

Abstract
Objectives
We describe the antimicrobial activity against Pseudomonas aeruginosa of the de novo-derived antimicrobial peptide WLBU2 in an animal model of infection.
Methods
For this study, an intravenous (iv) model of P. aeruginosa infection was established. The minimum lethal murine dose of P. aeruginosa strain PA01 was determined to be 3 × 107 cfu when bacteria were administered iv. Increasing concentrations of WLBU2 were instilled either prior to or following PA01 septic exposure.
Results
For the mice given peptide post-bacterial infection, in the 1 mg/kg group, nine of nine animals died because of Pseudomonas sepsis; in the 3 mg/kg group, only one of nine succumbed to infection and in the 4 mg/kg group, all mice were protected (P < 0.0001). Similar results were obtained when WLBU2 was given 1 h prior to Pseudomonas infection.
Conclusions
Although the therapeutic window in this model is narrow, the results nonetheless provide encouraging evidence for WLBU2 as a potential prophylactic or treatment of bacterial infection.
Keywords: antimicrobial peptide, WLBU2, sepsis
Introduction
Despite the development of safe and potent antibiotics, serious bacterial infections remain a worldwide health priority. An increasing concern is the emergence of multidrug-resistant (MDR) pathogens and their role as opportunistic pathogens.1 Resistant Pseudomonas aeruginosa has become particularly important in the setting of immunocompromised, cystic fibrosis, septic, burn and ventilated patients. Owing to the significant antibiotic resistance in MDR pathogens,2 there is a critical need for the development of more effective antimicrobials with unique bactericidal mechanisms to overcome resistance.
We have developed a novel class of engineered cationic antimicrobial peptides (eCAPs),3 which originate from discrete segments of the lentiviral transmembrane protein cytoplasmic tail. The prototype peptide is LLP1 that is derived from the C-terminal 28 residues encoded by HIV-1 gp41. A series of peptides based on a six amino acid residue motif that retains the cationic amphipathic character of the peptide were designed.4 These hexamers were designed by positioning key amino acids, Arg, Val or Trp, to maintain a cationic and hydrophobic face when modelled as an α-helix. It was reasoned that these hexapeptides could be oligomerized through solution-phase chemistry, significantly decreasing the cost of synthesis. Using these principles, a series of de novo antimicrobial peptides were designed4 and a lead 24-residue peptide, WLBU2, was identified. To characterize the toxicity of WLBU2 under these conditions, red blood cells, white blood cells and human skin fibroblasts co-cultured with P. aeruginosa were exposed to WLBU2 and viability assessed.4 To further evaluate toxicity, an intraperitoneal (ip) mouse model with P. aeruginosa infusion was established and WLBU2 given intravenously (iv) to prevent the progression of infection.4
To investigate the potential role of WLBU2 as a prophylactic or therapeutic agent, an iv P. aeruginosa sepsis mouse model was developed to control the systemic level of bacterial infusion into the circulatory system. Systemic administration of WLBU2, both at prophylactic and at therapeutic time points, increased the survival of P. aeruginosa-infected mice by preventing progression to bacterial sepsis.
Materials and methods
Peptide synthesis
The engineered peptide WLBU2 (RRWVRRVRRWVRRVVRVVRRWVRR) was synthesized, purified and quantified, as previously described.3–5
Bacteria and killing assays
P. aeruginosa PAO1 was used throughout these studies. Bacterial suspensions (1 × 106 cfu/mL) in 10 mM potassium phosphate buffer containing 150 mM NaCl (PBS), pH 7.2 were incubated with 2-fold dilutions of WLBU2 at 37°C for 1 h. Serial peptide dilutions were performed and plated on tryptic soy agar (TSA; Difco, Detroit, MI, USA). Surviving colonies were counted the next day to determine the MBC, defined as the molar concentration of peptide reducing the viable bacteria within a suspension by three orders of magnitude.
Distribution of bacteria in fluids and organs
Quantitative blood cultures were performed to determine bacterial loads over the course of the infection. Blood samples were obtained from the tail vein by aseptic percutaneous puncture 1–24 h after bacterial challenge and serially diluted. A 0.1 mL volume of each dilution was plated on TSA and incubated at 37°C overnight for enumeration of developed colonies. Toxicity was evaluated on the basis of the presence of peptide-related adverse effects such as signs of inflammation, weight loss and presence of bacteria in the blood and tissues. Throughout the course of the infection or at the disease endpoint, animals were euthanized and tissues weighed and homogenized using 70 µm cell strainers to determine bacterial cfu/g tissue.
iv bacterial inoculation and iv antibacterial therapy
Swiss Webster mice (25–30 g) (Taconic, Germantown, NY, USA) were maintained and procedures performed according to the protocol approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh. Suspensions of mid-log phase P. aeruginosa were centrifuged at 2000 g for 10 min. Supernatants were discarded and the bacteria suspended and diluted in sterile PBS to achieve a concentration of ∼2–4 × 108 cfu/mL. Mice were injected iv with 0.1 mL of the bacterial suspensions, estimated as the minimum lethal dose. The animals were then randomized to receive iv PBS (control group), or 1, 1.5, 3 and 4 mg/kg WLBU2 ∼60 min after bacterial challenge. The animals in each group, which included 7–11 mice, were returned to individual cages and subsequently monitored for up to 7–10 days for survival. The endpoints of the study were indicated either by 7–10 days of survival or by complete absence of motility as a sign of terminal illness. For the prophylactic portion, animals were randomized to receive iv PBS versus a 3 mg/kg WLBU2 bolus 1 h prior to infusion of bacterial suspension as described earlier. The group consisted of six mice and was monitored as described earlier.
Statistical analysis
Murine survival data for either the WLBU2-uninfected or WLBU2-infected group were analysed using GraphPad Prism version 3.00 for Windows (GraphPad Software, San Diego, CA, USA). Kaplan–Meier survival analysis was performed and log-rank test was used to compare survival between groups. Significance was accepted at a P value of less than 0.05.
Results
Murine iv pseudomonal sepsis model
The minimum iv-administered P. aeruginosa lethal dose (PLD) was determined by establishing the lowest iv bacterial dose (3 × 107 cfu) leading to 100% mortality (data not shown). After administration of an iv PLD (3–4 × 107 cfu), 11 mice were treated 1 h post-infection with 3 mg/kg WLBU2 (PAO1 + WLBU2) or PBS. At the experimental endpoint, 100% survival was observed for WLBU2-treated mice. Infusion 1 h pre-infection of 11 mice with 3 mg/kg WLBU2 (WLBU2 + PAO1) or PBS (PBS + PA01) led to 100% survival for WLBU2-treated mice. A log-rank test reveals a P value of less than 0.0001 when comparing survival of WLBU2-treated with that of PBS-treated mice. In comparison with the PBS-treated mice, no lethality was observed in prophylactically and therapeutically treated groups (Figure 1). These results provide evidence for a potential prophylactic and therapeutic role of WLBU2 in the treatment of P. aeruginosa sepsis.
Figure 1.

Therapeutic and prophylactic protection by WLBU2 in the bacterial sepsis model. Kaplan–Meier survival analysis comparing 1 h pre-infection infusion of WLBU2 (WLBU2 + PA01), 1 h post-infection infusion of WLBU2 (PA01 + WLBU2) and a PBS control group.
Murine dose dependence of WLBU2 protection from bacterial sepsis
Infected mice (nine per group) were treated therapeutically after 1 h with 0, 1, 1.5, 3 and 4 mg/kg peptide and monitored for 7 days post-treatment. WLBU2 effectively eradicated the infection at a minimum of 3 mg/kg (Figure 2a), with one fatality out of nine mice treated. These data demonstrate dose dependence of WLBU2 protection from P. aeruginosa sepsis. Mice were also euthanized at 4 and 24 h post-treatment to determine bacterial loads in the blood (Figure 2b) and kidney (Figure 2c). In the blood, the bacterial load decreased from 104 cfu/mL to 0 cfu/ml after treatment with 3 or 4 mg/kg WLBU2. Similar results were obtained for per gram of tissue when the kidney was sampled. These experiments show a dose dependence of WLBU2 treatment. This suggests that WLBU2 can play a therapeutic role in helping to control the bacterial infection in peptide-treated mice.
Figure 2.
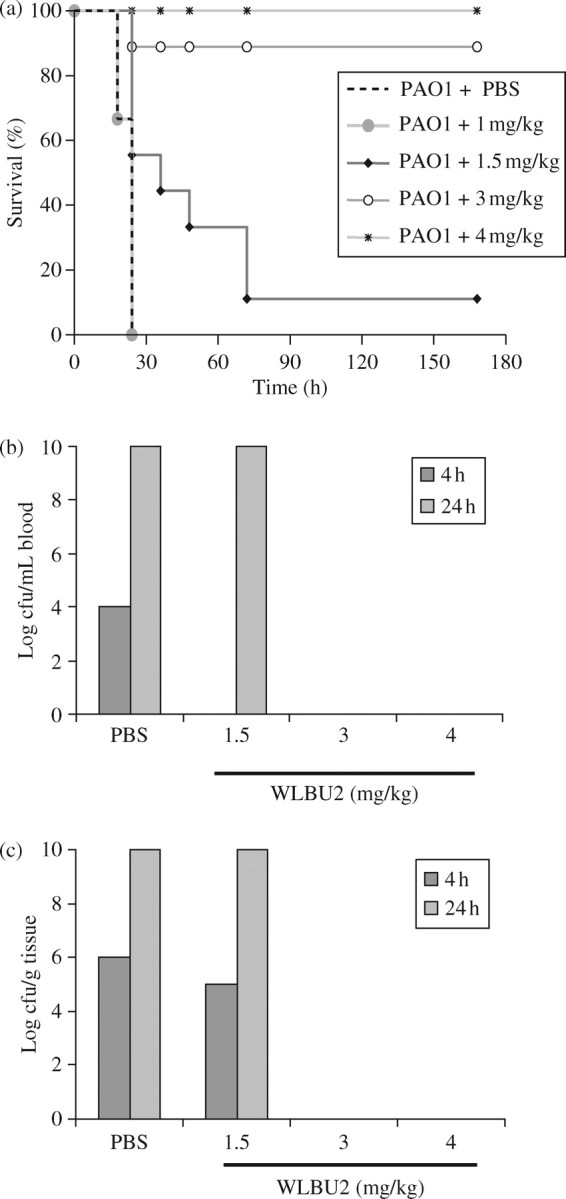
Survival and bacterial loads vary with peptide doses. (a) Kaplan–Meier survival analysis comparing varying doses of WLBU2 versus a PBS control group. (b) Log cfu/mL blood and (c) log cfu/g tissue at 4 and 24 h post-PA01 infusion.
Discussion
The role of antimicrobial peptides in innate immunity has been long established through various studies of the microbial killing mechanisms of neutrophils and macrophages.6 However, it was not until the last two decades that CAPs have been seriously considered as a potential therapeutic source. This is even more critical because of emerging multiple drug resistance among bacteria. Although numerous CAPs with potent antibacterial efficacy in vitro have been described, only few display significant activity in animal models.7,8 An important explanation is that CAP activity is suppressed in biological environments. To address this, a series of studies were initiated to develop de novo CAPs for clinical applications.4,9,10 From these studies, we have demonstrated that the peptide WLBU2 retained its activity against P. aeruginosa in the presence of physiological concentrations of monovalent and divalent cations, human serum and whole blood.4 These studies warranted further investigations of WLBU2 efficacy in vivo.
As can be seen from the experiments (Figure 1), the prophylactic timing of the dose for WLBU2 served a protective role against the infusion of PLD. This dose of 3 mg/kg was as effective as the two high treatment doses for the therapeutic experiments; therefore, the prophylactic window is within the 1 h prior to exposure of an infectious agent. As a therapeutic agent, WLBU2 at concentrations of 3 and 4 mg/kg given 1 h post-infusion of PLD demonstrated increased survival rates when compared with the lower concentration group as well as the PBS control group. This suggests that, at lower concentration, WLBU2 is not able to control the development of sepsis. Our maximal tolerated dose iv was 14 mg/kg and our effective therapeutic dose for 100% survival is 4 mg/kg. Therefore, our therapeutic index is 3.5. This index would be slightly more favourable than similar antimicrobial peptides (e.g. colistin) currently used in clinical scenarios with MDR pathogens.
Taken together, this study demonstrates the successful development of a P. aeruginosa sepsis model in the context of a competent immune system. Using this model, we show that the de novo-derived CAP was prophylactic and therapeutic against P. aeruginosa bacteraemia in mice. These results provide fundamental information that may be useful for evaluating in vivo efficacy of other CAPs. Further, the data underscore the need for comparative studies of WLBU2 to establish the potential of these proposed eCAPs for therapeutic and/or prophylactic antimicrobial advancement.
Acknowledgements
Support for this project was supplied in part by grants to the University of Pittsburgh Cystic Fibrosis Program Project Grant FRIZZE97R0 (Ray Frizzell), National Institutes of Health Minority Supplement grant 1 U19 AI51 661-01 (Sharon L. Hillier and Michael Parniak), NIH grants AR-99-005 #1P30 AR47 372-01, P01 AI039061-09 (Tim Mietzner) and a minority supplement to 3U19 AI 06 540-02 S1 (Phalguni Gupta).
Transparency declarations
None to declare.
References
- 1.Trnobranski PH. Are we facing a ‘post-antibiotic era’?—a review of the literature regarding antimicrobial drug resistance. J Clin Nurs. 1998;7:392–400. doi: 10.1046/j.1365-2702.1998.00181.x. [DOI] [PubMed] [Google Scholar]
- 2.Pitt TL, Sparrow M, Warner M, et al. Survey of resistance of Pseudomonas aeruginosa from UK patients with cystic fibrosis to six commonly prescribed antimicrobial agents. Thorax. 2003;58:794–6. doi: 10.1136/thorax.58.9.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tencza SB, Douglass JP, Creighton DJ, Jr, et al. Novel antimicrobial peptides derived from human immunodeficiency virus type 1 and other lentivirus transmembrane proteins. Antimicrob Agents Chemother. 1997;41:2394–8. doi: 10.1128/aac.41.11.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deslouches B, Phadke SM, Lazarevic V, et al. De novo generation of cationic antimicrobial peptides: influence of length and tryptophan substitution on antimicrobial activity. Antimicrob Agents Chemother. 2005;49:316–22. doi: 10.1128/AAC.49.1.316-322.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tencza SB, Creighton DJ, Yuan T, et al. Lentivirus-derived antimicrobial peptides: increased potency by sequence engineering and dimerization. J Antimicrob Chemother. 1999;44:33–41. doi: 10.1093/jac/44.1.33. [DOI] [PubMed] [Google Scholar]
- 6.Zeya HI, Spitznagel JK. Cationic proteins of polymorphonuclear leukocyte lysosomes. I. Resolution of antibacterial and enzymatic activities. J Bacteriol. 1966;91:750–4. doi: 10.1128/jb.91.2.750-754.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Navon-Venezia S, Feder R, Gaidukov L, et al. Antibacterial properties of dermaseptin S4 derivatives with in vivo activity. Antimicrob Agents Chemother. 2002;46:689–94. doi: 10.1128/AAC.46.3.689-694.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braunstein A, Papo N, Shai Y. In vitro activity and potency of an intravenously injected antimicrobial peptide and its dl amino acid analog in mice infected with bacteria. Antimicrob Agents Chemother. 2004;48:3127–9. doi: 10.1128/AAC.48.8.3127-3129.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phadke SM, Deslouches B, Hileman SE, et al. Antimicrobial peptides in mucosal secretions: the importance of local secretions in mitigating infection. J Nutr. 2005;135:1289–93. doi: 10.1093/jn/135.5.1289. [DOI] [PubMed] [Google Scholar]
- 10.Phadke SM, Islam K, Deslouches B, et al. Selective toxicity of engineered lentivirus lytic peptides in a CF airway cell model. Peptides. 2003;24:1099–107. doi: 10.1016/j.peptides.2003.07.001. [DOI] [PubMed] [Google Scholar]