

Abstract
Increasing evidence shows that estrogens are involved in lung cancer proliferation and progression, and most human lung tumors express estrogen receptor β (ERβ) as well as aromatase. To determine if the aromatase inhibitor anastrozole prevents development of lung tumors induced by a tobacco carcinogen, alone or in combination with the ER antagonist fulvestrant, ovariectomized female mice received treatments with the tobacco carcinogen 4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanone (NNK) along with daily supplements of androstenedione, the substrate for aromatase. Placebo, anastrozole and/or fulvestrant were administered in both an initiation and a promotion protocol of lung tumorigenesis. The combination of fulvestrant and anastrozole given during NNK exposure resulted in significantly fewer NNK-induced lung tumors (mean = 0.5) compared with placebo (mean = 4.6, P < 0.001), fulvestrant alone (mean = 3.4, P < 0.001) or anastrozole alone (mean = 2.8, P = 0.002). A significantly lower Ki67 cell proliferation index was also observed compared with single agent and control treatment groups. Beginning antiestrogen treatment after NNK exposure, when preneoplastic lesions had already formed, also yielded maximum antitumor effects with the combination. Aromatase expression was found mainly in macrophages infiltrating preneoplastic and tumorous areas of the lungs, whereas ERβ was found in both macrophages and tumor cells. Antiestrogens, especially in combination, effectively inhibited tobacco carcinogen-induced murine lung tumorigenesis and may have application for lung cancer prevention. An important source of estrogen synthesis may be inflammatory cells that infiltrate the lungs in response to carcinogens, beginning early in the carcinogenesis process. ERβ expressed by inflammatory and neoplastic epithelial cells in the lung may signal in response to local estrogen production.
Introduction
There is accumulating evidence that estrogen is a major driver of lung cancer. Non-small cell lung cancer (NSCLC) has been shown in many studies to express estrogen receptors (ERs), especially ERβ (1–3), and both genomic (3,4) and non-genomic (3,5) signaling through ERβ has been reported in NSCLC. Proliferative signaling induced by β-estradiol has been reported in NSCLC cells, such as induction of cyclin D1 (6) and activation of mitogen-activated protein kinase (3,5). β-Estradiol causes release of ligands for epidermal growth factor receptor (EGFR) and EGFR activation in NSCLC (5). The enzyme CYP19A1, aromatase, which catalyzes the last step in 17β-estradiol synthesis, is frequently expressed in both normal lung and NSCLC lung tumors from men and women (1,7,8). Aromatase was observed in NSCLC tumor cells themselves as well as in inflammatory cells. Lung tumors thus may be capable of both producing and responding to β-estradiol, and estrogenic signaling may be up-regulated compared with normal lung (1). Local production of estrogens may be part of the chronic inflammatory reaction occurring in lung tumors.
Exposure to exogenous estrogens through hormone replacement therapy (HRT) has negative effects on lung cancer survival. Ganti et al. have reported a significant association between both a younger median age at lung cancer diagnosis and a shorter median survival time in women who used HRT around the time of diagnosis compared with those who did not (9). The Women’s Health Initiative also recently reported a strong adverse effect on survival after a lung cancer diagnosis in women who took HRT containing both β-estradiol and a progesterone (10). In the Women’s Health Initiative randomized trial, more than 16 000 post-menopausal women received placebo or daily HRT for over 5 years. There was a trend toward more lung cancer diagnoses in the HRT group compared with placebo, which did not reach statistical significance. However, the HRT group experienced a significantly greater likelihood of dying from lung cancer. These observations strongly suggest that HRT provides a tumor growth advantage in lung cancer, as it does in breast cancer. Additionally, breast cancer patients who received antiestrogen treatment had significantly lower subsequent lung cancer mortality (11), whereas an increased incidence of lung cancer with HRT use has been reported (12). Together, these studies support the idea of estrogen acting as a promoter of lung cancer aggressiveness and possibly formation. Thus, the estrogen pathway may be a target for lung cancer prevention.
Several aromatase inhibitors (AIs), drugs that block estrogen production, are in current clinical use, including anastrozole (Arimidex), letrozole (Femara) and exemestane (Aromasin). A decreased incidence of primary lung cancer in breast cancer patients treated with exemestane following 2–3 years of tamoxifen therapy has been reported, compared with those patients with continued tamoxifen therapy without an AI (13). Furthermore, Weinberg et al. have shown a 90% decrease in human lung tumor xenograft growth with anastrozole therapy (7), whereas we reported a 44% decrease in lung tumor xenograft growth by the ER antagonist fulvestrant (4).
AIs are known to have a strong inhibitory effect against breast cancer. Data from the Arimidex, Tamoxifen, alone or in combination trial, found that anastrozole in the adjuvant setting decreased the risk of development of contralateral breast cancer by 58% (14). In a randomized, placebo-controlled, double-blind prevention clinical trial, exemestane was demonstrated to significantly reduce primary breast cancers by 65% in healthy post-menopausal women at moderate risk for breast cancer (15). Other large AI prevention trials for breast cancer are ongoing (16). Because AIs are active in breast cancer prevention, we sought to determine if lung cancer development in response to a tobacco carcinogen could be inhibited by an AI, alone or in combination with an ER antagonist. Use of endocrine agents with different modes of action has the potential to more effectively block estrogen signaling, especially because ligand-independent actions of steroid hormone receptors are possible. Here, we report that both anastrozole and fulvestrant alone were effective in blocking lung tumorigenesis in female mice, whereas the combination of anastrozole and fulvestrant had maximum antitumor effects, using a lung carcinogenesis prevention model with two different time points of therapeutic treatment. These antihormonal treatments could potentially reduce the risk of developing lung cancer in women exposed to tobacco products.
Materials and methods
Chemicals and reagents
4-(Methylnitrosoamino)-1-(3-pyridyl)-1-butanone (NNK) was purchased from Toronto Research Chemicals (Toronto, ON) and was dissolved in 0.9% saline. Androstenedione and fulvestrant were purchased from Sigma (St. Louis, MO). Anastrozole was obtained under a Material Transfer Agreement from AstraZeneca.
Mouse model of lung cancer prevention
Female ovariectomized FVB/N mice (6 weeks of age) were exposed to the tobacco carcinogen, NNK [3mg per intraperitoneal (i.p.) injection; twice per week] from weeks 1–4. We have shown previously that this mouse strain is susceptible to lung carcinogenesis induced by NNK (17). All mice received daily subcutaneous (s.c.) supplementation with 0.1mg androstenedione, the substrate for aromatase, for the entire experiment. This experimental model has been used extensively to study AIs in breast cancer models (18). Under these conditions, estrogen production is dependent on conversion of androstenedione by extra-ovarian aromatase and a compensatory increase in aromatase expression by the ovaries following AI administration does not occur. The dose of androstenedione used yields approximately 70 pg/ml circulating plasma estrogen (19), which is within the physiological level for female mice. Each treatment group started with 10 mice based on power calculations to achieve at least a 50% inhibition in number of tumors between treatment groups and to account for random deaths throughout the experiment.
For the first treatment protocol of prevention of lung tumor initiation, treatments and final surviving group sizes were the following: anastrozole (0.1mg/kg; daily oral gavage (p.o.) p.o. ; n = 8), fulvestrant (30mg/kg; 2x/week, s.c.; n = 9), combination (n = 8) or placebo control (peanut oil/3% dimethyl sulfoxide; n = 10). These agents were administered from week 3 until week 15. At week 15, mice were killed, the lungs were inflated with formalin under 25cm intra-alveolar pressure and removed. Tumors were counted under a dissecting microscope and tumor size (defined as surface area, mm2) was measured using Motic Images 2000 software. All histological interpretation was performed under blinded experimental conditions, and results were verified by a lung pathologist (S.D.). Lungs were paraffin embedded and sectioned. Animal care was in strict compliance with the institutional guidelines established by the University of Pittsburgh. We did not observe any systemic toxicity of either the NNK treatments or the therapeutic treatments. Mice did not show weight loss, hair loss, bruising or other general toxicity during these experiments, and at autopsy, all organs outside of the lung appeared normal. For the second protocol for the prevention of lung tumor promotion, NNK was administered for the first 4 weeks as in the prevention of initiation protocol. However, drug treatments were initiated at a later time point (week 9) to allow the formation of lung preneoplasia before start of treatments and to avoid any possible confounding effects of antiestrogens on uptake or metabolism of NNK. We documented presence of preneoplasias [atypical adenomatous hyperplasia (AAH)]) at 9, 15 and 21 weeks following NNK [confirmed by our lung pathologist (S.D.)] in 5 µm H&E sections through the entire lung containing all the lobes, by scanning the entire slide at ×20 power. One slide from each mouse in the treatment groups was evaluated. Anastrozole (0.1mg/kg; daily p.o.; n = 9), fulvestrant (30mg/kg; 2x/week, s.c.; n = 7), combination (n = 10) or placebo control (n = 9) was administered from week 9 until week 21, at which time mice were killed and lung tumors were evaluated. Lungs were prepared as described above.
Immunohistochemistry
Lungs were fixed in 10% buffered formalin. Lung samples were paraffin embedded, sliced and mounted on slides. Slides were stained and evaluated for expression of aromatase, β-estradiol, ERβ and Ki67 as described previously (1,20). β-estradiol has recently been shown to be detectable via immunohistochemistry (21). Antibodies used and dilutions are as follows: ERβ (1:25 overnight at 4°C; H-150; Santa Cruz), CYP19A1 aromatase (1:50 overnight at 4°C; MCA2077; AbD Serotec), β-estradiol (prediluted from BioGenex, AR038-5R as described) (21), Ki67 (1:100 overnight at 4°C; DakoCytomation) and F4/80 macrophage marker (1:25 dilution at 4°C). All antibodies reacted with mouse antigens. Heat-induced antigen retrieval was performed using 10 mmol/l of citrate buffer (pH 6) using a pressure cooker. Endogenous peroxidase was quenched with 3% hydrogen peroxide for 5min at room temperature. Blocking was performed with non-immune normal serum. Immunoreactivity for immunohistochemistry was detected using biotinylated IgG secondary antibodies specific for each primary antibody followed by incubation with diaminobenzidine chromogenic substrate. Ki67-positive cells were counted from 3–5 tumors or AAHs per experimental treatment and presented as the mean number of Ki67-positive cells per field. For dual immunofluorescence experiments, slides were labeled with F4/80 (1:15 dilution at 4°C) and aromatase (1:25 dilution at room temperature) primary antibodies and visualized using Qdot® (Invitrogen) streptavidin conjugates. For each experimental run, the following controls were performed: positive control for ERβ and β-estradiol was breast cancer tissue; placenta was the positive control for aromatase. Representative images are shown in Supplementary Figure 1, available at Carcinogenesis Online. Negative controls were performed for all markers by eliminating the primary antibody. Slides were read and interpreted by a lung pathologist (S.D.). Images were obtained using a Leica DMI3000B inverted microscope at ×20 or ×40 magnification with Leica LAS software, version 3.5.0.
K-ras mutation analysis
Laser capture microdissection, DNA isolation and K-ras mutation analysis were performed as described previously (20). Nested PCR was performed to amplify K-ras (codons 12/13 and 61) followed by denaturing gel electrophoresis to separate mutant from wild-type alleles. Direct sequencing was performed to confirm the mutations.
Statistical analysis
Poisson regression was used to compare the mean number of tumors per mouse in each treatment group, and linear contrasts were used to determine which groups differed significantly from one another. To examine the differences in tumor sizes between treatment groups, a linear mixed effects regression model was utilized, allowing for repeated measures (tumors) within individual mice. For immunohistochemical quantitation, analysis of variance was used. All statistical tests were two-sided with the threshold for statistical significance defined as P < 0.05. Analyses were conducted with SAS v. 9.2 (Cary, NC).
Results
Murine preneoplastic areas and lung tumors that arise from NNK exposure express targets of antiestrogen therapy
We have found previously that ERα and aromatase are expressed in human lung tumors (1). ERα protein using antibodies developed for breast cancer analysis is rarely detected in primary lung tumors and is thought to mainly consist of truncated proteins that may not be functional because ERα−specific agonists do not show significant effects on gene transcription or cell proliferation in lung cancer cells (3). Additionally, high ERβ expression in human lung cancer was an independent negative prognostic predictor of overall survival, whereas ERα was not, and the contribution of aromatase expression resulted in further negative effects on survival (1). To confirm that murine preneoplastic areas and lung tumors induced by NNK also expressed these targets of antiestrogens, we examined expression of aromatase and ERβ as well as β-estradiol, in the lungs of NNK-treated female mice.
Figure 1A–C shows representative immunohistochemical staining of aromatase (Figure 1A), β-estradiol (Figure 1B) and ERβ (Figure 1C), respectively, in preneoplastic areas of the lungs of mice treated with NNK for 4 weeks followed by an additional 5 weeks of no treatment before killing. Preneoplastic areas displayed AAH (Figure 1A–C) and were not present without carcinogen exposure (data not shown). At 9 weeks after beginning NNK treatment, numerous preneoplasias were present throughout the lungs, but no lung tumors were observed. Mean number of AAHs found at 9 weeks in a cross section of the entire lungs was 2.8. The AAHs express two targets of endocrine therapy, aromatase (Figure 1A) and ERβ (Figure 1C). Aromatase positivity was confined almost exclusively to inflammatory cells that had infiltrated preneoplastic areas of the lungs, whereas the abnormal epithelial cells were largely negative (Figure 1A). Most infiltrating inflammatory cells highly positive for aromatase within or near preneoplastic areas of the lungs showed the cytology of macrophages based on large cell size with abundant vacuolated cytoplasm and a central round nucleus (arrow). We confirmed macrophage infiltration of AAHs using the marker F4/80 alone (Supplementary Figure 2A, available at Carcinogenesis Online) and confirmed co-localization of aromatase and the macrophage marker F4/80 with dual immunofluorescence. Most cells positive for F4/80 green fluorescent staining also showed strong aromatase staining (red fluorescence) (Supplementary Figure 3, available at Carcinogenesis Online). We also demonstrated presence of β-estradiol in macrophages within and around preneoplastic areas by immunohistochemistry (Figure 1B). Normal areas of the lungs near AAHs also showed infiltrating macrophages that are highly positive for β-estradiol (Figure 1B, arrow). Positive staining for ERβ was observed in preneoplastic epithelial cells within each lesion (Figure 1C). We also observed that the infiltrating macrophages were ERβ positive (Figure 1C, arrow). Macrophages have been documented previously to express aromatase and ERs (22–29).
Fig. 1.

Hormonal signaling markers are expressed in (A–C) preneoplasias and (D–F) lung tumors from carcinogen treated mice. A and D are representative aromatase staining. B and E are representative β-estradiol staining. C and F are representative ERβ staining. Scale bar= 50μm.
Lung tumors with adenomatous differentiation were present by week 15. Figure 1D–F shows representative immunohistochemical staining of aromatase (Figure 1D), β-estradiol (Figure 1E) and ERβ (Figure 1F), respectively, in adenomas from mice treated with NNK for 4 weeks and killed at 15 weeks. As observed in preneoplasias, adenomatous lung tumors arising from NNK treatment express the targets for antiestrogen therapy, providing the rationale to inhibit the estrogen pathway in this carcinogen-susceptible mouse strain. Similar to preneoplasias, aromatase positivity was again almost exclusively confined to macrophages infiltrating the murine lung tumors (Figure 1D, arrow), and tumor areas with high numbers of macrophages showed the most intense staining for β-estradiol (Figure 1E, arrows), whereas other tumor areas were largely negative. We confirmed presence of macrophages in lung tumor sections with the marker F4/80 (Supplementary Figure 2B, available at Carcinogenesis Online). Both neoplastic epithelial cells and macrophages within tumors were positive for ERβ (Figure 1F, arrow), as was observed in preneoplasias.
Maximum antitumor effects are observed with combined anastrozole and fulvestrant treatment in a prevention of lung tumor initiation protocol
Experiments in human NSCLC xenografts suggest that AIs, which are approved for breast cancer treatment, may also have a role in treatment of lung cancer (7). To determine if blocking estrogen synthesis with an AI with or without blocking the ER with an antiestrogen would prevent lung tumor formation, we tested these agents singly and in combination in the NNK murine lung tumorigenesis model. In order to place estrogen production under the control of tissue aromatase in each animal and to minimize inter-mouse variation in estrogen levels, we used ovariectomized female mice and administered androstenedione on a daily basis, as has been carried out in breast cancer protocols (30). AIs also cannot be administered in the premenopausal state because the ovaries will compensate by over-expressing aromatase (30). NNK was administered during weeks 1–4, with antiestrogen agents administered for 13 weeks beginning at week 3, after which lungs were assessed for presence of tumors. The experimental results are summarized in Supplementary Table IA, available at Carcinogenesis Online. A total of 46 lung tumors were found in the control group, 22 in the anastrozole group, 31 in the fulvestrant group and only 4 in the combination group. Tumors were analyzed for K-ras activating mutations and 35.3% of resulting tumors harbored the K-ras activating mutation G12D, without differences among the groups.
There was a significant relationship between treatment group and median number of tumors per animal (Figure 2A). Anastrozole and the combination treatment resulted in significantly fewer tumors per animal compared with control (anastrozole versus control, P < 0.05; combination versus control, P < 0.001), whereas the number of tumors from fulvestrant treatment was not significantly different from the control group (fulvestrant versus control, P = 0.213). Additionally, the combination group had significantly fewer tumors compared with each single agent group (anastrozole versus combination, P = 0.002; fulvestrant versus combination, P < 0.001). The mean number of tumors per animal in the control group was 4.6, whereas it was 2.8 in the anastrozole treated group and 3.4 in the fulvestrant treated group. There was a mean of only 0.5 tumors per animal in the combination treatment group (an 89% decrease compared with control). The distribution of the number of tumors observed in each treatment group is shown in the histogram in Figure 2A. The maximum number of tumors per animal (eight) was only observed in the control group. About 50% of the mice in the combination treatment group had no visible tumors, whereas the other 50% had only one tumor. Mean number of tumors in the control group in this experiment was not significantly different from the mean number of tumors observed previously per animal in intact FVB/N female mice using this same NNK protocol (average of 5.0±1.3 tumors per mouse; P > 0.05). This suggests that substituting extra-ovarian estrogen production for ovarian estrogen did not itself alter the risk of developing lung cancer in these mice.
Fig. 2.

A) Histogram of number of animals divided by treatment groups and number of tumors found per animal in the prevention of lung tumor initiation protocol. *P < 0.05; **P < 0.005; ***P < 0.001; n.s. = non-significant. Poisson regression was used for analysis of number of tumors. B) Boxplot of the log (tumor size, mm2) by treatment group. A mixed effects model was used for analysis of tumor size. Horizontal line in box of boxplot represents the median. + represents the mean.
In this experiment, there was also no significant effect on mean or median tumor size (P = 0.558) or log (mean or median tumor size) (P = 0.346) with treatment (Figure 2B), even though the range of tumor size in the combination group was reduced compared with other treatments. Although this experimental design was primarily testing for an effect on tumor initiation, there may not have been sufficient time to observe a significant effect of antiestrogens on resulting tumor size.
To confirm an effect on tumor initiation, we examined lung sections histologically for presence of AAH. At the 15 week time point, AAHs are still present along with grossly observed adenomas. Although AAH is an early stage of tumor development, it is an indicator of whether tumor initiation is affected by antiestrogens. The number of AAHs observed by microscopic evaluation of a 5 µm section through the lungs containing all lobes (one section per animal) was significantly decreased with anastrozole (n = 9, mean 2.6; P = 0.010), fulvestrant (n = 8, mean 2.9; P = 0.035) and combination treatment (n = 8, mean 2.5; P = 0.011) compared with control treatment (n = 10, mean 4.7), suggesting the preneoplastic stage of tumorigenesis is inhibited by antiestrogens. However, at this time point, combination treatment was not superior to the single agents in reducing hyperplasias, suggesting the superior effect of combined treatment observed on adenomas might be more related to growth rate of AAHs than on their initial formation.
Ki67 proliferation index is decreased in tumors treated with anastrozole and fulvestrant in the prevention of tumor initiation model
Although a significant decrease in median tumor size was not observed among treatment groups, a significant decrease in the cell proliferation index within the tumors was observed (Figure 3). Anastrozole and fulvestrant treatment alone resulted in a 57.8% (P < 0.001) and 44.1% (P < 0.001) decrease, respectively, in the number of Ki67-positive cells in the tumors compared with control treatment. Further inhibition of 80.2% was observed with the two inhibitors combined (P < 0.001, P < 0.01 and P < 0.001, compared with control, anastrozole and fulvestrant, respectively). This suggests that over an extended time period, the tumor size could potentially be affected due to the lower proliferative rate observed. Decreased Ki67 expression with treatment was also observed in AAHs at this time point. Anastrozole, fulvestrant and the combination resulted in 45.0% (P < 0.001), 19.4% (P = 0.078) and 56.5% (P < 0.001) decreases in Ki67 labeling, respectively, compared with control, providing evidence for suppression of growth of early lesions in addition to decreased growth of adenomas (Supplementary Figure 4A, available at Carcinogenesis Online).
Fig. 3.
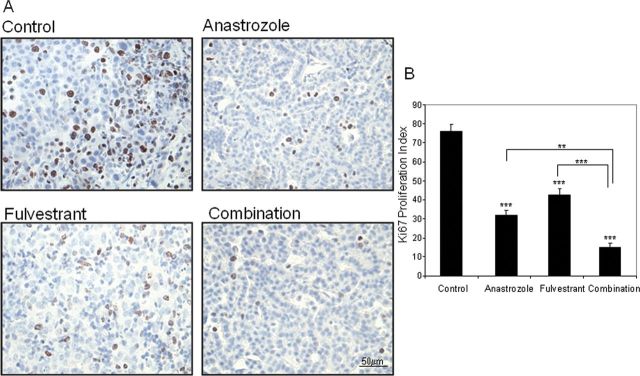
Cell proliferation is significantly decreased in tumors treated with hormonal treatment in the prevention of lung tumor initiation protocol. A) Representative Ki67 staining from lung tumor from each treatment groups. B) Quantitation of Ki67-positive tumor cells in five high-powered fields for 3–5 tumors per treatment group. Analysis of variance; **P < 0.01; ***P < 0.001.
Maximum antitumor effects are observed with combined anastrozole and fulvestrant treatment in a prevention of tumor promotion protocol
We next used the same FVB/N ovariectomized mouse model that was used in the first prevention experiment and introduced a lag period between carcinogen treatment and experimental treatment to allow for the formation of AAH lesions in the lung and to rule out the possibility that inhibition of uptake or changes in metabolism of NNK were responsible for the prevention effect seen in the first protocol. In this protocol, mice are equivalent to ex-smokers who may have preneoplastic airway changes. The total time of the tumorigenesis experiment was lengthened to 21 weeks to allow for observing a treatment effect with the same duration of treatment as in the previous design. The longer time period also may allow for observing effects on tumor growth rate. Results are summarized in Supplementary Table IB, available at Carcinogenesis Online. With this longer time period for tumor development, a total of 117 tumors were found in the control group, 63 in the anastrozole group, 59 in the fulvestrant group and only 39 in the combination group. In this experimental model, K-ras activating mutations (G12D) were detected in 77% of the tumors analyzed with no significant difference observed among treatment groups.
As in the prevention of initiation experimental protocol, we observed a significant relationship between treatment group and median number of tumors per animal (Figure 4A). Each single agent and the combination treatment resulted in significantly fewer adenomatous tumors per animal compared with control (anastrozole versus control, P < 0.001; fulvestrant versus control, P < 0.001; combination versus control, P < 0.001). Additionally, the combination treatment group had significantly fewer tumors compared with each single agent treatment group (P < 0.001). The mean number of tumors per animal in the control group was 16.5, whereas it was 7.3 in the anastrozole treated group and 9.2 in the fulvestrant treated group. There was a mean of 4.2 tumors per animal in the combination treatment group, a 74.5% decrease from control. The distribution of the tumors observed in each treatment group is shown in the histogram of Figure 4A, with a shift from a greater proportion of animals that are tumor-bearing with more tumors in the lungs in the control group, toward a lesser proportion of animals that are tumor-bearing, with fewer tumors in the lungs in the combination treatment group.
Fig. 4.

A) Histogram of number of animals divided by treatment groups and number of tumors found per animal in the prevention of lung tumor promotion protocol. ***P < 0.001. Poisson regression was used for analysis of number of tumors. B) Boxplot of the log (tumor size) by treatment group. A mixed effects model was used for analysis of tumor size. Horizontal line in box of boxplot represents the median. + represents the mean.
We observed similar decreases in the incidence of AAHs (which were also present at 21 weeks along with adenomas in this protocol) with antiestrogen treatments. Counting the number of lesions in sections of the lungs containing all lobes (one section per animal) showed a significant decrease with anastrozole (n = 9, mean 3.1; P = 0.008), fulvestrant (n = 7, mean 3.6; P = 0.048) and combination treatment (n = 10, mean 1.9; P < 0.001) compared with control treatment (n = 9, mean 5.8). The lower number of AAHs found in the combination group trended toward significance compared with the anastrozole group (P = 0.097) and was significant compared with the fulvestrant group (P = 0.038). This confirms the finding in the first protocol that some of the antiestrogen effect is due to suppression of formation of hyperplasias; however, the maximum inhibition of AAH (47% with combination treatment) is lower than the 74.5% inhibition observed for adenoma formation, suggesting that blocking proliferation of lesions is the stronger effect.
Unlike the prevention of initiation experiment, there was a significant effect on both mean and median tumor size with treatment using the promotion protocol (Figure 4B). The mean tumor size was 0.45mm2 in the control group, 0.25mm2 in the anastrozole group and 0.24mm2 in the fulvestrant group. The mean tumor size in the combination group was 0.17mm2. Each group had a significantly decreased mean tumor size compared with control treated mice (P < 0.001 for all comparisons). The combination group had the smallest tumors and was significantly different compared with each single agent group (P < 0.001 all combinations).
Ki67 proliferative index is maximally decreased by combined therapy in the prevention of tumor promotion protocol
Figure 5A shows representative immunostaining of Ki67 in each treatment group and the quantitative results in Figure 5B. A decrease in Ki67 immunostaining of 18.2% (P < 0.05) and 10.22% (P = 0.3230) was observed with anastrozole and fulvestrant treatment alone, respectively. Ki67-positive cells in the combination treatment resulted in a further decrease of 34.8% compared with control (P = 0.0031) (fulvestrant versus combination, P < 0.05; anastrozole versus combination, P = 0.0701). A decreased growth rate over a longer treatment probably resulted in significantly smaller tumors with antiestrogen treatments. Furthermore, large decreases in Ki67 expression with treatment were observed in AAHs at the 21 week time point. Anastrozole, fulvestrant and the combination resulted in a 45% (P < 0.001), 22% (P < 0.05) and 80% (P < 0.001) decrease in Ki67 labeling, respectively, compared with control, providing additional evidence of the strong effect of these drugs on suppressing proliferation of the nascent lesions (Supplementary Figure 4B, available at Carcinogenesis Online).
Fig. 5.
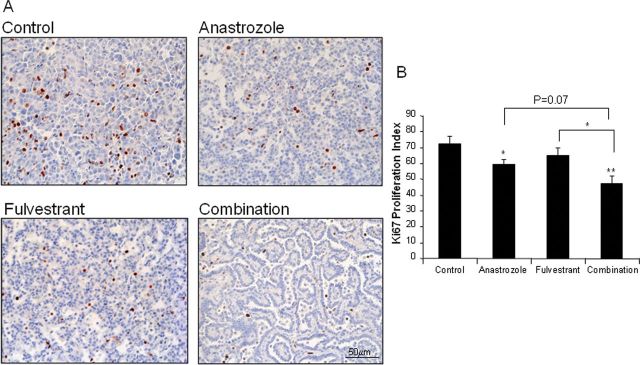
Cell proliferation is significantly decreased in tumors treated with hormonal treatment in the prevention of lung tumor promotion protocol. A) Representative Ki67 staining from lung tumor from each treatment groups. B) Quantitation of Ki67-positive tumor cells in five high-powered fields for 3–5 tumors per treatment group. Analysis of variance; *P < 0.05; **P < 0.01.
We also examined apoptosis using the Tunel assay and found that little or no staining was observed in the tumors from any of the treatment groups, suggesting that the effect of antiestrogens in this model is not due to inducing tumor cell death. This supports the observation with Ki67 labeling that the effect appears to involve prevention of proliferation. In both experimental models, expression of aromatase and ERβ was not changed in the limited number of tumors that arose despite these treatments (data not shown). These markers were expressed in the tumors to the same extent in all treatment groups, suggesting that up-regulation of ERβ or aromatase did not occur in the tumors that were resistant to treatment.
Discussion
A hormonal role in the pathology of lung cancer is well documented. Both intra-tumoral aromatase and ERs have been demonstrated to play important roles in growth pathways in NSCLC and have been studied as potential therapeutic targets for this disease (1,3,7,31). We report here for the first time using a preclinical animal model of lung cancer prevention that modulating both axes of the estrogen signaling pathway (receptor and ligand) has profound antitumorigenic effects in the lung. The tobacco carcinogen NNK has been widely utilized in mouse models of lung tumorigenesis because of its direct relevance to tobacco-induced human lung cancer. Fujimoto et al. have recently demonstrated that NNK-induced lung tumors in mice are highly comparable to human lung adenocarcinomas based on gene expression signatures (32). Tumors arising from NNK treatment in these experiments had an adenomatous histology and an incidence of the activating G12D K-ras mutation (a common mutation found in human lung adenocarcinoma) of 35–77%, similar to reported previously K-ras mutation rates in the FVB/N mouse strain (20). The G12D mutation has also been reported to be the most frequent K-ras mutation found in human lung tumors from non-smokers, thus these results may be relevant to non-smokers as well (33).
The number of tobacco carcinogen-induced lung tumors was dramatically decreased with down-modulation of the estrogen pathway. Strong preventive effects were observed in an experimental protocol where the endocrine therapies were administered concurrent with the tobacco carcinogen NNK (to inhibit initiation of carcinogenesis as well as subsequent proliferation) as well as when the carcinogen was given first, followed by therapy (to inhibit promotion of preneoplasia). The effect, therefore, appears to be primarily due to direct inhibition of estrogen pathways rather than to inhibition of NNK uptake or changes in its metabolism that might be caused by modulating estrogens. The inhibitory effects involve both a suppression of preneoplasia formation as well as an inhibition of proliferation. Inhibition of proliferation in nascent preneoplastic lesions by antiestrogens appears to be stronger relative to inhibition of lesion formation. Tumor proliferative index was also suppressed by endocrine therapies, and the cumulative effect was a reduction in tumor size over time. These results suggest that targeting estrogens can inhibit lung cancer development during induction of damage to the airways induced by tobacco carcinogens, as well as after airway damage from tobacco exposure has occurred. However, a greater overall antitumor effect was seen when intervention was carried out as preneoplasias were forming, rather than after their formation was permitted.
The greatest antitumor effects were observed with the simultaneous administration of both an AI and an antiestrogen. We observed a greater effect on tumor number and proliferative index with anastrozole than with fulvestrant; however, only a single dose of each drug was used. The dose of fulvestrant used in this study is comparable to other studies in breast cancer (34,35), although below the 5mg/week maximum tolerated dose in animals. It is possible that the maximally tolerated dose might have increased the effect of fulvestrant, but fulvestrant at a higher dose was still inferior to an AI in a breast cancer model (36,37). Preclinical models of breast cancer prevention have also shown superior results with the combination of an AI and an antiestrogen compared with single agents (36,37), consistent with our findings.
We reported previously that ERβ is expressed in 96% of lung tumors and aromatase in 61%, from both male and female patients, whereas full-length ERα is rarely expressed (1). Additionally, in an analysis of overall survival, a hazards ratio of 1.64 was observed for patients with high ERβ compared with low ERβ in their lung tumors. When expression of other hormonal and growth factors was taken into account, including aromatase, progesterone receptor and EGFR expression, the hazards ratio increased to 6.60. In an additional report, high aromatase expression in lung tumors conferred a worse prognosis for survival; however, this was only observed in women aged 65 years and above (38). When aromatase was combined with ERβ expression levels, the survival rate was worse than with either marker alone, and this was found in both male and female patients (38). These clinical data add support to our observations reported here in a murine model.
The prevention of lung cancer by antiestrogens reported here is consistent with studies that showed an increase in lung cancer incidence or mortality in women with exposure to HRT (9,10,12). Some reports, however, suggest that HRT use before lung cancer diagnosis could actually protect women from developing lung cancer, particularly if they smoked (39). In post-menopausal women with ER-positive lung tumors, an inverse relationship was observed between HRT use and NSCLC risk (40). This association was not observed in women with ER-negative lung tumors. The reasons for different reported effects of HRT on lung cancer incidence are not known. Timing or duration of HRT use could be factors because estrogen can stimulate the immune system, which might produce protective effects against the emergence of neoplastic changes. It is also possible that exogenous hormone use reduced local estrogen production in the lungs or inflammatory cells by negative feedback regulation. Catabolism of tobacco carcinogens and estrogens occur through the same inducible cytochrome p450 enzymes, so HRT might also accelerate tobacco carcinogen catabolism. Estrogen is also known to induce apoptosis in some cell types, so the balance between cell division and cell death in lung epithelia might be altered by HRT use. Contradictory and counter-intuitive effects of HRT use on incidence of heart disease have been observed (41), despite evidence that estrogen protects against heart disease, suggesting that the systemic effect of estrogen supplementation (particularly in combination with progesterone) is complex.
An unexpected finding of our mouse experiments was that expression of aromatase and β-estradiol was mainly localized to inflammatory cells in the lungs, many of which appeared to be of the macrophage lineage. We cannot exclude the possibility that other types of inflammatory cells were present that are aromatase positive because lymphocytes are also reported to express aromatase (42,43). Tobacco carcinogens elicit strong inflammatory reactions in response to cellular and DNA damage, which can magnify the carcinogenesis process through the production of reactive oxygen species and the pro-tumorigenic activity of released cytokines (44). We observed no differences in the number of inflammatory cells infiltrating the lungs with antiestrogen treatment, suggesting the antitumor effects of these agents are downstream of inflammatory cell actions, rather than preventing infiltration. Although macrophages have been described as being capable of expressing aromatase and both ERα and β (22–29), to our knowledge, this is the first report of local estrogen production being part of the inflammatory reaction in lungs exposed to tobacco carcinogens. Autocrine response of macrophages to estrogen production could be mediated by both ERα and β. This finding is similar to observations in breast cancer, in which crown-like structures that consist of necrotic adipocytes surrounded by macrophages within murine mammary glands and visceral fat were associated with increased aromatase expression and elevated estrogen levels (23). In the breast cancer model, the release of cytokines by activated macrophages was thought to stimulate aromatase expression in preadipocytes (23). However, it is also possible that the macrophages themselves could be a source of aromatase activity in crown structures. Although we observed previously that aromatase expression in the malignant epithelial cell compartment in tumors of NSCLC patients (1), we also observed aromatase localization in macrophages infiltrating these tumors. In the animal model, aromatase expression was rarely observed in lung tumor cells that formed by 21 weeks, so aromatase expression by tumor cells in mice may not be induced until late stages of lung tumorigenesis. For practical reasons, we killed mice to quantify lung tumors at the adenoma stage, before the tumors had become highly vascularized and before metastasis to lymph nodes or other organs. Neither aromatase expression nor ERβ expression were modulated in murine lungs with antiestrogen treatment, suggesting that up-regulation in response to estrogen pathway inhibition did not occur.
There are several ongoing clinical trials combining anastrozole and fulvestrant in breast cancer treatment such as SoFEA, FACT, Southwestern Oncology Group SWOG-S0226 and D6997C00057 (45). Till date, superior clinical activity of an antiestrogen and an AI has not been observed in FACT (46), whereas results from SWOG-S0226 are not yet available. Whether the combination that we found superior in lung cancer prevention in mice would also show superiority to single agents in treatment or prevention settings in humans is unknown. The extent to which blocking both the ER and estrogen synthesis would be superior may depend on many factors. For example, the extent of ligand-independent signaling of the ER might allow for ER signaling despite blocking of estrogen synthesis.
Hormonal strategies are now being rapidly translated to the clinic for lung cancer treatment. We demonstrated previously that the combination of fulvestrant and the EGFR TKI gefitinib in post-menopausal women with NSCLC was well-tolerated (47). Such an approach is designed to interrupt ER-EGFR cross-activation (5). A phase II clinical trial for combination therapy with fulvestrant and erlotinib versus erlotinib alone is currently underway (48). A phase II randomized trial of fulvestrant and anastrozole as consolidation therapy in post-menopausal women with advanced NSCLC is also in progress (49). Fulvestrant competes with β-estradiol for binding to the ER and reduced estrogen levels with an AI might act to increase fulvestrant binding efficacy. Our data suggest that primary lung cancer prevention or early stage lung cancer intervention with these strategies as secondary prevention may be beneficial and remains unexplored at this time. Because tumors from both male and female lung cancer patients express ERs and aromatase, and because intra-tumoral estrogen may be produced in inflammatory cells and has been found in tumors from men as well as women, antiestrogen therapies could potentially benefit all patients. Experiments to test whether antiestrogens could inhibit lung tumorigenesis in male mice are warranted. Further investigation in the clinic, including identification of those most probably to benefit, is warranted.
Supplementary material
Supplementary Table 1 and Figures 1–4 can be found at http://carcin.oxfordjournals.org/
Funding
Supported by P50 CA090440, SPORE in Lung Cancer, to J.M.S. and R21 CA129260 to P.K.. This project used the UPCI Animal Facility and Tissue and Research Pathology Services Facility and was supported in part by the Comprehensive Cancer Center award P30CA047904.
Supplementary Material
Acknowledgements
We thank Ms Lisa Chedwick and Ms Marie Acquafondata for immunohistochemical processing.
Glossary
Abbreviations:
- AAH
atypical adenomatous hyperplasia
- AI
aromatase inhibitor
- ER
estrogen receptor
- HRT
hormone replacement therapy
- NNK
4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanone
- NSCLC
non-small cell lung cancer
Footnotes
Conflict of Interest Statement: None declared.
References
- 1. Stabile L.P, et al. (2011). Combined analysis of estrogen receptor beta-1 and progesterone receptor expression identifies lung cancer patients with poor outcome Clin. Cancer Res. 17 154–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schwartz A.G, et al. (2005). Nuclear estrogen receptor beta in lung cancer: expression and survival differences by sex Clin. Cancer Res. 11 7280–7287 [DOI] [PubMed] [Google Scholar]
- 3. Hershberger P.A, et al. (2009). Estrogen receptor beta (ERbeta) subtype-specific ligands increase transcription, p44/p42 mitogen activated protein kinase (MAPK) activation and growth in human non-small cell lung cancer cells J. Steroid Biochem. Mol. Biol. 116 102–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stabile L.P, et al. (2002). Human non-small cell lung tumors and cells derived from normal lung express both estrogen receptor alpha and beta and show biological responses to estrogen Cancer Res. 62 2141–2150 [PubMed] [Google Scholar]
- 5. Stabile L.P, et al. (2005). Combined targeting of the estrogen receptor and the epidermal growth factor receptor in non-small cell lung cancer shows enhanced antiproliferative effects Cancer Res. 65 1459–1470 [DOI] [PubMed] [Google Scholar]
- 6. Hershberger P.A, et al. (2005). Regulation of endogenous gene expression in human non-small cell lung cancer cells by estrogen receptor ligands Cancer Res. 65 1598–1605 [DOI] [PubMed] [Google Scholar]
- 7. Weinberg O.K, et al. (2005). Aromatase inhibitors in human lung cancer therapy Cancer Res. 65 11287–11291 [DOI] [PubMed] [Google Scholar]
- 8. Mah V, et al. (2007). Aromatase expression predicts survival in women with early-stage non small cell lung cancer Cancer Res. 67 10484–10490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ganti A.K, et al. (2006). Hormone replacement therapy is associated with decreased survival in women with lung cancer J Clin Oncol 24 59–63 [DOI] [PubMed] [Google Scholar]
- 10. Chlebowski R.T, et al. (2009). Oestrogen plus progestin and lung cancer in postmenopausal women (Women’s Health Initiative trial): a post-hoc analysis of a randomized controlled trial Lancet 374 1243–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bouchardy C et al. (2011)Lung cancer mortality risk among breast cancer patients treated with anti-estrogens Cancer 117 1288–1295 [DOI] [PubMed] [Google Scholar]
- 12. Slatore C.G et al. (2010)Lung cancer and hormone replacement therapy: association in the vitamins and lifestyle study J. Clin. Oncol. 28 1540–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Coombes R.C, et al. (2004). A randomized trial of exemestane after two to three years of tamoxifen therapy in postmenopausal women with primary breast cancer N. Engl. J. Med. 350 1081–1092 [DOI] [PubMed] [Google Scholar]
- 14. Cuzick J et al. (2010)Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 10-year analysis of the ATAC trial Lancet Oncol. 11 1135–1141 [DOI] [PubMed] [Google Scholar]
- 15. Goss P.E et al. (2011)Exemestane for breast-cancer prevention in postmenopausal women N. Eng. J. Med. 364 2381–2391 [DOI] [PubMed] [Google Scholar]
- 16. Cuzick J. (2005). Aromatase inhibitors for breast cancer prevention J. Clin. Oncol. 23 1636–1643 [DOI] [PubMed] [Google Scholar]
- 17. Stabile L.P, et al. (2006). Transgenic mice overexpressing hepatocyte growth factor in the airways show increased susceptibility to lung cancer Carcinogenesis 27 1547–1555 [DOI] [PubMed] [Google Scholar]
- 18. Chumsri S, et al. (2011). Aromatase inhibitors and xenograft studies Steroids 76 30–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Foster P.A, et al. (2008). A new therapeutic strategy against hormone-dependent breast cancer: the preclinical development of a dual aromatase and sulfatase inhibitor Clin. Cancer Res. 14 6469–6477 [DOI] [PubMed] [Google Scholar]
- 20. Stabile L.P, et al. (2008). Therapeutic targeting of human hepatocyte growth factor with a single neutralizing monoclonal antibody reduces lung tumorigenesis Mol. Cancer Ther. 7 1913–1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meireles S.I, et al. (2010). Early changes in gene expression induced by tobacco smoke: evidence for the importance of estrogen within lung tissue Cancer Prev. Res. 3 707–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mor G, et al. (1998). Macrophages, estrogen and the microenvironment of breast cancer J. Steroid Biochem. Mol. Biol. 67 403–411 [DOI] [PubMed] [Google Scholar]
- 23. Subbaramaiah K, et al. (2011). Obesity is associated with inflammation and elevated aromatase expression in the mouse mammary gland Cancer Prev. Res. 4 329–346 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24. Lambert K.C, et al. (2004). Estrogen receptor-▯ deficiency promotes increased TNF-a secretion and bacterial killing by murine macrophages in response to microbial stimuli in vitro J. Leukocyte Biol. 75 1166–1172 [DOI] [PubMed] [Google Scholar]
- 25. Piesta A, et al. (2009). The influence of mating on estrogen receptor alpha protein level in spleen and uterine macrophages in female mice Reproductive Biol. 9 225–240 [DOI] [PubMed] [Google Scholar]
- 26. Calippe B, et al. (2008). Chronic estradiol administration in vivo promotes the proinflammatory response of macrophages to TLR4 activation: involvement of the phosphatidylinositol kinase pathway J. Immunol. 180 7980–7988 [DOI] [PubMed] [Google Scholar]
- 27. Kawano N, et al. (2004). Identification and localization of estrogen receptor a- and b-positive cells in adult male and female mouse intestines at various estrogen levels Histochem. Cell Biol. 121 399–405 [DOI] [PubMed] [Google Scholar]
- 28. Kramer P.R, et al. (2002). 17-Beta-estradiol regulates expression of genes that function in macrophage activation and cholesterol homeostasis J. Steroid Biochem. Mol. Biol. 81 203–216 [DOI] [PubMed] [Google Scholar]
- 29. Subbaramaiah K, et al. (2009). Oestrogen modulates human macrophage apoptosis via differential signaling through oestrogen receptor-▯ and β. J. Cell Mol. Med. 13 2317–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lu Q, et al. The effects of aromatase inhibitors and antiestrogens in the nude mouse model Breast Cancer Res. Treat 50 63–71 [DOI] [PubMed] [Google Scholar]
- 31.Marquez–Garban D.C, et al. Targeting aromatase and estrogen signaling in human non-small cell lung cancer. Ann. NY Acad. Sci. (2009);1155:194, 205. doi: 10.1111/j.1749-6632.2009.04116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fujimoto J, et al. (2010). Comparative functional genomics analysis of NNK tobacco-carcinogen induced lung adenocarcinoma development in Gprc5a-knockout mice PloS One 5: e11847. doi:10.1371/journal.pone.0011847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Riely G.J, et al. (2008). Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma Clin. Cancer Res. 14 5731–5734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yoneya T, et al. (2009). Effects of CH4893237, a new orally active estrogen receptor downregulator, on breast cancer xenograft models with low serum estrogen levels Oncol. Rep. 21 747–755 [PubMed] [Google Scholar]
- 35.Hoffman J, et al. Characterization of new estrogen receptor destabilizing compounds: effects on estrogen-sensitive and tamoxifen-resistant breast cancer. J. Natl Cancer Inst. (2004);96:210, 208. doi: 10.1093/jnci/djh022. [DOI] [PubMed] [Google Scholar]
- 36. Jelovac D, et al. (2005). Additive antitumor effect of aromatase inhibitor and antiestrogen fulvestrant in a postmenopausal breast cancer model Cancer Res. 65 5439–5444 [DOI] [PubMed] [Google Scholar]
- 37.Macedo L.F, et al. Combination of anastrozole with fulvestrant in the intratumoral aromatase xenograft model. Cancer Res. (2008);68:3516, 3522. doi: 10.1158/0008-5472.CAN-07-6807. [DOI] [PubMed] [Google Scholar]
- 38. Mah V, et al. (2011). Expression levels of estrogen receptor beta in conjunction with aromatase predict survival in non-small cell lung cancer Lung Cancer 74 318–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ramnath N, et al. (2007). Hormone replacement therapy as a risk factor for non-small cell lung cancer: results of a case-control study Oncology 73 305–310 [DOI] [PubMed] [Google Scholar]
- 40. Schwartz A.G, et al. (2007). Reproductive factors, hormone use, estrogen receptor expression and risk of non-small cell lung cancer in women J. Clin. Oncol. 25 5785–5792 [DOI] [PubMed] [Google Scholar]
- 41.Harman S.M, et al. Timing and duration of menopausal hormone treatment may affect cardiovascular outcomes. Am. J. Med. (2011);124:199, 205. doi: 10.1016/j.amjmed.2010.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Berstein L.M, et al. (1998). Ability of lymphocytes infiltrating breast-cancer tissue to convert androstenedione Int. J. Cancer 77 485–487 [DOI] [PubMed] [Google Scholar]
- 43. Berstein L.M, et al. (2002). Aromatase (CYP19) expression in tumor-infiltrating lymphocytes and blood mononuclears J. Cancer Res. Clin. Oncol. 128 173–176 [DOI] [PubMed] [Google Scholar]
- 44. Coussens L.M, et al. (2002). Inflammation and cancer Nature 420 860–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Leary A, et al. (2006). Combination therapy with aromatase inhibitors: the next era of breast cancer treatment? Br. J. Cancer 95 661–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bergh J, et al. (2012). FACT: an open-label, randomized phase III study of fulvestrant and anastrozole in combination compared with anastrozole alone as first-line therapy for patients with receptor-positive postmenopausal breast cancer J. Clin. Oncol. 30 1919–1925 [DOI] [PubMed] [Google Scholar]
- 47. Traynor A.M, et al. (2009). Pilot study of gefitinib and fulvestrant in the treatment of post-menopausal women with advanced non-small cell lung cancer Lung Cancer 64 51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Garon E.B, et al. (2011). Randomized multicenter phase II study of erlotinib (E) + fulvestrant (F) in previously treated advanced non-small cell lung cancer (NSCLC) J. Clin. Oncol. 29, (suppl; abstr TPS216) [Google Scholar]
- 49. McLaughlin B.T, et al. (2011). A phase II randomized trial of anastrozole (A) and fulvestrant (F) as consolidation therapy in postmenopausal women with advanced non-small cell lung cancer who have received first-line platinum-based chemotherapy with or without bevacizumab J. Clin. Oncol. 29, (suppl; abstr TPS212) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.