostatic cancer progression and targeting therapies. Carcinogenesis 27 1–22 [DOI] [PubMed] [Google Scholar]
- 48. Pollack A., et al. (1997). Quiescence in R3327-G rat prostate tumors after androgen ablation. Cancer Res. 57 2493–2500 [PubMed] [Google Scholar]
- 49. Westin P., et al. (1995). Castration therapy rapidly induces apoptosis in a minority and decreases cell proliferation in a majority of human prostatic tumors. Am. J. Pathol. 146 1368–1375 [PMC free article] [PubMed] [Google Scholar]
- 50. Bruckheimer E.M., et al. (2000). Apoptosis in prostate carcinogenesis. A growth regulator and a therapeutic target. Cell Tissue Res. 301 153–162 [DOI] [PubMed] [Google Scholar]
- 51.Sapieha P., et al. 5-Lipoxygenase metabolite 4-HDHA is a mediator of the antiangiogenic effect of ω-3 polyunsaturated fatty acids. Sci. Transl. Med. (2011);3:69ra12. doi: 10.1126/scitranslmed.3001571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Birbach A., et al. (2011). Persistent inflammation leads to proliferative neoplasia and loss of smooth muscle cells in a prostate tumor model. Neoplasia 13 692–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vasto S., et al. (2008). Inflammation and prostate cancer. Future Oncol. 4 637–645 [DOI] [PubMed] [Google Scholar]
- 54. Huss W.J., et al. (2001). Angiogenesis and prostate cancer: identification of a molecular progression switch. Cancer Res. 61 2736–2743 [PubMed] [Google Scholar]
- 55.Aragon-Ching J.B., et al. Angiogenesis inhibition in prostate cancer: current uses and future promises. J. Oncol. (2010);2010:361836. doi: 10.1155/2010/361836. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.