

Background: San1 is a ubiquitin-protein ligase that recognizes exposed hydrophobicity in misfolded nuclear proteins.
Results: San1 prefers a window of exposed hydrophobicity that causes a particular level of protein insolubility.
Conclusion: The hydrophobicity/insolubility threshold suggests that San1 evolved to target highly aggregation-prone proteins.
Significance: Identifying the specific parameters of misfolding recognized by ubiquitin-protein ligases allows us to understand the nature of substrate targeting.
Keywords: Proteasome, Protein Degradation, Ubiquitin, Ubiquitin Ligase, Ubiquitination, Ubiquitylation, San1, Hydrophobicity, Insolubility
Abstract
Misfolded proteins present an escalating deleterious challenge to cells over the course of their lifetime. One mechanism the cell possesses to prevent misfolded protein accumulation is their destruction by protein quality control (PQC) degradation systems. In eukaryotes, PQC degradation typically proceeds via multiple ubiquitin-protein ligases that act throughout the cell to ubiquitinate misfolded proteins for proteasome degradation. What the exact feature of misfolding that each PQC ubiquitin-protein ligase recognizes in their substrates remains an open question. Our previous studies of the budding yeast nuclear ubiquitin-protein ligase San1 indicated that it recognizes exposed hydrophobicity within its substrates, with the threshold of hydrophobicity equivalent to that of 5 contiguous hydrophobic residues. Here, we uncover an additional parameter: the nature of the exposed hydrophobicity that confers San1-mediated degradation correlates with significant protein insolubility. San1 particularly targets exposed hydrophobicity that leads to insolubility and aggregation above a certain threshold. Our studies presented here provide additional insight into the details of misfolded nuclear protein recognition and demonstrate that there is selectivity for the type of exposed hydrophobicity.
Introduction
The continuous stochastic production of misfolded proteins is a problem faced by all cells. Proteins that have lost their native structures expose hydrophobicity that is normally buried within the core of their structures. The exposed hydrophobicity can lead to protein insolubility and the formation of intracellular aggregates (1). Aggregate formation and the long term cellular persistence of aggregates are thought to be the underlying causes of many devastating human pathologies including Alzheimer, Parkinson, Huntington, and amyotrophic lateral sclerosis (2).
Due to the deleterious nature of misfolded proteins, cells have evolved several mechanisms to prevent their accumulation. One central means is the elimination of misfolded proteins via protein quality control (PQC)2 degradation. Eukaryotic PQC degradation is generally mediated by ubiquitin-protein ligases that ubiquitinate misfolded proteins for subsequent proteasome degradation (3). PQC ubiquitin-protein ligases have been identified in most cellular compartments including the endoplasmic reticulum (4–9), cytoplasm (10–13), and nucleus (14–18). Multiple PQC ligases often exist in the same compartment. For example, Hrd1 and Doa10 operate in the yeast endoplasmic reticulum (4, 6, 9, 19, 20), whereas San1 and Doa10 act in the yeast nucleus (15, 21). Although a cellular compartment can contain multiple PQC ubiquitin-protein ligases, they generally do not target the same substrates (20, 22–26), indicating that the compartment-specific PQC ligases are, for the most part, not functionally redundant.
The lack of functional redundancy suggests that each PQC ubiquitin-protein ligase might recognize a unique feature of misfolding in its substrates. This likely depends upon the specific cellular compartment and the spatial organization of the PQC ligases within that compartment. With this consideration in mind, it remains an open question as to which feature(s) of misfolding an individual PQC ubiquitin-protein ligase targets. If each PQC ligase recognizes exposed hydrophobicity, are the extent of exposed hydrophobicity, the types of hydrophobic residues, and/or the context in which the hydrophobicity is presented different for each PQC ligase? To answer these questions, we require detailed information as to what each individual PQC ubiquitin-protein ligase recognizes in its misfolded substrates.
We have been probing this by exploring the features of misfolding recognized by the yeast nuclear PQC ubiquitin-protein ligase San1. Recently, we found that San1 targets exposed hydrophobicity in its substrates, with a minimal window of 5 exposed hydrophobic residues necessary for substrate recognition by San1 (27). Here, we find that San1 prefers particular types of hydrophobic residues within that window, and this preference can be explained by the strength of the residues' ability to cause insolubility and aggregation.
EXPERIMENTAL PROCEDURES
Yeast Strains and Plasmids
Yeast strains used in this study are BY4741 (met15Δ0 his3Δ1 ura3Δ0 leu2Δ0) (28), RGY506 (BY4741 san1Δ), RGY565 (BY4741 pdr5Δ), RGY1794 (BY4741 san1Δ pdr5Δ), and RGY1273 (his3Δ1 ura3Δ0 leu2Δ0 lys 2Δ0 DBP5-dsRED::HIS3 san1Δ::HygMX) (27). Standard yeast growth media and yeast genetic methods were employed (29). Plasmids used in this study are listed in Table 1. Standard cloning methods were used to construct each plasmid. The relevant portion of each plasmid was sequenced to verify the fusions. Oligonucleotide sequences and cloning details will be provided upon request.
TABLE 1.
Plasmids used in this study
| Plasmid | Figure(s) | Plasmid description |
|---|---|---|
| pRG3789 | 1 | PGAL1-GFPNLS-TSTFVLYIII, LEU2, 2μ |
| pRG3790 | 1 | PGAL1-GFPNLS-TSTSVLYIII, LEU2, 2μ |
| pRG3791 | 1 | PGAL1-GFPNLS-TSTSTLYIII, LEU2, 2μ |
| pRG3792 | 1 | PGAL1-GFPNLS-TSTSTSYIII, LEU2, 2μ |
| pRG3793 | 1 | PGAL1-GFPNLS-TSTSTSTIII, LEU2, 2μ |
| pRG3384 | 2 | PGAL1-GFPNLS-TSTSTYVILL, LEU2, 2μ |
| pRG3385 | 2 and 3 | PGAL1-GFPNLS-TSTSTLLLLL, LEU2, 2μ |
| pRG3386 | 2 and 3 | PGAL1-GFPNLS-TSTSTAAAAA, LEU2, 2μ |
| pRG3387 | 2 and 3 | PGAL1-GFPNLS-TSTSTFFFFF, LEU2, 2μ |
| pRG3388 | 2–5 | PGAL1-GFPNLS-TSTSTVVVVV, LEU2, 2μ |
| pRG3436 | 2, 3, 5, and 6 | PGAL1-GFPNLS-TSTSTIIIII, LEU2, 2μ |
| pRG3437 | 2 and 3 | PGAL1-GFPNLS-TSTSTWWWWW, LEU2, 2μ |
| pRG3438 | 2 and 3 | PGAL1-GFPNLS-TSTSTYYYYY, LEU2, 2μ |
| pRG3545 | 2 and 3 | PGAL1-GFPNLS-TSTSTMMMMM, LEU2, 2μ |
| pRG3650 | 5 | PGAL1-GFPNLS-TSTSTVVVIV, LEU2, 2μ |
| pRG3785 | 5 | PGAL1-GFPNLS-TSTSTVIVVV, LEU2, 2μ |
| pRG3786 | 5 | PGAL1-GFPNLS-TSTSTVVIVV, LEU2, 2μ |
| pRG3991 | 6 | PGAL1-GFPNLS-TSTSRIIIII, LEU2, 2μ |
| pRG3993 | 6 | PGAL1-GFPNLS-TSTSRFFFFF, LEU2, 2μ |
| pRG3610 | 7 | PGAL1-GFPNLS-TSTSTVVVVVVV, LEU2, 2μ |
| pRG3611 | 7 | PGAL1-GFPNLS-TSTSTLLLLLLL, LEU2, 2μ |
| pRG3613 | 7 | PGAL1-GFPNLS-TSTSTIIIIIII, LEU2, 2μ |
| pRG3619 | 7 | PGAL1-GFPNLS-TSTSTAAAAAAA, LEU2, 2μ |
| pRG3620 | 7 | PGAL1-GFPNLS-TSTSTYYYYYYY, LEU2, 2μ |
| pRG3621 | 7 | PGAL1-GFPNLS-TSTSTWWWWWWW, LEU2, 2μ |
| pRG3622 | 7 | PGAL1-GFPNLS-TSTSTMMMMMMM, LEU2, 2μ |
Degradation Assays
Cycloheximide-chase assays were performed as described previously (15). Briefly, cells were grown at 30 °C in liquid synthetic media with 3% raffinose to a culture density of ∼1 × 107 cells/ml. Galactose was added (final concentration of 3%), and the cells were incubated for 2 h at 30 °C. Cycloheximide was added (final concentration of 50 μg/ml), and the cultures were further incubated for 0–3 h. In certain cases, MG132 (final concentration of 10 μg/ml) was added 1 h prior to cycloheximide addition. 2 ml of cells was harvested and lysed at the appropriate time points after cycloheximide addition in 200 μl SUMEB (8 m urea, 1% SDS, 10 mm MOPS, pH 6.8, 10 mm EDTA, 0.01% bromphenol blue) by vortexing 5 min with 100 μl of 0.5 mm acid-washed glass beads. Lysates were incubated at 65 °C for 10 min and then clarified for 5 min by centrifugation at 12,800 × g. Proteins were resolved on SDS-polyacrylamide gels, transferred to nitrocellulose, and immunoblotted with anti-GFP (Sigma) antibodies.
Solubility Assays
Solubility assays were adapted from a protocol described previously (30). Cells were grown in liquid synthetic medium with 3% raffinose to a culture density of ∼1 × 107 cells/ml. Galactose was added (final concentration of 3%), and cultures were incubated at 30 °C for 2 h. 4 ml of cells was harvested and lysed in 200 μl of lysis buffer (100 mm Tris, pH 7.5, 200 mm NaCl, 1 mm EDTA, 1 mm DTT, 5% glycerol, and 0.1% Nonidet P-40) + PMSF by vortexing 5 min at 4 °C with 100 μl of 0.5-mm acid-washed glass beads. To remove unlysed cells, lysates were centrifuged at 700 × g for 1 min at 4 °C. 50 μl of lysate, representing the “total lysate,” was removed and added to 50 μl of SUMEB. Remaining lysate was centrifuged at 12,800 × g for 15 min at 4 °C. 100 μl of supernatant, representing the “soluble fraction,” was added to 100 μl of SUMEB. The pellet, representing the “insoluble fraction,” was resuspended in 100 μl of lysis buffer plus 100 μl of SUMEB. All samples were incubated at 65 °C for 10 min and then clarified for 5 min by centrifugation at 12,800 × g. Proteins were resolved on SDS-polyacrylamide gels, transferred to nitrocellulose, and immunoblotted with anti-GFP antibodies.
Nondenaturing Polyacrylamide Gel Electrophoresis (PAGE)
The soluble fractions generated from the solubility assays, with or without SDS (final concentration of 2%) added to the loading buffer, were loaded onto 8–16% Tris-glycine polyacrylamide gels (Lonza Rockland Inc). Running buffer was 25 mm Tris and 192 mm glycine. Gels were run at 4 °C. Proteins were transferred to nitrocellulose and immunoblotted with anti-GFP antibodies.
Microscopy
Cells (san1Δ DBP5-dsRed) that expressed each GFPNLS-peptide fusion protein were grown at 30 °C in 3% raffinose medium to ∼ 0.5 × 107 cells/ml. Galactose was added (final concentration of 3%), and the cells were incubated for 8 h at 30 °C. Cells were harvested, fixed in 4% paraformaldehyde in 0.1 m sucrose for 15 min, washed in wash buffer (1.2 m sorbitol, 0.4 m KPO4), stained with DAPI for 10 min in wash buffer plus 2% Triton X-100, and washed two times in wash buffer. Cells were imaged on a Nikon Eclipse 90i microscope with a 100× objective, filters for GFP (excitation wavelength (450–490 nm), dichroic mirror (495 nm), and emission filter (500–550 nm)), DAPI-stained DNA (excitation wavelength (325–375 nm), dichroic mirror (400 nm), and emission filter (435–485 nm)), and dsRed (excitation wavelength (540–580 nm), dichroic mirror (585 nm), and emission filter (593–668 nm)), and a Photometrics Cool Snap HQ2 cooled CCD camera with NIS-Elements acquisition software.
Image Processing
Western blots were scanned using an Epson Perfection V350 Photo scanner at 300 dpi. All images were processed with a Mac iMac or Pro computer (Apple) using Photoshop CS (Adobe). Decay curves for degradation assays and quantification for solubility assays were determined using ImageJ.
RESULTS
San1 Recognition of Hydrophobicity Correlates with Substrate Insolubility
Previously, we found that San1 recognizes short hydrophobic peptides as degrons when fused to reporter proteins such as GFP possessing a nuclear localization sequence (GFPNLS) or the Gal4 activating domain used in two-hybrid analyses (27, 31). Although the peptide degrons are not misfolded proteins by themselves, they likely allow San1 recognition of the reporter proteins by mimicking the feature San1 recognizes in a misfolded protein, exposed hydrophobicity. As such, we can use these small peptides in a controlled way to determine the biochemical parameters of the exposed hydrophobicity that leads to San1 targeting. In our previous study exploring the minimal number of contiguous hydrophobic residues in the peptide degrons necessary for San1-mediated degradation, we found ≥5 contiguous hydrophobic residues were required in the peptide for recognition by San1 and ≤4 contiguous hydrophobic residues were insufficient (Ref. 27 and Fig. 1A). Intrigued by this sharp delineation, we wanted to understand the biochemical nature of this limit.
FIGURE 1.

San1-mediated degradation correlates with substrate insolubility. A, cycloheximide-chase degradation assays were performed to assess the stability of GFPNLS-peptide fusions containing 3–7 contiguous hydrophobic residues in the peptide degron. Stability was examined in cells with (SAN1) or without (san1Δ) the SAN1 gene intact. The time after cycloheximide addition is indicated above the blots. GFPNLS-peptide fusions in all Western blots were detected using anti-GFP antibodies. Decay curves for degradation assays were determined using ImageJ. B, solubility assays were performed to determine how the indicated GFPNLS-peptide fusion partitions between the insoluble pellet fraction (I) and the soluble supernatant fraction (S) from SAN1 cells. Total lysate (T) indicates the total amount of GFPNLS-peptide fusion in cell lysates. Note that all samples from the solubility assays were run on the same SDS-polyacrylamide gel so that direct comparisons could be made. Percentage of protein in the insoluble versus soluble fractions is listed below each appropriate lane. Relative levels of the insoluble and soluble fractions were determined using ImageJ. C, nondenaturing PAGE shows GFPNLS-peptide fusions containing 3–7 contiguous hydrophobic residues in the peptide degron. Soluble fractions were isolated as in B and run on a nondenaturing 8–16% Tris-glycine gel without or with SDS in the loading buffer. D, microscopy shows san1Δ cells expressing GFPNLS-peptide fusions containing 3–7 contiguous hydrophobic residues in the peptide degron.
Because insolubility and aggregate formation are detrimental results of protein misfolding, we examined the in vivo solubility of GFPNLS-peptide fusions containing 3, 4, 5, 6, or 7 contiguous hydrophobic residues. We found that the threshold for San1-mediated degradation, ≥5 contiguous hydrophobic residues, correlated with the GFPNLS-peptide fusions becoming highly insoluble (Fig. 1B). The GFPNLS-peptide fusions containing ≤4 contiguous hydrophobic residues were largely soluble (Fig. 1B).
The GFPNLS-peptide fusion proteins present in the insoluble fraction are likely part of large inclusions because this fraction represents the cellular debris that pellets at 12,800 × g. Therefore, we also examined whether the GFPNLS-peptide fusions in the soluble fraction formed high molecular mass species typical of aggregates using nondenaturing PAGE (32, 33). Similar to the solubility assays, we found that the GFPNLS-peptide fusions with ≥5 contiguous hydrophobic residues formed mostly higher molecular mass species compared with the GFPNLS-peptide fusions with ≤4 contiguous hydrophobic residues that ran primarily as monomers (Fig. 1C, left). These higher molecular mass species were sensitive to the addition of SDS to the loading buffer (Fig. 1C, right), indicating that they are likely not SDS-insensitive amyloids (34).
Finally, we examined whether the GFPNLS-peptide fusions formed visible cellular inclusions in vivo. Using fluorescence microscopy, we found that the GFPNLS-peptide fusions with ≥5 contiguous hydrophobic residues were predominantly nuclear localized and formed a single visible inclusion in the nucleus (Fig. 1D), indicating that they do aggregate in vivo. By contrast, the GFPNLS-peptide fusions with ≤4 contiguous hydrophobic residues showed more uniform localization throughout the cell, although there was some enrichment in the nucleus (Fig. 1D). Furthermore, there was no inclusion formation in cells expressing the ≤4 contiguous hydrophobic residue GFPNLS-peptide fusions (Fig. 1D), which is consistent with their solubility and lack of aggregate formation (Fig. 1, B and C). The difference in overall cellular localization between the GFPNLS-peptide fusions with ≥5 contiguous hydrophobic residues, which primarily localize to the nucleus, and the GFPNLS-peptide fusions with ≤4 contiguous hydrophobic residues, which localize throughout the cytoplasm and nucleus, is likely due to the aggregated versus monomeric state of the GFPNLS-peptide fusion in relation to the passive diffusion limit of the yeast nuclear pore. The GFPNLS-peptide fusions are ∼30 kDa in size and thus have a monomeric molecular mass below the passive <40-kDa diffusion limit of the yeast nuclear pore (35, 36). Although the GFPNLS-peptide fusions with ≥5 contiguous residues share the same monomeric molecular mass as those with ≤4 contiguous residues, the formation of an aggregate with ≥2 molecules in the nucleus would render an aggregate above the size of the nuclear pore passive diffusion limit. Thus, all of the GFPNLS-peptide fusions can be trafficked into the nucleus via classic NLS-dependent import, but the GFPNLS-peptide fusions with ≥5 contiguous hydrophobic residues cannot passively diffuse back into the cytoplasm as can the GFPNLS-peptide fusions with ≤4 contiguous hydrophobic residues.
Altogether from the combined solubility, nondenaturing PAGE, and microscopy results, it appears that San1 recognizes exposed hydrophobicity that crosses a particular threshold of insolubility. This threshold coincides with aggregation and inclusion formation in the nucleus. This intriguing result prompted further experiments to dissect the parameters of the hydrophobicity nature of a San1 substrate.
Unexpected Selectivity of San1 for the Types of Hydrophobic Residues Recognized in a 5-Residue Window
Hydrophobicity is a general term that encompasses multiple amino acids. Algorithms have been developed that score amino acid residues either on their hydrophobicity, such as the well known Kyte-Doolittle hydropathy algorithm (37), or their propensity to occur in protein regions prone to aggregation, such as the AGGRESCAN algorithm (38). In Fig. 2, A and B, we have ranked the different hydrophobicity and aggregation propensity scores for each amino acid residue according to Kyte-Doolittle hydropathy or AGGRESCAN measures.
FIGURE 2.

Homopentameric stretches of hydrophobic residues confer different San1 dependences. A and B, Kyte-Doolittle hydrophobicity and AGGRESCAN aggregation propensity scores for the indicated amino acid. C–E, cycloheximide-chase degradation assays were performed to assess the stability of GFPNLS-peptide fusions containing homopentameric stretches of hydrophobic residues in the peptide degron. Stability was examined in cells with (SAN1) or without (san1Δ) the SAN1 gene intact. Time after cycloheximide addition is indicated above the blots. GFPNLS-peptide fusions in the Western blots were detected using anti-GFP antibodies. Solubility assays were performed to determine how the indicated GFPNLS-peptide fusion partition between the insoluble pellet fraction (I) and the soluble supernatant fraction (S). Total lysate (T) indicates the total amount of GFPNLS-peptide fusion in cell lysates. Percentage of protein in the insoluble versus soluble fractions is listed below each appropriate lane. Relative levels of the insoluble and soluble fractions were determined using ImageJ.
Because 5 contiguous hydrophobic residues are the minimal feature recognized by San1 (Fig. 1), we wanted to examine the ability of homopentameric amino acid stretches to function as San1 degrons. Our initial prediction was that homopentameric stretches of the most hydrophobic residues (Ile, Val, Leu, and Phe) would allow San1-mediated degradation, and there might be variability with homopentameric stretches of lesser hydrophobic residues (Tyr, Trp, Met, and Ala). In actuality, the results differed unexpectedly from our predictions. When we examined the degradation of each hompentameric GFPNLS-peptide fusion, we found three distinct groups. The first group consisted of 5Phe and 5Ile stretches, which were degraded primarily in a San1-dependent manner as we predicted (Fig. 2C). The second group was composed of 5Leu, 5Val, and 5Ala stretches, which were degraded, but in a primarily San1-independent manner (Fig. 2D). This was surprising as we had anticipated that the 5Leu and 5Val stretches, which are highly hydrophobic by both scoring measures (Fig. 2, A and B), would cause San1-mediated degradation similar to the 5Ile and 5Phe stretches. The final group encompassed 5Tyr, 5Met, and 5Trp stretches, which were stable (Fig. 2E).
The correlation between San1-mediated degradation and the insolubility of the original GFPNLS-peptide fusions (Fig. 1B) prompted us to examine the insolubility of each homopentameric GFPNLS-peptide fusion. As expected from their San1-mediated degradation, the 5Ile and 5Phe GFPNLS-peptide fusions showed the highest levels of insolubility (Fig. 2C). Also as expected from their complete stability, the 5Tyr, 5Met, and 5Trp GFPNLS-peptide fusions were primarily soluble (Fig. 2E). Interestingly, the 5Leu, 5Val, and 5Ala GFPNLS-peptide fusions showed a range of solubilities from complete or near complete solubility (5Ala and 5Val) to insolubility close to that observed with 5Ile (5Leu) (Fig. 2D).
Given the variability in the insolubility of the homopentameric GFPNLS-peptide fusions, we further characterized the fusions using fluorescence microscopy because this parameter separated San1 substrates from nonsubstrates by the propensity for in vivo inclusion formation. As expected from their San1-dependent degradation and large degree of insolubility, the 5Ile and 5Phe GFPNLS-peptide fusions were predominantly nuclear and formed a single nuclear inclusion (Fig. 3). In contrast, every other homopentameric GFPNLS-peptide fusion localized throughout the nucleus and cytoplasm and did not form inclusions (Fig. 3). Overall, these results again demonstrated that San1-dependent degradation generally correlates with the degree of insolubility and formation of inclusions in vivo. However, the results were surprising if the conventional hydrophobicity score of a residue was the sole initial consideration.
FIGURE 3.

In vivo inclusion formation of homopentamer GFPNLS-peptide fusions correlates with San1-dependent degradation. Microscopy shows san1Δ cells expressing GFPNLS-peptide fusions containing the indicated homopentameric hydrophobic residues in the peptide degron.
Degradation of the GFPNLS-5Val Peptide Does Not Occur via Ubr1 or Doa10
The San1-independent degradation of the 5Leu, 5Val, and 5Ala GFPNLS-peptide fusions indicated that there is another degradation pathway that likely recognizes a lower threshold of insolubility than San1. Because the 5Val GFPNLS-peptide fusion was degraded the most rapidly, we used it to explore some of the features of the San1-independent pathway(s). First, we examined whether the San1-independent degradation of the 5Val GFPNLS-peptide fusion required the proteasome. Addition of the proteasome inhibitor MG132 significantly stabilized the 5Val GFPNLS-peptide fusion (Fig. 4A), indicating that the bulk of its degradation was through the proteasome.
FIGURE 4.
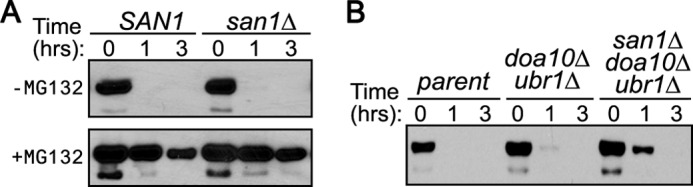
Degradation of the 5Val GFPNLS-peptide fusion is proteasome-dependent but Ubr1- and Doa10-independent. A, cycloheximide-chase assays were performed to assess stability of the 5Val GFPNLS-peptide fusion in the presence or absence of the proteasome inhibitor MG132. Stability was examined in cells with (SAN1) or without (san1Δ) the SAN1 gene intact. B, cycloheximide-chase assays were performed to assess stability of the 5Val GFPNLS-peptide fusion in the presence or absence of DOA10, UBR1, and/or SAN1. The time after cycloheximide addition is indicated above the blots. GFPNLS-peptide fusions were detected using anti-GFP antibodies.
It was shown previously that some San1 substrates are also targeted by the ubiquitin-protein ligase Ubr1 (10, 39). In addition, the ubiquitin-protein ligase Doa10 is localized to the nuclear membrane in part and functions in the degradation of nuclear proteins (21, 40). Therefore, we tested whether either of these ubiquitin-protein ligases were involved in the proteasome degradation of the 5Val GFPNLS-peptide fusion. Double deletion of both UBR1 and DOA10 had a very slight stabilizing effect on the degradation of the 5Val GFPNLS-peptide fusion (Fig. 4B), indicating that they are not the major degradative pathways. Deletion of SAN1 in ubr1Δdoa10Δ cells had an additional modest stabilizing effect (Fig. 4B), revealing that San1 does recognize the 5Val GFPNLS-peptide fusion to a minor degree. Because the major known cytoplasmic and nuclear PQC degradation pathways do not appreciably target the 5Val GFPNLS-peptide fusion, it appears that another, as-yet-unknown, proteasome-dependent PQC degradation pathway exists and operates at a lower insolubility threshold than San1.
Substitution of a Single Ile Residue in the 5Val GFPNLS-Peptide Fusion Causes San1-dependent Degradation in a Position-dependent Manner
Because the 5Val GFPNLS-peptide fusion was very modestly stabilized after deletion of SAN1 in ubr1Δdoa10Δ cells, we reasoned that, if we increased the hydrophobicity of the 5Val stretch, then we might be able to shift the degradation of the GFPNLS-peptide fusion toward increased San1 dependence. To do this, we replaced one Val within the homopentameric stretch with an Ile to create VVVIV, VVIVV, and VIVVV stretches fused to GFPNLS. As we anticipated, the VVVIV stretch resulted in the GFPNLS-peptide fusion being subject to primarily San1-dependent degradation (Fig. 5, A and C). Surprisingly, however, the closer the Ile substitution was to the hydrophilic portion of the peptide fusion, the less effect it had on converting the degradation of the GFPNLS-peptide fusion to San1 dependence (Fig. 5, A and C). The San1 dependence of degradation did correlate with the insolubility of the GFPNLS-peptide fusions containing the single Ile (Fig. 5B), indicating the rules of hydrophobicity/insolubility still applied. From these results, we suspected that the hydrophilic linker sequence positioned near the hydrophobic residue stretch could influence the strength of the hydrophobic residues and mitigate the effect of hydrophobicity, which would alter the ability of San1 to recognize its substrates. In support of this hypothesis, flanking sequences to aggregation-prone regions have been shown to influence protein aggregation (41–47).
FIGURE 5.

Substitution of a single Ile residue in the 5Val GFPNLS-peptide fusion causes San1-dependent degradation in a position-dependent manner. A, cycloheximide-chase degradation assays were performed to assess the stability of GFPNLS-peptide fusions with the indicated hydrophobic residue stretch in the peptide degron. Stability was examined in cells with (SAN1) or without (san1Δ) the SAN1 gene intact. The time after cycloheximide addition is indicated above the blots. GFPNLS-peptide fusions were detected using anti-GFP antibodies. B, solubility assays were performed to determine how the indicated GFPNLS-peptide fusion partition between the insoluble pellet fraction (I) and the soluble supernatant fraction (S). Total lysate (T) indicates the total amount of GFPNLS-peptide fusion in cell lysates. Note that all samples from the solubility assays were run on the same SDS-polyacrylamide gel so that direct comparisons could be made. Percentage of protein in the insoluble versus soluble fractions is listed below each appropriate lane. Relative levels of the insoluble and soluble fractions was determined using ImageJ. C, decay curves for degradation data in A were determined using ImageJ.
To test further the idea that flanking hydrophilic residues can affect San1 recognition, we focused on the 5Ile and 5Phe GFPNLS-peptide fusions, which undergo predominantly San1-dependent degradation (Fig. 2C). To increase the flanking hydrophilicity to its maximum potential, we substituted the charged residue Arg into the position immediately proximal to the 5Ile or 5Phe stretches in their respective GFPNLS-peptide fusions (Fig. 6). We chose to add an Arg residue because it is the most hydrophilic by the Kyte-Doolittle hydophobicity measure (Fig. 2A) and thus should have the best capacity to reveal if increasing hydrophilicity alters San1-dependent degradation. Indeed, substitution of an Arg residue in the proximal position did increase the degradation rate of both the 5Ile and 5Phe GFPNLS-peptide fusions in SAN1 cells, and this was due to acquisition of partial San1-independent degradation observed in san1Δ cells (Fig. 6). Altogether, we think the combined observations suggest that, not only must the insoluble nature of the exposed hydrophobicity be taken into account, but so too must the nature of flanking sequences.
FIGURE 6.
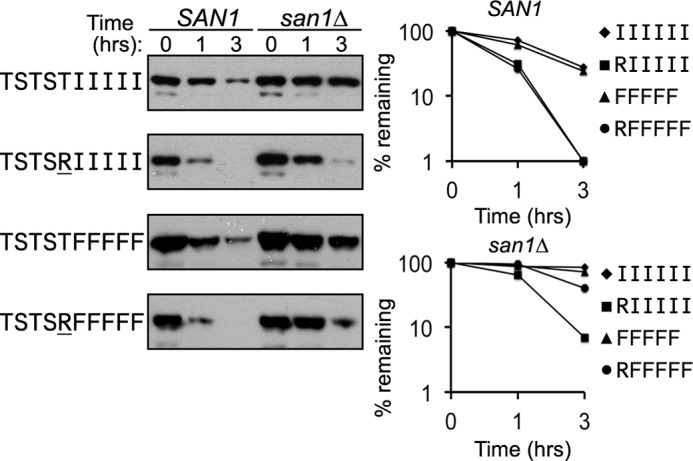
Substitution of a single Arg residue immediately proximal to the 5Ile or 5Phe stretches causes partial San1-independent degradation of the GFPNLS-peptide fusions. Cycloheximide-chase degradation assays were performed to assess the stability of GFPNLS-peptide fusions with the indicated hydrophobic residue stretch with or without an Arg residue substituted immediately proximal. Stability was examined in cells with (SAN1) or without (san1Δ) the SAN1 gene intact. The time after cycloheximide addition is indicated above the blots. GFPNLS-peptide fusions were detected using anti-GFP antibodies. Decay curves for degradation assays were determined using ImageJ.
Homoseptameric Stretches Show Increased Dependence on San1 for Degradation
The results from substituting a single Ile in the 5Val stretch and addition of a single Arg before the 5Ile and 5Phe stretches suggested that the hydrophobicity of the homopentameric stretch is important but is likely also affected by adjacent residues. Accordingly, we surmised that increasing the homopentameric stretches to homoseptameric stretches, creating a greater hydrophobic stretch adjacent to the hydrophilic sequence, might now lead to San1-dependent degradation of the 7Val and 7Leu GFPNLS-peptide fusions and possibly others. This was the case as we found that most homoseptameric stretches resulted in the acquisition of at least some San1-dependent degradation ranging from strong (7Tyr) to moderate (7Val, 7Leu) to weak (7Met) (Fig. 7A). The exceptions were the 7Trp stretch, which remained stable, and the 7Ala stretch, which continued to be degraded in a San1-independent manner (Fig. 7A). Comparison of the degradation rates between the homopentameric and the homoseptameric GFPNLS-peptide fusions in SAN1 and san1Δ cells are plotted in Fig. 7C. The change in San1 dependence in terms of degradation also prompted us to look at solubility of the homoseptameric GFPNLS-peptide fusions. We observed that the acquisition of San1-dependent degradation also generally correlated with increased insolubility (Fig. 7B). The observations suggest a hierarchy of hydrophobic residues in terms of their strength to cause insolubility and San1 recognition with Ile and Phe being the strongest, Tyr, Val, and Leu being the next strongest, and Met being the weakest.
FIGURE 7.

Homoseptameric stretches show increased dependence on San1 for degradation. A, cycloheximide-chase degradation assays performed to assess the stability of GFPNLS-peptide fusions containing homoseptameric stretches of hydrophobic residues in the peptide degron. Stability was examined in cells with (SAN1) or without (san1Δ) the SAN1 gene intact. The time after cycloheximide addition is indicated above the blots. GFPNLS-peptide fusions were detected using anti-GFP antibodies. B, solubility assays performed to determine how the indicated GFPNLS-peptide fusion partition between the insoluble pellet fraction (I) and the soluble supernatant fraction (S). Total lysate (T) indicates the total amount of GFPNLS-peptide fusion in cell lysates. Percentage of protein in the insoluble versus soluble fractions is listed below each appropriate lane. Relative levels of the insoluble and soluble fractions were determined using ImageJ. C, decay curves for data in A and Fig. 2, B–D. The amount of protein remaining at each time point of the degradation assays in A and Fig. 2, B–D, was determined using ImageJ.
DISCUSSION
PQC degradation systems preferentially target misfolded proteins over normally folded ones, which means that they must recognize a feature of structural abnormality that differentiates the misfolded protein from its normal counterpart. Furthermore, because each cellular compartment typically possesses multiple PQC degradation systems that appear to be functionally distinct, each PQC system likely recognizes a unique feature of misfolding. Identifying what these features are for each PQC degradation system and how they differ is key to understanding their substrate selectivity.
In our initial examination of what San1 recognizes within its misfolded protein substrates, we discovered that San1 prefers a particular threshold of exposed hydrophobicity, which could be defined as the amount of hydrophobicity conferred by a window of ≥5 contiguous hydrophobic residues (Ref. 27 and Fig. 1A). Here, we have presented evidence that it is not simply exposed hydrophobicity, but that the hydrophobicity also correlates with a particular threshold of insolubility to allow San1 recognition. If we think about this in terms of substrate-binding affinity, the mechanism of San1 substrate binding likely requires a similar degree of hydrophobicity necessary for two hydrophobic sequences to associate strongly with each other to form an aggregate. Thus, the degree of San1 substrate insolubility serves as a proxy measure for how “strong” the exposed hydrophobicity must be to trigger San1 recognition. How San1 achieves substrate binding with the necessary selectivity and affinity for the appropriate hydrophobicity is not yet clear.
For simplicity, we can think of San1 operating within a particular spectrum of hydrophobicity that confers strong insolubility leading to aggregation (Fig. 8). Protein aggregation is exceptionally deleterious in the nucleus (48), so the selectivity of San1 makes sense from an evolutionary perspective of misfolded protein recognition. It would be physiologically advantageous for a robust nuclear PQC degradation system like San1 to target the types of misfolded proteins that are likely to be the most prone to insolubility and toxic aggregation. Supporting this, we found in an early study, in which we identified new San1 substrates using a two-hybrid analysis, that 11 of 14 substrates destroyed predominantly in a San1-dependent manner were highly toxic in the absence of San1 (31).
FIGURE 8.

Model for San1 substrate recognition. The spectrum of protein solubility is represented with exposed hydrophobicity mapped to increasing insolubility. San1 appears to target misfolded proteins on the far end of the insolubility spectrum. Another unknown PQC degradation pathway that recognizes less exposed hydrophobicity and insolubility than San1 is noted.
It is important to note that we do not think San1 alone has the ability to target proteins once they aggregate. This thought is based on our previous studies examining the ability of San1 to ubiquitinate denatured luciferase in vitro (31). We found that San1 was unable to ubiquitinate denatured luciferase if San1 was added after the denaturation step, but San1 was able to ubiquitinate denatured luciferase when San1 was added during the denaturation step. We interpreted this to mean that San1 can only recognize exposed hydrophobicity that is not buried within an aggregate. Based on this interpretation and our studies here, we do not think that San1 targets a certain type of aggregate but instead recognizes misfolded proteins that have not yet aggregated.
If San1 itself cannot recognize aggregated proteins, the question is how San1 mediates the degradation of highly insoluble proteins in vivo once they form aggregates. Previous studies have demonstrated that the Hsp70 chaperones Ssa1 and Ssa2 are required for the degradation of some San1 substrates in vivo (10, 39, 49), suggesting that the solubility and accessibility of San1 substrates could be maintained upstream of San1 by the action of protein chaperones. It is also important to understand how San1 substrates maintain their solubility after ubiquitination by San1 because the proteasome has difficulty degrading aggregated proteins (50–52). We recently found that the maintenance of San1 substrate solubility upstream of the proteasome requires the AAA-ATPase Cdc48.3 Thus, disaggregation capabilities can be employed both upstream and downstream of San1 to maintain the solubility of highly aggregation-prone San1 substrates.
We do note that whereas hydrophobicity and insolubility above a particular threshold generally correlated with San1-mediated degradation, there were a few deviations. For example, the 5Leu GFPNLS-peptide fusion is not targeted by San1 but has insolubility similar to the 5Ile GFPNLS-peptide fusion (Fig. 2). Also, the degradation of the 7Tyr GFPNLS-peptide fusion is primarily San1-dependent, but it is more soluble than the 7Val and 7Leu GFPNLS-peptide fusions, which are subject to degradation that is less San1-dependent (Fig. 7). The deviations from general insolubility could be due to the assay we used, which is a qualitative assay that cannot distinguish fine quantitative differences in solubility. In fact, we caution against comparing solubility/insolubility percentages between figures because of the day-to-day experimental variability in performing the assay. However, because the solubility assays for each figure were performed at the same time and run on the same gels, comparisons are valid within each figure. In addition to the qualitative nature of the solubility assay, there could be additional parameters to San1 recognition that we have yet to discover. Perhaps the shape of the residue side chains or the overall complexity of the residues in the hydrophobic window play a role in how San1 recognizes a substrate.
Our results also revealed that there is another potential nuclear PQC degradation pathway that appears to recognize a lower threshold of hydrophobicity and insolubility than San1. The pathway targets substrates for proteasome degradation, but it is not the previously described PQC degradation systems of Ubr1 or Doa10. That there might be two nuclear PQC degradation pathways operating in different ranges of exposed hydrophobicity/insolubility is intriguing in terms of how the cell might have evolved triage hierarchies for the repair and degradation of misfolded proteins (53, 54). It could be that having separate PQC systems based on the degree of exposed hydrophobicity and insolubility allows the cell to repair misfolded proteins that have not exposed sufficient hydrophobicity to be in danger of rapid aggregation, yet quickly and robustly destroy those misfolded proteins that have. Identification and characterization of the additional nuclear PQC degradation pathway(s) will allow for in depth comparison of the targeting capabilities with San1.
Acknowledgment
We thank Fred Goldberg for discussions on hydrophobicity and alternative degradation pathways.
This worked was supported, in whole or in part, by National Institutes of Health Training Grant 5T32GM007750 through the NIGMS (to E. K. F.) and Grant R01AG031136 through the NIA (to R. G. G.). This work was also supported by an Ellison Medical Foundation New Scholar Award in Aging (to R. G. G.) and a Marian E. Smith Junior Faculty Award (to R. G. G.).
P. S. Gallagher and R. G. Gardner, unpublished observations.
- PQC
- protein quality control
- NLS
- nuclear localization signal.
REFERENCES
- 1. Hartl F. U., Bracher A., Hayer-Hartl M. (2011) Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 [DOI] [PubMed] [Google Scholar]
- 2. Wang S. S., Wu J. W., Yamamoto S., Liu H. S. (2008) Diseases of protein aggregation and the hunt for potential pharmacological agents. Biotechnol. J. 3, 165–192 [DOI] [PubMed] [Google Scholar]
- 3. Fredrickson E. K., Gardner R. G. (2012) Selective destruction of abnormal proteins by ubiquitin-mediated protein quality control degradation. Semin. Cell Dev. Biol. 23, 530–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bordallo J., Plemper R. K., Finger A., Wolf D. H. (1998) Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded luminal and integral membrane proteins. Mol. Biol. Cell 9, 209–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fang S., Ferrone M., Yang C., Jensen J. P., Tiwari S., Weissman A. M. (2001) The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 98, 14422–14427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hampton R. Y., Gardner R. G., Rine J. (1996) Role of 26 S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol. Cell 7, 2029–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kaneko M., Ishiguro M., Niinuma Y., Uesugi M., Nomura Y. (2002) Human HRD1 protects against ER stress-induced apoptosis through ER-associated degradation. FEBS Lett. 532, 147–152 [DOI] [PubMed] [Google Scholar]
- 8. Nadav E., Shmueli A., Barr H., Gonen H., Ciechanover A., Reiss Y. (2003) A novel mammalian endoplasmic reticulum ubiquitin ligase homologous to the yeast Hrd1. Biochem. Biophys. Res. Commun. 303, 91–97 [DOI] [PubMed] [Google Scholar]
- 9. Swanson R., Locher M., Hochstrasser M. (2001) A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matα2 repressor degradation. Genes Dev. 15, 2660–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heck J. W., Cheung S. K., Hampton R. Y. (2010) Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc. Natl. Acad. Sci. U.S.A. 107, 1106–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang J., Ballinger C. A., Wu Y., Dai Q., Cyr D. M., Höhfeld J., Patterson C. (2001) CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J. Biol. Chem. 276, 42938–42944 [DOI] [PubMed] [Google Scholar]
- 12. Murata S., Minami Y., Minami M., Chiba T., Tanaka K. (2001) CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2, 1133–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nillegoda N. B., Theodoraki M. A., Mandal A. K., Mayo K. J., Ren H. Y., Sultana R., Wu K., Johnson J., Cyr D. M., Caplan A. J. (2010) Ubr1 and Ubr2 Function in a quality control pathway for degradation of unfolded cytosolic proteins. Mol. Biol. Cell 21, 2102–2116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fu L., Gao Y. S., Tousson A., Shah A., Chen T. L., Vertel B. M., Sztul E. (2005) Nuclear aggresomes form by fusion of PML-associated aggregates. Mol. Biol. Cell 16, 4905–4917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gardner R. G., Nelson Z. W., Gottschling D. E. (2005) Degradation-mediated protein quality control in the nucleus. Cell 120, 803–815 [DOI] [PubMed] [Google Scholar]
- 16. Iwata A., Nagashima Y., Matsumoto L., Suzuki T., Yamanaka T., Date H., Deoka K., Nukina N., Tsuji S. (2009) Intranuclear degradation of polyglutamine aggregates by the ubiquitin proteasome system. J. Biol. Chem. 284, 9796–9803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Janer A., Martin E., Muriel M. P., Latouche M., Fujigasaki H., Ruberg M., Brice A., Trottier Y., Sittler A. (2006) PML clastosomes prevent nuclear accumulation of mutant ataxin-7 and other polyglutamine proteins. J. Cell Biol. 174, 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Z., Prelich G. (2009) Quality control of a transcriptional regulator by SUMO-targeted degradation. Mol. Cell. Biol. 29, 1694–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bays N. W., Gardner R. G., Seelig L. P., Joazeiro C. A., Hampton R. Y. (2001) Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat. Cell Biol. 3, 24–29 [DOI] [PubMed] [Google Scholar]
- 20. Huyer G., Piluek W. F., Fansler Z., Kreft S. G., Hochstrasser M., Brodsky J. L., Michaelis S. (2004) Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J. Biol. Chem. 279, 38369–38378 [DOI] [PubMed] [Google Scholar]
- 21. Deng M., Hochstrasser M. (2006) Spatially regulated ubiquitin ligation by an ER/nuclear membrane ligase. Nature 443, 827–831 [DOI] [PubMed] [Google Scholar]
- 22. Hill K., Cooper A. A. (2000) Degradation of unassembled Vph1p reveals novel aspects of the yeast ER quality control system. EMBO J. 19, 550–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wilhovsky S., Gardner R., Hampton R. (2000) HRD gene dependence of endoplasmic reticulum-associated degradation. Mol. Biol. Cell 11, 1697–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taxis C., Hitt R., Park S. H., Deak P. M., Kostova Z., Wolf D. H. (2003) Use of modular substrates demonstrates mechanistic diversity and reveals differences in chaperone requirement of ERAD. J. Biol. Chem. 278, 35903–35913 [DOI] [PubMed] [Google Scholar]
- 25. Carvalho P., Goder V., Rapoport T. A. (2006) Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 126, 361–373 [DOI] [PubMed] [Google Scholar]
- 26. Kanehara K., Xie W., Ng D. T. (2010) Modularity of the Hrd1 ERAD complex underlies its diverse client range. J. Cell Biol. 188, 707–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fredrickson E. K., Rosenbaum J. C., Locke M. N., Milac T. I., Gardner R. G. (2011) Exposed hydrophobicity is a key determinant of nuclear quality control degradation. Mol. Biol. Cell 22, 2384–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brachmann C. B., Davies A., Cost G. J., Caputo E., Li J., Hieter P., Boeke J. D. (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132 [DOI] [PubMed] [Google Scholar]
- 29. Guthrie C., Fink G. R. (1991) Guide to yeast genetics and molecular biology. Methods Enzymol. 194, 1–863 [PubMed] [Google Scholar]
- 30. Theodoraki M. A., Nillegoda N. B., Saini J., Caplan A. J. (2012) A network of ubiquitin ligases is important for the dynamics of misfolded protein aggregates in yeast. J. Biol. Chem. 287, 23911–23922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rosenbaum J. C., Fredrickson E. K., Oeser M. L., Garrett-Engele C. M., Locke M. N., Richardson L. A., Nelson Z. W., Hetrick E. D., Milac T. I., Gottschling D. E., Gardner R. G. (2011) Disorder targets misorder in nuclear quality control degradation: a disordered ubiquitin ligase directly recognizes its misfolded substrates. Mol. Cell 41, 93–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lefebvre B. G., Comolli N. K., Gage M. J., Robinson A. S. (2004) Pressure dissociation studies provide insight into oligomerization competence of temperature-sensitive folding mutants of P22 tailspike. Protein Sci. 13, 1538–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jung C. H., Na Y. R., Im H. (2004) Retarded protein folding of deficient human alpha 1-antitrypsin D256V and L41P variants. Protein Sci. 13, 694–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kryndushkin D. S., Alexandrov I. M., Ter-Avanesyan M. D., Kushnirov V. V. (2003) Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J. Biol. Chem. 278, 49636–49643 [DOI] [PubMed] [Google Scholar]
- 35. Shulga N., Goldfarb D. S. (2003) Binding dynamics of structural nucleoporins govern nuclear pore complex permeability and may mediate channel gating. Mol. Cell. Biol. 23, 534–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shulga N., Mosammaparast N., Wozniak R., Goldfarb D. S. (2000) Yeast nucleoporins involved in passive nuclear envelope permeability. J. Cell Biol. 149, 1027–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kyte J., Doolittle R. F. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 [DOI] [PubMed] [Google Scholar]
- 38. Conchillo-Solé O., de Groot N. S., Avilés F. X., Vendrell J., Daura X., Ventura S. (2007) AGGRESCAN: a server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinformatics 8, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Prasad R., Kawaguchi S., Ng D. T. (2010) A nucleus-based quality control mechanism for cytosolic proteins. Mol. Biol. Cell 21, 2117–2127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Furth N., Gertman O., Shiber A., Alfassy O. S., Cohen I., Rosenberg M. M., Doron N. K., Friedler A., Ravid T. (2011) Exposure of bipartite hydrophobic signal triggers nuclear quality control of Ndc10 at the endoplasmic reticulum/nuclear envelope. Mol. Biol. Cell 22, 4726–4739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Angeli S., Shao J., Diamond M. I. (2010) F-actin binding regions on the androgen receptor and huntingtin increase aggregation and alter aggregate characteristics. PLoS One 5, e9053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beerten J., Jonckheere W., Rudyak S., Xu J., Wilkinson H., De Smet F., Schymkowitz J., Rousseau F. (2012) Aggregation gatekeepers modulate protein homeostasis of aggregating sequences and affect bacterial fitness. Protein Eng. Des. Sel. 25, 357–366 [DOI] [PubMed] [Google Scholar]
- 43. Duennwald M. L., Jagadish S., Muchowski P. J., Lindquist S. (2006) Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc. Natl. Acad. Sci. U.S.A. 103, 11045–11050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ignatova Z., Thakur A. K., Wetzel R., Gierasch L. M. (2007) In-cell aggregation of a polyglutamine-containing chimera is a multistep process initiated by the flanking sequence. J. Biol. Chem. 282, 36736–36743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lakhani V. V., Ding F., Dokholyan N. V. (2010) Polyglutamine induced misfolding of huntingtin exon1 is modulated by the flanking sequences. PLoS Comput. Biol. 6, e1000772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Robertson A. L., Horne J., Ellisdon A. M., Thomas B., Scanlon M. J., Bottomley S. P. (2008) The structural impact of a polyglutamine tract is location-dependent. Biophys. J. 95, 5922–5930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Siwach P., Sengupta S., Parihar R., Ganesh S. (2011) Proline repeats, in cis- and trans-positions, confer protection against the toxicity of misfolded proteins in a mammalian cellular model. Neurosci. Res. 70, 435–441 [DOI] [PubMed] [Google Scholar]
- 48. Woulfe J. M. (2007) Abnormalities of the nucleus and nuclear inclusions in neurodegenerative disease: a work in progress. Neuropathol. Appl. Neurobiol. 33, 2–42 [DOI] [PubMed] [Google Scholar]
- 49. Prasad R., Kawaguchi S., Ng D. T. (2012) Biosynthetic mode can determine the mechanism of protein quality control. Biochem. Biophys. Res. Commun. 425, 689–695 [DOI] [PubMed] [Google Scholar]
- 50. Holmberg C. I., Staniszewski K. E., Mensah K. N., Matouschek A., Morimoto R. I. (2004) Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J. 23, 4307–4318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Snyder H., Mensah K., Theisler C., Lee J., Matouschek A., Wolozin B. (2003) Aggregated and monomeric α-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J. Biol. Chem. 278, 11753–11759 [DOI] [PubMed] [Google Scholar]
- 52. Verhoef L. G., Lindsten K., Masucci M. G., Dantuma N. P. (2002) Aggregate formation inhibits proteasomal degradation of polyglutamine proteins. Hum. Mol. Genet. 11, 2689–2700 [DOI] [PubMed] [Google Scholar]
- 53. McClellan A. J., Tam S., Kaganovich D., Frydman J. (2005) Protein quality control: chaperones culling corrupt conformations. Nat. Cell Biol. 7, 736–741 [DOI] [PubMed] [Google Scholar]
- 54. Wickner S., Maurizi M. R., Gottesman S. (1999) Posttranslational quality control: folding, refolding, and degrading proteins. Science 286, 1888–1893 [DOI] [PubMed] [Google Scholar]