

Background: Potent inhibitors of the tuberculosis drug target KasA are needed.
Results: Three-position analogs of the natural product thiolactomycin (TLM) were designed based on transient one-dimensional NOEs that reveal the relative orientation of TLM and a pantetheine analog bound simultaneously to KasA.
Conclusion: Three-position analogs of TLM bind to KasA with increased potency.
Significance: Optimization of TLM will lead to improved inhibitors of KasA.
Keywords: Drug Design, Enzyme Inhibitors, Fatty Acid Synthase, Mycobacterium tuberculosis, Nuclear Magnetic Resonance, KasA, Lead Optimization, Structure Activity Relationships, Thiolactomycin, Transient one-dimensional NOE
Abstract
Thiolactomycin (TLM) is a natural product inhibitor of KasA, the β-ketoacyl synthase A from Mycobacterium tuberculosis. To improve the affinity of TLM for KasA, a series of TLM analogs have been synthesized based on interligand NOEs between TLM and a pantetheine analog when both are bound simultaneously to the enzyme. Kinetic binding data reveal that position 3 of the thiolactone ring is a suitable position for elaboration of the TLM scaffold, and the structure-activity relationship studies provide information on the molecular features that govern time-dependent inhibition in this enzyme system. These experiments also exemplify the utility of transient one-dimensional NOE spectroscopy for obtaining interligand NOEs compared with traditional steady state two-dimensional NOESY spectroscopy.
Introduction
The β-ketoacyl-AcpM synthase (KasA)5 from Mycobacterium tuberculosis is an essential enzyme in the mycobacterial fatty acid biosynthesis (FAS-II) pathway (Fig. 1A) and is thought to be a promising target for antibacterial discovery (1, 2). The isolation of several natural product inhibitors of the KAS enzymes, including thiolactomycin (TLM), platensimycin, and cerulenin (Fig. 1B), has spurred a renewed interest in developing KasA inhibitors (3–9). TLM, a natural product thiolactone, shows broad spectrum antibacterial activity against Gram-positive and Gram-negative bacteria and mycobacteria (4, 10, 11). Despite moderate Minimum Inhibitory Concentration (62.5 μm against M. tuberculosis), TLM is a promising lead molecule for the development of potent KasA inhibitors because of its favorable physicochemical properties, low cytotoxicity, high bioavailability, and activity in animal infection models (10, 12). Because TLM inhibits wild-type KasA with a Kd of only ∼200 μm (11), there is significant interest in optimizing the interactions between TLM and the enzyme to improve both affinity and selectivity.
FIGURE 1.

A, the type II fatty acid biosynthesis pathway in Mycobacterium tuberculosis. B, natural product KAS inhibitors.
Interligand NOEs (ILOEs) between small molecule ligands can be used as a powerful tool to aid and guide fragment-based drug discovery (13–15). If two or more small molecules bind to a macromolecule in close proximity to each other, the strong negative ILOEs that develop in their bound complex geometries can be observed even in the presence of substoichiometric amounts of the target, provided there is a rapid exchange between the bound and free state (15). Pairs of suspected weak inhibitors can be chosen as prospects for binding to a protein either based on structural characteristics or by screening chemical libraries. Protein-mediated ILOEs can then assist in pharmacophore identification and guide the design and synthesis of bidentate ligands using the weak binding fragments as building blocks.
Two-dimensional NOESY techniques are the methods of choice to investigate structural relationships in large biological molecules, primarily because all of the data are collected at once, and the expected NOEs are large and negative. However, ILOEs between small molecules can be very weak and difficult to detect and differentiate because of chemical shift overlaps and background issues typical of two-dimensional NOESY experiments (16). Such issues can adversely limit the application of the method and the ability to obtain and interpret NOE data. Overcoming these limitations would require longer mixing times (beyond 500 ms), thereby excluding the early time points of the NOE buildup that are crucial for distance measurements.
In partial mitigation of these issues, we sought to extend traditional ILOE NMR by use of the selective one-dimensional NOE technique pioneered by Shaka and co-workers (16, 17) and later refined by Hu and Krishnamurthy (18). Here, as opposed to the conventional steady state approach, the transient NOEs arising only from selectively inverted resonances are detected. Pairs of selective pulses and pulsed field gradients are used in a double pulsed field gradient spin echo (DPFGSE) sequence to cleanly select and invert specific resonances such that only those signals related to NOEs originating from the inverted signal are detected. Background and chemical shift overlap issues are therefore removed (16, 17). Selective one-dimensional NOE experiments enable significantly increased sensitivity per unit of data collection time, effectively extending NOE detection and distance limits, and better supporting systems with short lifetimes. In addition, NOE buildup curves can easily be constructed to include shorter mixing times.
In this article, we demonstrate the use of this technique for detecting ILOEs between two ligands bound to KasA. We have previously shown that TLM is a slow onset inhibitor of the KasA acyl enzyme (11). This is consistent with the knowledge that TLM mimics the malonyl group of malonyl-AcpM, the second substrate in the ping-pong reaction catalyzed by KasA. Structural data suggest that TLM might bind to KasA in the presence of ligands that occupy the pantetheine-binding channel (19). To test this hypothesis, we synthesized a pantetheine analog (PK940) and used ILOE NMR spectroscopy to analyze the interaction of this compound with TLM and KasA. Because malonyl-AcpM and TLM interact preferentially with the KasA acyl-enzyme, the C171Q KasA mutant was used for many of the experiments because this mutation has previously been shown to lead to structural changes in the active site that mimic acylation of Cys-171 (3, 20). Based on these studies, we then synthesized TLM analogs that have higher affinity for KasA than the parent compound.
EXPERIMENTAL PROCEDURES
Chemicals
5(R,S)-Thiolactomycin was obtained from Sigma. Chemicals for analog synthesis were obtained from commercial sources and used without further purification.
5(R)-Thiolactomycin and TLM Analogs
Enantiomerically pure 5(R)-TLM was synthesized using a reported protocol (6, 21, 22). This method was also used for the preparation of the position 3 analogs shown in Table 1. Details of the synthesis and analytical data for 5(R)-TLM are given in supplemental Fig. S1, whereas the synthesis of the analogs will be reported in a separate publication.
TABLE 1.
Kinetic and thermodynamic constants for the interaction of the TLM analogs with wild-type and C171Q KasA
Dissociation constants were determined using the fluorescence based assay described previously (11). The inhibitor stock solutions, dissolved in Me2SO or 50 mm Tris-HCl, 150 mm NaCl, pH 8.5, were titrated into the enzyme (1 μm) in 50 mm Tris-HCl, 150 mm NaCl, 1 mm DTT, pH 8.5. Changes in fluorescence were monitored using excitation and emission wavelengths of 280 and 337 nm, respectively. By convention, Kd is used to identify the dissociation constant for wild-type KasA, whereas Ki and Ki* are used for the initial and final equilibrium constants for inhibitors binding to C171Q KasA.

a Reported by Machutta et al. (11). The ClogP values were calculated by Chemdraw.
Synthesis of PK940
PK940 (Fig. 2) was synthesized using a published protocol (22, 23). Details of the synthesis and analytical data are given in supplemental Fig. S2.
FIGURE 2.

1H NMR spectrum of TLM and PK940 in D2O at 700 MHz. TLM (1): 1H NMR (D2O) 6.23 (dd, 1H, H10), 5.45 (s, 1H, H8), 5.1 (d, 1H, H11, J = 18Hz), 4.9 (d, 1H, H11, J = 10.4 Hz), 4.7 (H2O), 1.50 (s, 3H, H7), 1.44 (s, 3H, H12), 1.41 (s, 3H, H6). The TLM methyls at positions 6, 7s and 12 are at 1.41, 1.50s and 1.44 ppms respectively. PK940: 1H NMR (D2O) 3.81 (s, 1H, H6′), 3.35 (d, 1H, H8′), 3.22 (d, 1H, H8′), 3.06 (dd, 2H, H4′), 1.32 (dd, 2H, H3′), 1.15 (dd, 2H, H2′), 0.75 (s, 3H, prochiral H9′/H10′), 0.714 (s, 3H, prochiral H9′/H10′), 0.708 (dd, 3H, H1′). Stacked doublet of doublets (H1′, 0.708 ppm) were resolved from geminal methyl peaks (H9′ and H10′, 0.714 and 0.75 ppm) by examining Heteronuclear Multiple-Quantum Correlation, Heteronuclear Multiple Bond Correlation data, and DPFGSE line shapes.
Enzymes
C171Q KasA and wild-type KasA were expressed in Mycobacterium smegmatis mc2155 and purified as described previously (11). Following purification, the enzymes were stored at 4 °C in 50 mm Tris-HCl, 150 mm NaCl, pH 8.5 buffer.
Preparation of NMR Samples
C171Q KasA was exchanged into 50 mm sodium phosphate, 150 mm sodium chloride D2O, pD 8.5 buffer. 5(R,S)-TLM and PK940 were dissolved in the phosphate buffer to give final concentrations in the experiment of 8 and ∼4 mm, respectively. In the case of enantiomerically pure 5(R)-TLM, equimolar concentrations of the two ligands were used at ∼4 mm. The concentration of enzyme in the experiments was 30 μm.
Two- and One-dimensional NOE Spectroscopy
All NMR data were acquired on standard bore 700 or 900 MHz Bruker Avance NMR instruments at 15 °C using samples prepared in D2O solvents. The spectra were processed using Bruker TOPSPIN 2.1 software.
Two-dimensional NOESY spectra were collected at 700 MHz over a range of mixing times (50–900 ms) using a spectral width of 7716.05 Hz in F2 and 2048 complex data points for an acquisition time of 0.133 s with a 2-s recycle delay. 256 points were collected in the indirect F1 dimension for a 0.0166-s acquisition time. 40 scans were collected per F1 increment, and F1 quadrature detection was achieved using the States-TPPI method (24). Time domain data were apodized using squared sine bell functions in both dimensions and zero-filled in the indirect dimension to a final data matrix size of 1024 (F1) × 2048 (F2) after Fourier transformation.
Selective one-dimensional DPFGSE NOE data were collected using a spectral width of 14005.6 Hz and 32768 complex data points for a 1.17-s acquisition time with a 2-s recycle delay. Selective Gaussian pulses of 120 ms were used to invert the target resonances in the PK940 methyl cluster to observe interligand transfer of NOEs to the TLM methyl groups (16, 25).
Direct Binding Fluorescence Titrations
Binding of TLM and the TLM analogs to KasA was quantified by monitoring changes in the intrinsic tryptophan fluorescence of the enzyme using 280-nm excitation and 337-nm emission as described (11). The data were collected using a Quanta Master fluorimeter (Photon Technology International) with excitation and emission slit widths of 4.0 and 8.0 nm, respectively. Inhibitors were dissolved in Me2SO or buffer (50 mm Tris-HCl, 150 mm NaCl, 1 mm dithiothreitol, pH 8.5) and titrated into 1 μm solutions of enzyme in the same Tris buffer. Titration curves were corrected for the inner filter effect, and the Kd values were calculated using the quadratic Morrison equation (Grafit 4.0). To quantify the slow onset binding kinetics of analogs to C171Q KasA, the fluorescence emission was monitored as a function of time for 40 min. The change in fluorescence intensity was fit to Equation 1 (11), which includes a second term (kb) to account for the slow photobleaching of the chromophore.
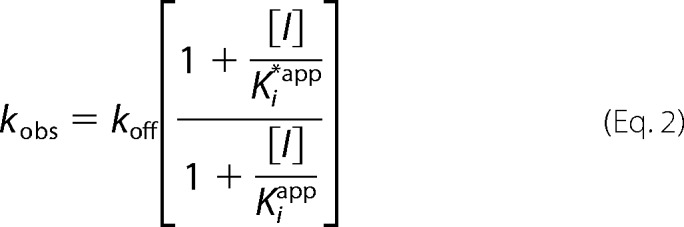
The values of kobs obtained from Equation 1 were dependent on the concentration of the inhibitor and were subsequently fit to Equation 2 (26) to obtain the rate constant for formation of free enzyme from the enzyme inhibitor complex (koff). Equation 3 (26) was then used to calculate the kon values.
 |
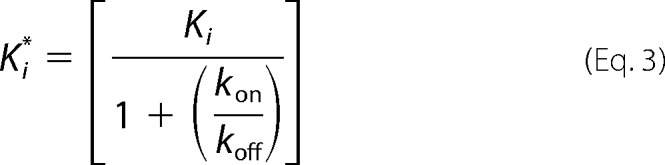 |
RESULTS
Conflicting 1H NMR assignments have been reported for some of the TLM resonances in earlier literature (25); therefore the 1H and 13C NMR spectra for TLM and TLM analogs were reassigned using values obtained from our 1H one-dimensional, 1H-1H COSY, 1H-13C Heteronuclear Multiple-Quantum Correlation, and Heteronuclear Multiple Bond Correlation NMR data. These assignments agree with values presented in more recent literature (27). The methyl peaks of TLM at C6, C7, and C12 (Fig. 2) were assigned to the resonances at 1.41, 1.50, and 1.44 ppm respectively. For the PK940 ligand, nonprochiral assignments of the singlet proton peaks at 0.714 and 0.75 ppm were made to the geminal methyl groups at C9′ and C10′, and the doublet of doublets centered near 0.708 ppm was assigned to the terminal methyl protons at C1′ (Fig. 2). The overlap of the PK940 C1′ methyl resonance with the upfield C9′/C10′ methyl singlet hinders the analysis of the standard one-dimensional 1H and two-dimensional NOESY spectra.
After revisiting the assignments of TLM, we performed a standard two-dimensional NOE experiment similar to that performed in other ILOE NMR experiments (15). Close inspection of the two-dimensional NOE spectra in Fig. 3 shows apparent negative ILOE cross-peaks between the C6, C12, and C7 TLM methyl resonances at 1.41, 1.44, and 1.50 ppm, respectively, and the PK940 methyl cluster peaks spanning 0.71–0.75 ppm. The cross-peaks have chemical shifts equal to the other C2′ and C3′ NOE signals observed from the K940 methylene groups at 1.15 and 1.32 ppm, respectively. Given the close proximity of the geminal C9′ and C10′ methyls, we expect that they should have similar NOEs to any point on TLM. The presence of only one associated ILOE eliminates the geminal methyls as the interacting partners with TLM.
FIGURE 3.

Two-dimensional NOESY spectra in the presence of C171Q KasA, TLM, and PK940 recorded on a 700-MHz Bruker Avance instrument at a mixing time of 700 ms. Negative (red) interligand NOE cross-peaks can be observed between the TLM methyls at 1.41, 1.44, and 1.50 ppm and the terminal PK940 methyl doublet. Positive (blue) intraligand NOE cross-peaks can be seen between terminal PK940 methyl and C2′ and C3′ methylenes.
In our experiments, we were able to observe ILOEs between TLM and the pantetheine analog (PK940) only at mixing times longer than 500 ms and were unable to detect ILOE signals at the shorter mixing times required for distance assessment. The lack of sensitivity and the inability to detect and differentiate signal in the presence of overlapping chemical shifts and high background noise have previously been reported to hinder ILOE detection involving small molecules (16). Consequently, we acquired one-dimensional NOE data using the DPFGSE method, inverting the PK940 methyl cluster signal at ∼0.75 ppm. Because of enhanced sensitivity, the one-dimensional experiment (16, 17) allowed detection of the ILOE signal even at very low mixing times (∼100 ms), giving us very clean and easily interpretable spectra. Moreover, the acquisition times were greatly reduced for the one-dimensional experiment, which also improved the quality of the data given that the enzyme was not very stable for long periods of time under the experimental conditions employed. The one-dimensional NOE experiment also indicated the presence of transient interligand NOEs between the three TLM methyls and the PK940 methyl cluster in the presence of the enzyme (Fig. 4). The simultaneous selective inversion of the three TLM methyl groups allowed us to specifically assign these ILOEs to interactions between the three TLM methyl groups and the terminal C1′ methyl of PK940.
FIGURE 4.

Overlay of 1H NMR DPFGSE one-dimensional NOE spectra of the ligands in the presence of C171Q KasA with mixing times of 100 and 500 ms. Negative interligand NOEs can be seen with the TLM m7ethyl resonances upon inversion of the PK940 methyl cluster at ∼0.75 ppm with a 120-ms shaped Gaussian pulse. Anti-phase contributions are observed in the line shapes of the C2′ and C3′ methylene protons of PK940 at 1.15 and 1.32 ppm because of possible strong coupling or Zero-Quantum artifact that was not removed by the DPFGSE pulse sequences that were used (16). The spectra were recorded on a 700 MHz Bruker Avance instrument.
NOE buildup curves were obtained for the three TLM methyls over a range of mixing times (Fig. 5A). The NOE intensities, normalized with respect to the inverted peak intensity, were plotted against the mixing time to obtain buildups that, to a large extent, canceled the effect of external relaxation at moderate mixing times (18) (Fig. 5B). Using the distance between the TLM vinyl protons on C11 (1.85 Å) as a reference, the distances between the C1′ PK940 methyl and TLM C12, C6, and C7 methyls were calculated to be 3.0, 3.4, and 3.4 Å, respectively.
FIGURE 5.

A, NOE buildup curves for the TLM methyls C6, C7, and C12 upon inversion of the PK940 methyl cluster at ∼0.75 ppm are shown. B, NOE buildups for TLM methyls 6, 7, and 12 normalized with respect to the inverted peak at ∼0.75 ppm show the same extended utility of the longer mixing times for estimating distances because of the lack of an appropriate scale mentioned in the text.
To refine the relative positions of the two ligands, we attempted to invert the three TLM methyls individually to observe the ILOE with the terminal methyl of PK940. This turned out to be a challenging task for a number of reasons. Initially, the chemical shifts of the three TLM methyls are very close, making individual inversion problematic. Use of a higher magnetic field (900 MHz) increased the separation of the three peaks sufficiently to provide selectivity in the absence of the enzyme. However, upon addition of enzyme, we observed a loss of selectivity, wherein inversion of all three methyls was observed upon inversion of any one methyl, especially at higher mixing times. This observation can be explained by proposing that the dipolar coupling between KasA and TLM provides a route for cross-relaxation between the KasA methyls/methylenes and TLM methyls by spin diffusion (28). Attempts to target the PK940 methyl cluster at ∼0.71 ppm and the singlet at 0.75 ppm exclusively on a 900-MHz field also resulted in a loss of selectivity that worsened at higher mixing times. Additionally, the expected line broadening caused by modulation in the transverse relaxation time (T2) in the bound and the unbound state of the ligands could contribute to the observed loss of selectivity. Despite these issues, we were able to selectively invert the downfield TLM methyl (C7) using a 180-ms Gaussian pulse at 900 MHz. Moreover, we observed similar rates of ILOE buildup (supplemental Fig. S3) compared with inversion of the PK940 methyl cluster.
Several control experiments were performed to confirm that the observed signal was an ILOE and not a transferred NOE with the protein. First, the experiments were repeated in the absence of the enzymes as a control. No ILOE was observed between the two ligands. Second, no signal was observed at ∼1.45 ppm when the bandwidth at ∼0.75 ppm was irradiated in the presence of only TLM and enzyme, whereas negative ILOEs were observed when PK940 was also present. Similarly, no signal was observed at ∼0.75 ppm when the methyl bandwidth at ∼1.45 ppm was irradiated in the presence of only PK940 and enzyme. These controls confirmed the presence of ILOEs between TLM and PK940 with the terminal methyl of PK940 oriented toward the TLM methyl groups. Using this information, we modeled PK940 into the active site of KasA using the x-ray structure of the TLM-C171Q KasA complex (19). To satisfy the constraints imposed by the NMR data, PK940 must occupy the putative KasA pantetheine-binding pocket (Fig. 6A). This structural analysis suggested that elaboration of the TLM thiolactone ring at either the 3 or the 4 position would result in an increase in affinity of the inhibitor for the enzyme. This is shown conceptually in Fig. 6B where DOCK6 was used to build a model of a hypothetical TLM-pantetheine conjugate (yellow) bound to KasA (29, 30).
FIGURE 6.

A, proposed orientation of TLM and PK940 in the C171Q KasA active site. The figure was made using PyMOL (36). B, the structure of two ligands docked to KasA have been overlaid. In yellow is a hypothetic compound generated by linking TLM and PK940 through the TLM C3 position. Compound 12 is shown in blue. The bound ligand structures were calculated using the DOCK6 suite of dock programs with default parameters (29, 31). The substituents occupy the pantetheine-binding pocket consistent with the ILOE data.
As a first step in using the structural data to develop TLM analogs with increased affinity for KasA, we undertook the synthesis of compounds in which the methyl at the 3 position was replaced with a variety of substituents. We then determined the ability of these compounds to bind to wild-type KasA and to the C171Q acyl-enzyme mimic. TLM binds to the wild-type enzyme with a Kd value of 226 μm and is a slow onset inhibitor of C171Q KasA with Ki and Ki* values of 175 and 1.9 μm, respectively (11).
To explore the sensitivity of TLM to modification at C3, we initially synthesized several analogs of TLM in which the C3 methyl was replaced with hydrogen (compound 2), ethyl (compound 3), and propyl (compound 4) groups (Table 1). Previously, Townsend and co-workers (6) reported that a demethyl TLM analog had improved affinity for the FAS-I ketoacyl synthase. However, compounds 2–4 all had Ki values with the free enzyme within a factor of 2 of that for TLM. In addition, whereas compounds 3 and 4 were slow onset inhibitors of C171Q KasA, with Ki* values of 7 and 16 μm, respectively, demethyl TLM (compound 2) was no longer a slow onset inhibitor of C171Q, albeit with a ∼4-fold improvement in Ki. In agreement with previous proposals for the human fatty acid synthase (6), our modeling data suggested that the introduction of a hydrogen bond acceptor into the C3 substituent might lead to favorable interactions in the active site. We consequently synthesized acetyl and trifluoroacetyl-substituted TLM analog compounds 5 and 6, which bound significantly more tightly to wild-type KasA than TLM (9-fold) and were slow onset inhibitors of C171Q KasA with Ki* values of 0.9 and 0.5 μm, respectively. Thus, introduction of the keto group significantly improved the affinity of TLM for KasA (Table 1).
To provide additional structure-activity relationship, we also synthesized several TLM analogs with larger substituents at C3. This included analogs with keto groups (compounds 7–11), as well as compounds with phenyl and biphenyl groups attached to the thiolactone ring by linkers of variable lengths (compounds 11–16). Although none of these analogs were found to be slow onset inhibitors of C171Q KasA, several had significantly improved affinity for KasA compared with TLM. Moreover, as observed for the smaller acyl substituents at C3 (compounds 5 and 6), a keto group with larger substituents (compounds 7–11) resulted in improved binding affinities with both the free enzyme and the acyl mimic. In addition, compounds with larger, more hydrophobic groups were generally found to have higher affinity for wild-type and C171Q KasA enzymes, in agreement with previous studies in which lipophilicity was identified as a key feature of KAS inhibitor design (7–9). For example, ethyl-chlorophenyl analog compound 12 had Kd and Ki values of 128 and > 400 μm, respectively, whereas butyl-chlorophenyl analog compounds 15 had Kd and Ki values of 68 and 77 μm, respectively. Of these latter compounds, hexoyl biphenyl analog compound 11 and butyl biphenyl analog compounds 16 and 17 showed the highest affinity for both the free enzyme and the acyl mimic with Kd and Ki values in the range of 8–32 μm. The improvement in affinity of compounds with the longer (C4) linker may reflect the improved ability of these compounds to bind in the pantetheine-binding channel. This is consistent with the structure of analog compound 16 docked to KasA (Fig. 6B, cyan).
Despite improvements in affinity, none of the more hydrophobic analogs (compounds 7–17) retained the slow onset binding that is seen for the interaction of the parent compound (TLM) with acyl-KasA (11). This is important because slow onset inhibitors can have long residence times on their targets resulting in prolonged pharmacodynamic effects in vivo (32–34). To understand the effect of the C3 substituents on the off rates (koff) of the inhibitors, the binding kinetics of compounds 3–6 were evaluated with the acyl-KasA mimic. The Ki* values for these compounds span that reported previously for TLM (1.9 μm) and ranged from 0.5 μm for the trifluoromethyl analog compound 6 to 16 μm for the propyl analog compound 4. However, analogs with larger Ki* values were found to have the longest residence times for C171Q KasA. Ethyl analog compound 3 had a drug-target complex residence time of 33 min, which is 4-fold longer than that observed for TLM (8 min). This increase in residence time is likely due to transition state destabilization because the Ki* value for compound 3 (7 μm) is ∼3-fold larger than that for TLM (2 μm). In contrast, the residence time of compound 6 was only 3 min, suggesting that stabilization of the ground state via hydrogen bonding to the C3 keto group is also accompanied by a correspondingly larger stabilization of the transition state leading to EI*. This observation is a reminder that changes in the transition state for the slow isomerization step can play a significant role in modulating the life time of the enzyme-inhibitor complex (34). Although the changes in residence time in the present series of compounds are modest, it is noteworthy that modifications at the TLM C3 position do not abolish time-dependent inhibition and can even lead to improvements in residence time. This is important because future lead optimization will seek to increase both the thermodynamic affinity and residence time of inhibitors for this enzyme.
DISCUSSION
ILOE is a powerful tool to aid fragment based drug discovery, and traditional applications of this method have relied on standard two-dimensional NOESY measurements. However, long data acquisition times, significant background noise, and a lack of sensitivity limit the application of this technique, especially for time-sensitive samples. Here, we have successfully used the DPFGSE pulse sequence to obtain NOEs between ligands bound to an enzyme drug target. This technique enables cleaner NOE spectra to be obtained more rapidly over a wider range of mixing times. We anticipate extensive use of this method as an improvement on the standard two-dimensional NOESY methods for rapid and more efficient detection of ILOEs to direct fragment-based drug discovery. The NOEs and the subsequent orientations obtained from this approach provide valuable constraints for subsequent model building by limiting the degrees of freedom of the fragments.
Using this method, we mapped the relative orientations of TLM and a pantetheine analog, PK940, bound to the M. tuberculosis enzyme KasA which is a validated drug target. TLM is a micromolar inhibitor of KasA, and the long term object of this project is to develop analogs of TLM with improved affinity for the enzyme and increased antibacterial activity.
Previous synthetic efforts have primarily explored modifications of the isoprene chain at position 5 of the TLM nucleus, which revealed that the isoprene tail is relatively intolerant to change (12, 30, 35). In the present work, the ILOE experiments indicate that elaboration of either the 3 or 4 position of the TLM thiolactone ring may be a profitable approach for improving affinity for KasA. Our initial structure-activity relationship studies support this concept, highlighting the importance of hydrogen bonding groups in the C3 substituent and demonstrating that time-dependent inhibition could be retained. These studies lay the foundation for additional inhibitor discovery for this important drug target.
Supplementary Material
Acknowledgments
We acknowledge Dr. Shibani Bhattacharya and Dr. Mike Goger at the New York Structural Biology Center and Dr. Martine Ziliox at the Stony Brook Center for Structural Biology.
This work was supported, in whole or in part, by National Institutes of Health Grants GM102864 and AI044639 (to P. J. T.).

This article contains supplemental data and Figs. S1–S3.
- KasA
- β-ketoacyl synthase A
- TLM
- thiolactomycin
- DPFGSE
- double pulse field gradient spin echo
- ILOE
- interligand NOE.
REFERENCES
- 1. Sassetti C. M., Boyd D. H., Rubin E. J. (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84 [DOI] [PubMed] [Google Scholar]
- 2. Bhatt A., Kremer L., Dai A. Z., Sacchettini J. C., Jacobs W. R., Jr. (2005) Conditional depletion of KasA, a key enzyme of mycolic acid biosynthesis, leads to mycobacterial cell lysis. J. Bacteriol. 187, 7596–7606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang J., Soisson S. M., Young K., Shoop W., Kodali S., Galgoci A., Painter R., Parthasarathy G., Tang Y. S., Cummings R., Ha S., Dorso K., Motyl M., Jayasuriya H., Ondeyka J., Herath K., Zhang C., Hernandez L., Allocco J., Basilio A., Tormo J. R., Genilloud O., Vicente F., Pelaez F., Colwell L., Lee S. H., Michael B., Felcetto T., Gill C., Silver L. L., Hermes J. D., Bartizal K., Barrett J., Schmatz D., Becker J. W., Cully D., Singh S. B. (2006) Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature 441, 358–361 [DOI] [PubMed] [Google Scholar]
- 4. Noto T., Miyakawa S., Oishi H., Endo H., Okazaki H. (1982) Thiolactomycin, a new antibiotic. III. In vitro antibacterial activity. J. Antibiot. 35, 401–410 [DOI] [PubMed] [Google Scholar]
- 5. Omura S. (1981) Cerulenin. Methods Enzymol. 72, 520–532 [PubMed] [Google Scholar]
- 6. McFadden J. M., Medghalchi S. M., Thupari J. N., Pinn M. L., Vadlamudi A., Miller K. I., Kuhajda F. P., Townsend C. A. (2005) Application of a flexible synthesis of (5R)-thiolactomycin to develop new inhibitors of type I fatty acid synthase. J. Med. Chem. 48, 946–961 [DOI] [PubMed] [Google Scholar]
- 7. Lee P. J., Bhonsle J. B., Gaona H. W., Huddler D. P., Heady T. N., Kreishman-Deitrick M., Bhattacharjee A., McCalmont W. F., Gerena L., Lopez-Sanchez M., Roncal N. E., Hudson T. H., Johnson J. D., Prigge S. T., Waters N. C. (2009) Targeting the fatty acid biosynthesis enzyme, β-ketoacyl-acyl carrier protein synthase III (PfKASIII), in the identification of novel antimalarial agents. J. Med. Chem. 52, 952–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Al-Balas Q., Anthony N. G., Al-Jaidi B., Alnimr A., Abbott G., Brown A. K., Taylor R. C., Besra G. S., McHugh T. D., Gillespie S. H., Johnston B. F., Mackay S. P., Coxon G. D. (2009) Identification of 2-aminothiazole-4-carboxylate derivatives active against Mycobacterium tuberculosis H37Rv and the β-ketoacyl-ACP synthase mtFabH. PLoS One 4, e5617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Singh S., Soni L. K., Gupta M. K., Prabhakar Y. S., Kaskhedikar S. G. (2008) QSAR studies on benzoylaminobenzoic acid derivatives as inhibitors of β-ketoacyl-acyl carrier protein synthase III. Eur. J. Med. Chem. 43, 1071–1080 [DOI] [PubMed] [Google Scholar]
- 10. Miyakawa S., Suzuki K., Noto T., Harada Y., Okazaki H. (1982) Thiolactomycin, a new antibiotic. IV. Biological properties and chemotherapeutic activity in mice. J. Antibiot. 35, 411–419 [DOI] [PubMed] [Google Scholar]
- 11. Machutta C. A., Bommineni G. R., Luckner S. R., Kapilashrami K., Ruzsicska B., Simmerling C., Kisker C., Tonge P. J. (2010) Slow onset inhibition of bacterial β-ketoacyl-acyl carrier protein synthases by thiolactomycin. J. Biol. Chem. 285, 6161–6169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim P., Zhang Y. M., Shenoy G., Nguyen Q. A., Boshoff H. I., Manjunatha U. H., Goodwin M. B., Lonsdale J., Price A. C., Miller D. J., Duncan K., White S. W., Rock C. O., Barry C. E., 3rd, Dowd C. S. (2006) Structure-activity relationships at the 5-position of thiolactomycin. An intact (5R)-isoprene unit is required for activity against the condensing enzymes from Mycobacterium tuberculosis and Escherichia coli. J. Med. Chem. 49, 159–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen J., Zhang Z., Stebbins J. L., Zhang X., Hoffman R., Moore A., Pellecchia M. (2007) A fragment-based approach for the discovery of isoform-specific p38α inhibitors. ACS Chem. Biol. 2, 329–336 [DOI] [PubMed] [Google Scholar]
- 14. Price A. C., Choi K. H., Heath R. J., Li Z., White S. W., Rock C. O. (2001) Inhibition of β-ketoacyl-acyl carrier protein synthases by thiolactomycin and cerulenin. Structure and mechanism. J. Biol. Chem. 276, 6551–6559 [DOI] [PubMed] [Google Scholar]
- 15. Becattini B., Pellecchia M. (2006) SAR by ILOEs. An NMR-based approach to reverse chemical genetics. Chemistry 12, 2658–2662 [DOI] [PubMed] [Google Scholar]
- 16. Stott K., Keeler J., Van Q. N., Shaka A. J. (1997) One-dimensional NOE experiments using pulsed field gradients. J. Magn. Reson. 125, 302–324 [Google Scholar]
- 17. Stott K., Stonehouse J., Keeler J., Hwang T.-L., Shaka A. J. (1995) Excitation sculpting in high-resolution nuclear magnetic resonance spectroscopy. Application to selective NOE experiments. J. Am. Chem. Soc. 117, 4199–4200 [Google Scholar]
- 18. Hu H., Krishnamurthy K. (2006) Revisiting the initial rate approximation in kinetic NOE measurements. J. Magn. Reson. 182, 173–177 [DOI] [PubMed] [Google Scholar]
- 19. Luckner S. R., Machutta C. A., Tonge P. J., Kisker C. (2009) Crystal structures of Mycobacterium tuberculosis KasA show mode of action within cell wall biosynthesis and its inhibition by thiolactomycin. Structure 17, 1004–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Witkowski A., Joshi A. K., Lindqvist Y., Smith S. (1999) Conversion of a β-ketoacyl synthase to a malonyl decarboxylase by replacement of the active-site cysteine with glutamine. Biochemistry 38, 11643–11650 [DOI] [PubMed] [Google Scholar]
- 21. Wang C. L., Salvino J. M. (1984) Total synthesis of (+/−)-thiolactomycin. Tetrahedron Lett. 25, 5243–5246 [Google Scholar]
- 22. Szabo A., Kunzle N., Mallat T., Baiker A. (1999) Enantioselective hydrogenation of pyrrolidine-2,3,5-triones over the Pt-cinchonidine system. Tetrahedron Asymmetr. 10, 61–76 [Google Scholar]
- 23. Spry C., Chai C. L., Kirk K., Saliba K. J. (2005) A class of pantothenic acid analogs inhibits Plasmodium falciparum pantothenate kinase and represses the proliferation of malaria parasites. Antimicrob. Agents Chemother. 49, 4649–4657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marion D., Ikura M., Tschudin R., Bax A. (1989) Rapid recording of 2D NMR-spectra without phase cycling. Application to the study of hydrogen-exchange in proteins. J. Magn. Reson. 85, 393–399 [Google Scholar]
- 25. Sasaki H., Oishi H., Hayashi T., Matsuura I., Ando K., Sawada M. (1982) Thiolactomycin, a new antibiotic. II. Structure elucidation. J. Antibiot. 35, 396–400 [DOI] [PubMed] [Google Scholar]
- 26. Copeland R. A. (2000) Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, 2nd Ed., John Wiley & Sons, New York [Google Scholar]
- 27. Brown M. S., Akopiants K., Resceck D. M., McArthur H. A., McCormick E., Reynolds K. A. (2003) Biosynthetic origins of the natural product, thiolactomycin. A unique and selective inhibitor of type II dissociated fatty acid synthases. J. Am. Chem. Soc. 125, 10166–10167 [DOI] [PubMed] [Google Scholar]
- 28. Edzes H. T., Samulski E. T. (1977) Cross relaxation and spin diffusion in the proton NMR or hydrated collagen. Nature 265, 521–523 [DOI] [PubMed] [Google Scholar]
- 29. Lang P. T., Brozell S. R., Mukherjee S., Pettersen E. F., Meng E. C., Thomas V., Rizzo R. C., Case D. A., James T. L., Kuntz I. D. (2009) DOCK 6. Combining techniques to model RNA-small molecule complexes. RNA 15, 1219–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jones A. L., Herbert D., Rutter A. J., Dancer J. E., Harwood J. L. (2000) Novel inhibitors of the condensing enzymes of the type II fatty acid synthase of pea (5Pisum sativum). Biochem. J. 347, 205–209 [PMC free article] [PubMed] [Google Scholar]
- 31. Mukherjee S., Balius T. E., Rizzo R. C. (2010) Docking validation resources. Protein family and ligand flexibility experiments. J. Chem. Inf. Model. 50, 1986–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Swinney D. C. (2004) Biochemical mechanisms of drug action. What does it take for success? Nat. Rev. Drug Discov. 3, 801–808 [DOI] [PubMed] [Google Scholar]
- 33. Copeland R. A., Pompliano D. L., Meek T. D. (2006) Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discov. 5, 730–739 [DOI] [PubMed] [Google Scholar]
- 34. Lu H., Tonge P. J. (2010) Drug-target residence time. Critical information for lead optimization. Curr. Opin. Chem. Biol. 14, 467–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jones S. M., Urch J. E., Brun R., Harwood J. L., Berry C., Gilbert I. H. (2004) Analogues of thiolactomycin as potential anti-malarial and anti-trypanosomal agents. Bioorg. Med. Chem. 12, 683–692 [DOI] [PubMed] [Google Scholar]
- 36. DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific, Palo Alto, CA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.