

Background: The molecular basis of calcium release by ryanodine receptors (RyRs) is incompletely understood.
Results: Mutations predicted by a computational model alter Ca2+-dependent single RyR channel activity.
Conclusion: An interface between a pore-lining helix and a linker helix has a critical role in RyR channel gating.
Significance: The results provide new insights into how RyR activity is regulated by Ca2+.
Keywords: Calcium, Molecular Dynamics, Mutagenesis in Vitro, Protein Structure, Skeletal Muscle, Cryo-EM, RyR1, S4-S5 Segment, S6 Segment, Transmembrane Domain
Abstract
Ryanodine receptor type 1 (RyR1) releases Ca2+ from intracellular stores upon nerve impulse to trigger skeletal muscle contraction. Effector binding at the cytoplasmic domain tightly controls gating of the pore domain of RyR1 to release Ca2+. However, the molecular mechanism that links effector binding to channel gating is unknown due to lack of structural data. Here, we used a combination of computational and electrophysiological methods and cryo-EM densities to generate structural models of the open and closed states of RyR1. Using our structural models, we identified an interface between the pore-lining helix (Tyr-4912–Glu-4948) and a linker helix (Val-4830–Val-4841) that lies parallel to the cytoplasmic membrane leaflet. To test the hypothesis that this interface controls RyR1 gating, we designed mutations in the linker helix to stabilize either the open (V4830W and T4840W) or closed (H4832W and G4834W) state and validated them using single channel experiments. To further confirm this interface, we designed mutations in the pore-lining helix to stabilize the closed state (Q4947N, Q4947T, and Q4947S), which we also validated using single channel experiments. The channel conductance and selectivity of the mutations that we designed in the linker and pore-lining helices were indistinguishable from those of WT RyR1, demonstrating our ability to modulate RyR1 gating without affecting ion permeation. Our integrated computational and experimental approach significantly advances the understanding of the structure and function of an unusually large ion channel.
Introduction
Uncovering molecular determinants of ryanodine receptor type 1(RyR1)6 function is essential due to the physiological importance of RyR1; it is indispensable for muscle contraction, and congenital mutations leading to dysfunctional RyR1 give rise to myopathies (central core disease and multi-minicore disease) and malignant hyperthermia. However, structural studies in RyR1 have been challenging because of the large molecular size of RyR1; the functional channel consists of four 565-kDa RyR1 polypeptides and numerous RyR1-associated proteins. RyR1 is characterized by a C-terminal segment with at least six transmembrane helices per monomer (the six-transmembrane domain (6-TMD)) and a large cytosolic domain spanning the gap between the sarcoplasmic reticulum (SR) membrane and the invagination of the plasmalemma known as the transverse tubule (1–5). RyR1 is regulated mainly via a direct interaction with a plasmalemmal voltage-sensing Ca2+ channel (Cav1.1) and by Ca2+ ions (6), both binding to the cytosolic domain. Binding at the cytosolic domain by these effectors controls gating at the 6-TMD that contains the ion pore, but the molecular pathway of this control is still unknown.
The pore region of RyR1 has been well characterized; RyR1 has a high ion conductance for both monovalent cations (∼800 picosiemens (pS) with 250 mm K+ as the conducting ion) and divalent cations (∼150 pS with 50 mm Ca2+) but is selective for Ca2+ (PCa/PK ∼ 7) (7, 8). Sequence comparison, extensive mutagenesis, and single channel studies (9–12) have provided evidence that RyR1 has a pore structure similar to that of K+ channels, whose structure is known (13–17). Furthermore, the residues between the last two C-terminal membrane-spanning segments of RyR1 are located on the SR luminal side and have an amino acid motif (GGGIG) that is similar to the selectivity filter motif (T(V/I)GYG) of K+ channels (9–12).
The structure of RyR1 has been characterized using cryo-EM (2, 3, 18). Ludtke et al. (18) determined the structure of closed (non-conducting) RyR1. In their structural model, the inner helix has a bend at a highly conserved glycine (RyR1 Gly-4934). In K+ channels, bending at a glycine in the inner helix has been associated with channel opening, suggesting that bending of the inner helix alone does not open the pore in RyR1. Samsó et al. (3) determined the cryo-EM structure of open and closed RyR1 both complexed with the associated 12-kDa subunit FKBP12. As observed for the high resolution structures of K+ channels, the inner helix has an outward bend at a glycine (Gly-4934 or Gly-4941) in the open but not closed RyR1. K+ channels also have four pore-lining transmembrane helices (one from each subunit) undergoing a coordinated change from a straight to a kinked conformation while going from the closed to the open state of the pore (13, 15), supporting the idea that the RyR1 pore may have a gating mechanism similar to that displayed by the K+ channels. However, the mapped selectivity filter and the pore-lining helix comprise only ∼20% of the residues that form the 6-TMD of RyR1. The rest of the 6-TMD is thought to be important in mediating signals from the cytosolic domain to control pore gating, but the location of these residues with respect to the RyR1 pore and their interactions with the pore remain unknown. Here, we report the structural determinants of RyR1 gating based on homology modeling and high resolution cryo-EM data, which have been successfully validated by single channel measurements of WT and mutant channels.
EXPERIMENTAL PROCEDURES
Materials
[3H]Ryanodine was obtained from PerkinElmer Life Sciences, and protease and inhibitor mixtures were from Sigma. Chemicals were from Sigma unless specified otherwise.
Sequence Analysis
The six transmembrane (TM) segments of RyR1 correspond to residues 4561–4948. However, unlike 6-TM potassium channels, there is an insertion of 100 residues between S2 and S3, which, according to the topology of the channel, will be present in the cytoplasmic side. The Pfam annotation (a database of protein families) (19) for the C-terminal region of RyR1 indicates that the last four TM helices (S3–S6, residues 4766–4952) are grouped as a domain belonging to the ion transport family (accession number PF00520), whereas the first two TM helices (residues 4381–4670) belong to another putative domain (accession number PF06459). We used fragment 4766–4952 of RyR1, which is predicted to form a contiguous domain, to perform PSI-BLAST (20–22). We performed PSI-BLAST for five iterations with stringent inclusion criteria (an E-value threshold of 0.001). By the sixth iteration, we observed no new hits, indicating convergence of PSI-BLAST in five iterations. The alignment between Kv1.2 and RyR1 was first obtained from PSI-BLAST output. Because earlier studies have shown that the 4894GGGIGD4899 segment in RyR1 (9–12) is functionally similar to the K+ channel (13–17) and MlotiK1 (23) selectivity filters, we adjusted the alignments between RyR1 and the K+ channels to ensure that the selectivity filters were matched (see Fig. 2). Manual adjustment of alignments based on functional features such as selectivity filters and structural cysteines is essential to ensure physically accurate alignments (24).
FIGURE 2.

Sequence alignments used in construction of structural models of open and closed states. A, alignment of RyR1 with the Kv1.2 sequence (UniProt accession number P63142 and Protein Data Bank code 3LUT) that was used for constructing the structural model of the RyR1 open state. B, alignment of RyR1 with the MlotiK1 channel (UniProt accession number Q98GN8 and Protein Data Bank code 3BEH) used for constructing the structural model of the RyR1 closed state. Asterisks indicate positions that have a single fully conserved residue. Double dots indicate conservation between groups of strongly similar properties. Single dots indicate conservation between groups of weakly similar properties. SE, selectivity filter.
We used composition-based (first principle) and statistics-based (hidden Markov model and neural networks) programs for TM predictions (25–30). The composition-based programs we used were SOSUI, TopPred2, and SCAMPI. The statistics-based programs we used were MEMSAT3, OCTOPUS, and PRODIV. We used Jpred (31) and PSIPRED (27) for determining secondary structure propensities.
Model Construction
The crystal structures of Kv1.2 (15) and MlotiK1 (23) channels along with the modified PSI-BLAST alignment were used for constructing the structural models of the open and closed states of RyR1. The alignment presented only insertions in the luminal loops of RyR1 between S5 and S6 and between S6 and the P-helix. We first introduced the insertions, which are all loops, using discrete molecular dynamics simulations (32–34), where all residues except the ones right next to the insertions were constrained to be static to ensure minimal changes in the structure at this stage. Next, the sequence of the template was changed to that of RyR1 using Medusa (35–37). We then used highly constrained discrete molecular dynamics simulations (38) to resolve steric clashes introduced by sequence changes. We ensured that the minimized structures did not have excessive steric clashes or other anomalies in their covalent geometry using the Gaia server (39).
Calculations of Change in Stability (ΔΔG)
For all of the mutants presented in this study, we calculated the change in stability of the open and closed states of RyR1 (ΔΔG) with the corresponding structural model using Medusa (35–37). Medusa calculations involve a Monte Carlo-based simulated annealing procedure that uses rotamer libraries of amino acids for fast minimization of its energy function while leaving the backbone fixed. Medusa uses a combination of physics-based terms (van der Waals, hydrogen bond, and solvation) and knowledge-based terms (backbone and side chain torsions). For each position we considered, residues within 10 Å (Cα − Cα distance) were allowed to sample all available native rotamers, whereas the rest of the residues were allowed to sample the sub-rotameric states of the starting rotamer. We averaged the free energy obtained from at least 100 Medusa calculations to obtain ΔΔG for each mutation. We define ΔΔG as ΔGMut − ΔGWT, where ΔGMut is the stability of the mutant, and ΔGWT is the stability of the wild-type channel. Thus, a destabilizing mutation would result in a positive ΔΔG. For the two mutations (G4834W and V4838W) that resulted in a very large increase in van der Waals repulsion energy, we have not reported ΔΔG because, for these mutations, there is the possibility of backbone relaxation, and we did not allow backbone movement in our calculations (these are labeled as ND in Table 1). To predict change in channel open probability (Po) upon mutation, we considered the thermodynamic cycle (Scheme 1).
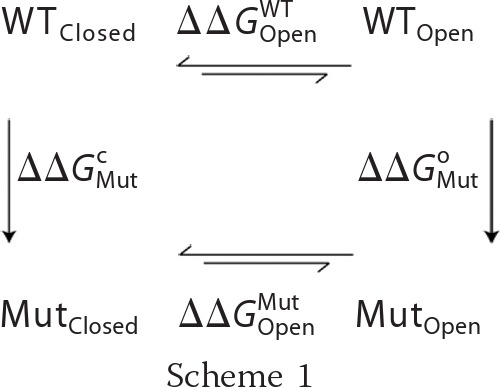 |
In the above schematic, ΔΔGOpenWT and ΔΔGOpenMut refer to the stability change upon channel opening for the WT and mutants, respectively, which were determined experimentally by Po measurements. ΔΔGMutc and ΔΔGMuto are the stability change upon mutation for the closed and open states, respectively, which were determined computationally using Medusa. The structural models for the open and closed states can be correlated with experimental measurements by summing two sides of the thermodynamic cycle (Equation 1).
Equation 1 can be rearranged to Equation 2.
The terms on the left side of Equation 2 could be determined experimentally using Po determined for WT and mutant RyR1. The terms on the right side of Equation 2 were determined with Medusa using structural models of the closed and open states of WT and mutant RyR1. In the case of homology models, such calculations are good predictors of the net effect of mutation (stabilization or destabilization), but the magnitude of ΔΔG is not highly accurate (40). Thus, in this study, we used the ΔΔG values to predict if there will be an increase or decrease in Po of mutant channels, but not the extent of change in Po.
TABLE 1.
Calculated stability changes of S4-S5 linker mutations in open and closed states of RyR1
The values denote means ± S.E. ND, not determined.
| Mutation | ΔΔG open state | ΔΔG closed state | Net ΔΔG |
|---|---|---|---|
| kcal/mol | kcal/mol | kcal/mol | |
| V4830W | 7.6 ± 0.9 | 12.6 ± 0.8 | −5.1 ± 1.1 |
| H4832W | 4.3 ± 0.8 | 2.3 ± 0.7 | 2.0 ± 1.1 |
| G4834W | ND | −2.3 ± 0.7 | ND |
| V4838W | ND | ND | ND |
| T4840W | 17.1 ± 0.9 | 24.6 ± 0.7 | −7.5 ± 1.1 |
| V4841W | 10.7 ± 0.9 | 4.0 ± 0.6 | 6.8 ± 1.1 |
| Q4947N | 2.4 ± 0.9 | −4.8 ± 0.7 | 7.3 ± 1.2 |
| Q4947T | 5.2 ± 0.9 | −3.6 ± 0.7 | 8.8 ± 1.2 |
| Q4947S | 0.8 ± 0.9 | −3.1 ± 0.7 | 4.0 ± 1.2 |
Cryo-EM Models of RyR1
The three-dimensional reconstructions of RyR1 in the closed and open states were obtained previously at a nominal resolution of 10.2 Å (Electron Microscopy Data Bank (EMDB) accession codes 1606 and 1607) (3). For Fig. 3 (B and C), 45L and S6 were further traced by following the high density of the helices through successive slices of the three-dimensional reconstruction. Docking of the structural models onto the cryo-EM map was performed first using the colores functionality in Situs (41). Colores aligns a generated Gaussian density map of the structural model (at 10 Å resolution) with the cryo-EM map through rigid-body fitting in six dimensions (three rotational and three translational). The best Situs fits were evaluated visually and refined using the fit-in-map functionality in Chimera.
FIGURE 3.

Structural models of RyR1 in open and closed states. A, the experimental structures of Kv1.2 and MlotiK1 channels were used to construct the structural models of RyR1 in the open and closed states. The RyR1 models are displayed in the ribbon representation using PyMOL. B, the open state viewed from the luminal side and the cytoplasmic side featuring the overlay of cryo-EM densities (transparent surface) and the open state structural model (ribbon representation). The overlap of S6 and 45L with the corresponding traced densities is shown on the right. C, an overlay of the closed state structure and cryo-EM densities shown as viewed from the luminal side and the cytoplasmic side. The overlay of the S6 and 45L helices with the traced densities is shown on the right. The two views represent a 180° rotation around the vertical axis. The scale bars shown in the overlay of the structural models and EM densities measure 10 Å. D, the interface between 45L and S6 in the open and closed states (corresponding to the boxed regions A) is shown to highlight the higher conservation of the 45L-S6 interface compared with the rest of the regions. Each residue is colored according to its conservation derived from multiple sequence alignment of PSI-BLAST hits, with blue representing 100% conservation and red representing 0% conservation. E, the residue propensities of 45L (left) and the C-terminal end of S6 (right) generated using the Berkeley WebLogo program.
Site-directed Mutagenesis
The full-length rabbit RyR1 cDNA was constructed as described previously (42). Mutations were introduced by Pfu PCR using mutagenic oligonucleotides following the QuikChange II site-directed mutagenesis kit protocol (Stratagene, La Jolla, CA). The C-terminal ClaI/XbaI fragment (14443/15276) of RyR1 cDNA cloned into the pBluescript vector served as the template for mutagenesis. Mutated sequences were confirmed by sequencing, and mutated C-terminal fragments were reintroduced into the ClaI and XbaI sites of the C-terminal fragment of RyR1. Mutated full-length expression plasmids were prepared by ligation of three fragments (ClaI/XhoI, XhoI/EcoRI, and EcoRI/XbaI containing the mutated sequence) and the expression vector pCMV5 (ClaI/XbaI) as described previously (42).
Expression and Preparation of Wild-type and Mutant Channels
Rabbit WT and mutant RyR1 cDNAs were transiently expressed in HEK 293 cells (11). Crude membrane fractions (42) and proteoliposomes containing purified recombinant WT and mutant RyR1 channels (43) were prepared as described.
Cellular Ca2+ Release
Stored Ca2+ release was determined using the fluorescent Ca2+ indicator dye Fluo-4 AM as described previously (44). HEK 293 cells grown on glass coverslips were washed three times with Krebs-Ringer-Henseleit buffer and loaded with 5 μm Fluo-4 AM for 30 min to 1 h at 37 °C. After loading and rinsing with Krebs-Ringer-Henseleit buffer to remove non-hydrolyzed Fluo-4 AM, cellular Ca2+ release was induced by ∼5 mm caffeine and measured in individual cells using the program EasyRatioPro (Photon Technology International, Lawrenceville, NJ).
SDS-PAGE and Immunoblot Analyses
WT and mutant RyR1 in crude membrane fractions were detected using 3–12% gradient SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blotted with 2% ECL Advance blocking reagent (Amersham Biosciences) in 0.5% Tween 20 and TBS (pH 7.4) at 24 °C and probed with primary anti-RyR1 polyclonal antibody and peroxidase-conjugated anti-mouse IgG (1:50,000; Calbiochem). Immunoblots were developed using enhanced chemiluminescence and quantified using ImageQuant TL analysis software.
[3H]Ryanodine Binding
The highly specific plant alkaloid ryanodine is widely used as a probe of RyR channel activity and content (45). Bmax values of [3H]ryanodine binding were determined by incubating crude membrane preparations for 4 h at 24 °C with a nearly saturating concentration of [3H]ryanodine (20 nm) in 20 mm imidazole (pH 7.0), 0.6 m KCl, 0.15 m sucrose, 100 μm Ca2+, and protease inhibitors. Nonspecific binding was determined using a 1000-fold excess of unlabeled ryanodine. Aliquots of samples were diluted 9-fold with ice-cold water and placed on Whatman GF/B filters saturated with 2% polyethyleneimine. Filters were washed with three 5-ml volumes of ice-cold 0.1 m KCl and 1 mm K-Pipes (pH 7.0). Radioactivity remaining on the filters was determined by liquid scintillation counting to obtain bound [3H]ryanodine.
Single Channel Recordings
Single channel measurements were performed using planar lipid bilayers containing a 5:3:2 mixture of bovine brain phosphatidylethanolamine, phosphatidylserine, and phosphatidylcholine (12). Proteoliposomes containing purified recombinant RyR1 were added to the cis (SR cytosolic side) chamber of a bilayer apparatus and fused in the presence of an osmotic gradient (250 mm cis KCl and 20 mm trans KCl in 20 mm K-Hepes (pH 7.4) and 2 μm Ca2+). After the appearance of channel activity, the trans (SR luminal side) KCl concentration was increased to 250 mm. A strong dependence of single channel activity on the cis Ca2+ concentration indicated that the cytosolic region faced the cis chamber of the bilayer. The trans side of the bilayer was defined as ground. Electrical signals were filtered at 2 kHz (0.5 kHz for Ca2+ currents at 0 mV), digitized at 10 kHz, and analyzed at a 50% threshold setting (12). Data acquisition and analysis were performed with a commercially available software package (pCLAMP, Axon Instruments). To determine permeability ratios, single channel activities were recorded in symmetrical 250 mm KCl solution with 10 mm Ca2+ on the trans side, and the reversal potential (Erev) was measured. The permeability ratio of Ca2+ versus K+ (PCa/PK) was calculated using a modified form of the Goldman-Hodgkin-Katz equation (Equation 3).
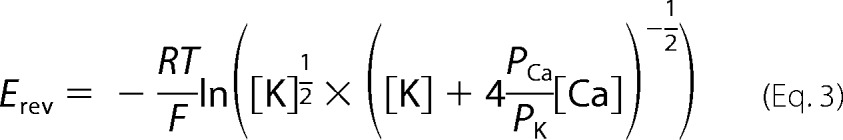 |
Biochemical Assays and Data Analysis
Free Ca2+ concentrations were established by adding the appropriate amounts of Ca2+ and EGTA calculated using stability constants and a published computer program (46). Free Ca2+ concentrations were verified with the use of a Ca2+-selective electrode. Differences between samples were analyzed using Student's t test; p < 0.05 was considered significant.
RESULTS
Sequence Analyses Point to a Conserved 6-TM Topology
To uncover the molecular determinants of RyR1 opening and closing, we sought to understand the role of residues outside the pore-forming region of the RyR1 6-TMD in controlling RyR1 gating by constructing and verifying structural models of the open and closed states of the 6-TMD. TM helix prediction suggested the presence of at least six TM segments (Fig. 1A) in the RyR1 C-terminal region (residues 4561–4948; UniProt accession number P11716). The presence of the six membrane-spanning segments is sufficient to support RyR1 ion channel activity as seen by single channel recordings of tryptic fragments (47) and deletion of the preceding putative TM segments (48). Overlaying the TM prediction on secondary structure prediction (Fig. 1B) further defined the boundaries of the six TM segments of RyR1. The boundaries of the TM segments combined with the known luminal or cytosolic location of intervening loops in between the TM segments (1, 48) enabled us to propose a TM topology for RyR1 (Fig. 1C), which underscores its similarity to the known structures of 6-TM cation channels (15, 23, 49). This topology for the 6-TMD features six TM segments (S1–S6), where the S4 segment is connected to the S5 segment through a S4-S5 linker helix (45L, 4830VTHNGKQLVMTV4841). The S5 segment is connected to a short pore helix (P-helix) through a luminal loop. The P-helix leads to the selectivity filter and then the pore-lining S6 segment.
FIGURE 1.

Bioinformatics-based predictions from RyR1 primary sequence. Shown are predictions of TM helices from various programs (A) and the secondary structure propensity of the TM segment of RyR1 calculated using Jpred (B). In the secondary structure (SS) propensity plot, the thick black lines represent TM helix prediction using MEMSAT3. In both the secondary structure and TM helix plots, the shaded areas represent the final assignment of the TM helices in RyR1 that was used to build the structural model, where L stands for the S4-S5 linker, P stands for the P-helix and SE stands for selectivity filter. C, topology of the RyR1 TM region. The boundaries of the TM helices are indicated.
We then directly identified structural homologs to RyR1 by querying protein sequence databases with PSI-BLAST (20–22), where we found the voltage-gated potassium channel Kv1.2 (UniProt accession number P63142 and Protein Data Bank code 3LUT) (Fig. 2A) to feature highly statistically significant (E-value of ≈2 × 10−4) alignment with RyR1, thus providing us with a structural template to model the RyR1 6-TMD. The structure of Kv1.2 has been solved in the open state (15), and the topology of its TM helices is identical to that of RyR1. For the closed state template, we utilized the structure of the cyclic nucleotide-gated K+ channel MlotiK1 (UniProt accession number Q98GN8 and Protein Data Bank code 3BEH) (Fig. 2B) (23), which has a membrane topology identical to that of RyR1 and Kv1.2. Besides the changes corresponding to open-closed transitions in the S1–S6 helices, the conformations of the P-helix and the selectivity filter in the open state Kv1.2 and closed state MlotiK1 structures are highly similar.
RyR1 Homology Structural Models Match the Corresponding RyR1 Conformation Determined by Cryo-EM
We used the alignments between Kv1.2 and RyR1 and between MlotiK1 and RyR1 to build structural models for the RyR1 membrane-spanning domains in the closed and open states (Fig. 3A) using Medusa (35–37) and discrete molecular dynamics (32–34). To analyze the correspondence between these homology models and the actual structure of RyR1 in the closed and open states, we docked our structural models to the 10 Å resolution cryo-EM densities of RyR1 in the closed and open states (Fig. 3, B and C) (3). We observed a high similarity between the structural models and the cryo-EM maps, with cross-correlation values of 0.89 and 0.90 for the closed-to-closed and open-to-open comparisons, respectively, when comparing the cryo-EM maps with the pseudo-atomic maps filtered at 10 Å resolution. The size of the pore of the structural models also fits the observed pore size in the cryo-EM maps, with distances of 17 and 21–22 Å between the centers of opposite inner helices for the closed and open states, respectively (Table 2). The lack of higher resolution EM densities prevented us from evaluating the fit of the S1–S4 helices, but we observed significant overlap between the EM densities and the structural model in the region starting at 45L helix and ending at the S6 helix for both the open and closed states (Fig. 3, B and C). Importantly, the distinct orientation of the 45L and S6 helices in the open and closed structural models fits the distinct densities of the corresponding regions traced in the cryo-EM maps (Fig. 3, B and C). The close fit of 45L and S6 in the closed and open state structural models with the corresponding cryo-EM maps demonstrates the suitability of our structural models to model interactions involved in RyR1 gating.
TABLE 2.
Center-to-center distance for opposite inner helices in the different RyR structural models
| RyR model | Distance |
|---|---|
| Å | |
| Cryo-EM (closed) | 17 |
| Structural model (closed) | 17 |
| Cryo-EM (open) | 22 |
| Structural model (open) | 21 |
45L Region Mediates RyR1 Gating
An important aspect of 6-TM K+ channels is the role of 45L in channel gating (50–55). The open-closed transition in the K+ channels is characterized by the movement of 45L while maintaining an interface with S6. In the closed state, S6 is a straight helix, whereas in the open state, S6 is bent at the gating hinge, which has been suggested to correspond to Gly-4934 in RyR1 (3). The position of 45L in the closed state precludes bending of S6, thus maintaining the closed state. We found that in our structural model, 45L partially overlaps with a cytoplasmic segment linking S4 and S5 according to the model of Du et al. (1, 48), where the use of GFP inserts predicted a S4-S5 loop of 10 amino acids in length (Thr-4825–Gly-4834). Earlier studies discovered that amino acid substitutions to alanine in the N-terminal half of this region (4825TILSS4829) result in changes in RyR1 gating (56). In our model, those residues are present in the C-terminal end of the S4 helix and would be accessible from the cytoplasmic side of the channel. The S4 helix in K+ channels is known to undergo structural transitions due to gating (53, 57). Hence, mutations in the S4 helix of RyR1 are also expected to affect channel gating.
If the predicted 45L-S6 interface were important for RyR1 gating, we would expect high evolutionary conservation of this interface. To explore the conservation of these regions, we generated a multiple sequence alignment from the PSI-BLAST hits that consisted of 368 protein sequences linked evolutionarily to rabbit RyR1. We observed nearly 100% conservation of residues in S6 that face 45L (Ile-4936, Phe-4940, Leu-4943, and Arg-4944), whereas the residues of 45L that face S6 are ∼70% conserved (Val-4830, Gly-4834, Leu-4837, Val-4838, and Val-4841) (Fig. 3, D and E). We also found that in addition to being ∼70% conserved, the S6-facing side of 45L also allows only conservative substitution (Fig. 3E). The high conservation of residues of the proposed 45L and S6 support the idea that they form a functional interface that is important in gating. Thus, through our evolutionary and structural analysis, we have discovered that a stretch of amino acids ∼70 residues away from the pore on the primary sequence may form an interface with the S6 helix to control its gating.
45L Mutants Relatively Stabilize Either the Open or Closed State of RyR1
To validate the proposed interface between 45L and S6 helices, we computationally determined the effect of tryptophan-scanning mutagenesis of 45L on RyR1 gating. The contacts between many of the residues in 45L with the S6 helix are different in the open and closed states of RyR1, implying mutations in 45L to have distinct effects on the closed and open states. The probability of channel opening (Po) measured experimentally can reveal whether a 45L mutant features a stabilized open or closed state relative to WT. We wanted to test the hypothesis that mutating residues in 45L would affect the relative stability of open and closed states, which would be reflected in the Po of the mutants being significantly higher or lower than that of WT RyR1. Hence, we first calculated the change in the relative stability of RyR1 open and closed states upon mutations in the 45L region using Medusa (35–37). We focused on four amino acid residues (Val-4830, Gly-4834, Val-4838, and Val-4841) that, according to our model, directly interacted with S6 and two residues (His-4832 and Thr-4840) that were not expected to directly interact with S6 (Fig. 3D). From our calculations (except for G4834W and V4838W, which featured a high extent of steric clashes upon mutation), we observed two effects upon performing these mutations: relative stabilization of the closed state or relative stabilization of the open state (Table 1), which translates to an expected decrease or increase in Po, respectively.
Designed 45L Mutants Display Altered Gating in Single Channel Measurements
Recombinant WT and six mutant RyR1 channels in 45L (V4830W, H4832W, G4834W, V4838W, T4840W, and V4841W) were expressed as functional RyRs as indicated by a cellular caffeine-induced Ca2+ response that, in most instances, correlated reasonably well with their RyR1 protein contents (Table 3). V4830W showed a caffeine response in a smaller number of cells compared with the other mutants. From ion conductance measurements using the planar lipid bilayer method, we observed that the mutations had no effect on K+ conductance and on Ca2+ selectivity (PCa/PK, in the presence of 10 mm SR luminal Ca2+) compared with WT (Table 3). Hence, we conclude that introduction of mutations in 45L does not markedly alter RyR1 ion conductance and selectivity.
TABLE 3.
Properties of 45L mutants
The values denote the mean ± S.E. NA, not applicable.
| RyR1 | Cells with caffeine response | Bmax of [3H]ryanodine bindinga | Intensity on immunoblots | γb | PCa/PKc |
|---|---|---|---|---|---|
| % WT | % WT | % WT | pS | ||
| WT | NA | NA | NA | 802 ± 8 (12) | 6.6 ± 0.1 (18) |
| V4830W | 22.5 ± 4.3 (20)d | 20.4 ± 11.1 (6)d | 51.5 ± 6.3 (14)d | 786 ± 6 (10) | 6.7 ± 0.2 (5) |
| H4832W | 67.2 ± 12.0 (9)d | 79.4 ± 13.5 (4) | 70.3 ± 6.2 (8)d | 785 ± 7 (14) | 6.4 ± 0.2 (5) |
| G4834W | 85.0 ± 10.5 (8) | 49.0 ± 14.5 (3)d | 123.6 ± 17.1 (7) | 791 ± 6 (15) | 6.6 ± 0.2 (6) |
| V4838W | 81.1 ± 10.9 (7) | 58.5 ± 11.4 (3)d | 104.5 ± 17.6 (13) | 772 ± 4 (17) | 6.8 ± 0.3 (4) |
| T4840W | 127.0 ± 8.5 (13)d | 30.0 ± 12.3 (3)d | 71.8 ± 8.9 (10)d | 770 ± 6 (7) | 6.7 ± 0.1 (5) |
| V4841W | 95.1 ± 5.3 (7) | 43.2 ± 13.3 (4)d | 63 ± 8.3 (8)d | 783 ± 7 (13) | 6.3 ± 0.2 (5) |
a Maximum binding capacity.
b K+ conductance.
c Selectivity of Ca2+ over K+.
d p < 0.05 compared with WT.
In contrast to ion conductance and selectivity, we observed major differences in Ca2+ sensitivity, Ca2+ dependence, and gating characteristics of mutant channels compared with WT (Fig. 4, A–C, and Table 4). V4830W exhibited a high Po (∼1) at 0.01 μm to 10 mm cytoplasmic Ca2+ (Fig. 4, A and B), which suggests that the mutant channel is constitutively active. T4840W also exhibited an increased Po at 0.01 μm to 10 mm cytoplasmic Ca2+ (Fig. 4, A and B), whereas H4832W and G4834W had a reduced Po compared with WT (Fig. 4, A and C). The two remaining mutant channels (V4838W and V4841W) exhibited a Po and Ca2+ dependence that were not significantly different from those of WT (Fig. 4, A and C). The effects of mutations on channel activity at 2 μm cytoplasmic Ca2+ were analyzed by time analysis of WT and mutant channel recordings. The marked increase in Po for V4830W and T4840W was accounted for by an increase in long open events (mean open times of 369.8 ± 70.8 and 19.9 ± 8.5 ms, respectively, versus 0.68 ± 0.06 ms for WT) (Table 4). In contrast, the reduced activities of H4832W and G4834W were associated with a decreased duration of the open events and an increased duration of the closed events. Consistent with no major change in Po, no significant differences in the mean open and closed time constants were obtained for V4838W and V4841W compared with WT. Our results indicate that the V4830W and T4840W mutations stabilize the open state, whereas the H4832W and G4834W mutations stabilize the closed state.
FIGURE 4.

Experimental characterization of 45L and S6 mutants. A, representative single channel traces for WT RyR1 and 45L mutants. B and C, effect of cytoplasmic Ca2+ on the Po of WT RyR1 and 45L mutants. D, representative single channel traces for S6 mutants. E, effect of cytoplasmic Ca2+ on the Po of S6 mutants.
TABLE 4.
Properties of 45L and S6 mutants
The values denote means ± S.E.
| RyR1 |
Po |
No. of events/min | Toa | Tcb | |
|---|---|---|---|---|---|
| 0.1 μm Ca2+ | 2 μm Ca2+ | ||||
| ms | ms | ||||
| WT | 0.005 ± 0.002 (24) | 0.27 ± 0.03 (29) | 23,821 ± 2001 (19) | 0.68 ± 0.06 (19) | 2.39 ± 0.37 (19) |
| V4830W | 0.99 ± 0.01 (16)c | 0.96 ± 0.03 (10)c | 318 ± 155 (9)c | 369.80 ± 70.8 (9)c | 1.73 ± 0.87 (9) |
| H4832W | 0.015 ± 0.014 (13) | 0.10 ± 0.03 (13)c | 13,647 ± 4235 (11)c | 0.32 ± 0.02 (11)c | 7.25 ± 1.80 (11)c |
| G4834W | 0.006 ± 0.005 (14) | 0.10 ± 0.04 (15)c | 9625 ± 2491 (10)c | 0.33 ± 0.02 (10)c | 11.69 ± 2.96 (10)c |
| V4838W | 0.026 ± 0.013 (8)c | 0.38 ± 0.07 (17) | 22,602 ± 2296 (14) | 1.72 ± 0.86 (14) | 2.22 ± 0.64 (14) |
| T4840W | 0.49 ± 0.22 (3)c | 0.83 ± 0.09 (7)c | 4120 ± 942 (6)c | 19.9 ± 8.5 (6)c | 2.26 ± 0.66 (6) |
| V4841W | 0.0003 ± 0.001 (3) | 0.20 ± 0.06 (13) | 15,982 ± 2516 (11) | 0.65 ± 0.14 (11) | 5.02 ± 1.25 (11) |
| Q4947N | 0.001 ± 0.001 (4) | 0.02 ± 0.01c (10) | 3355 ± 943c (10) | 0.36 ± 0.07c (10) | 107.63 ± 59.67c (10) |
| Q4947T | 0.003 ± 0.002 (3) | 0.03 ± 0.01c (10) | 5335 ± 1238c (10) | 0.35 ± 0.03c (10) | 24.76 ± 10.63c (10) |
| Q4947S | 0.004 ± 0.004 (4) | 0.07 ± 0.03c (11) | 10,182 ± 3534c (11) | 0.34 ± 0.03c (11) | 65.42 ± 41.82 (11) |
a Mean open time at cytoplasmic 2 μm Ca2+.
b Mean closed time at cytoplasmic 2 μm Ca2+.
c p < 0.05 compared with WT.
45L Mutants That Stabilize the Open State Also Affect Ca2+ Sensitivity of RyR1
RyR1 receptors are activated by the binding of Ca2+ to high affinity sites and are inhibited by Ca2+ binding to low affinity sites, giving rise to the bimodal Ca2+ dependence of channel activity (WT in Fig. 4, B and C). Mutagenesis and disease-causing mutations show that multiple regions located in the large cytosolic structure contribute to the regulation of RyRs (58, 59). We found that two of the six 45L mutants also altered Ca2+ regulation of RyR1. Whereas four of the mutants (H4832W, G4834W, V4838W, and V4841W) displayed a Ca2+ dependence of channel activity similar to that of WT (with a Po of ∼0 at 0.01 μm and 10 mm cytoplasmic Ca2+) (Fig. 4C), V4830W maintained a Po of ∼1.0 at 0.01 μm cytoplasmic Ca2+ (Fig. 4B). A decrease in Po was observed only at 10 mm Ca2+. These results indicate that the V4830W mutation leads to a decoupling of high affinity Ca2+ binding and channel opening and likely reduced Ca2+ binding to the inhibitory sites. The elevated channel open probabilities of T4840W at low and high cytoplasmic Ca2+ indicate partial decoupling of Ca2+ regulation and channel gating (Fig. 4B).
S6 Mutations Also Affect RyR1 Gating by Relatively Stabilizing the Closed State
Given the effect of the 45L mutations on RyR1 gating, we hypothesized that mutating residues in S6 that face the linker will also affect RyR1 gating. We found that mutations of Gln-4947 relatively stabilized the closed state (Table 1), which we characterized experimentally. Three mutants (Q4947N, Q4947T, and Q4947S) were expressed as functional RyRs as indicated by a robust cellular caffeine response (Table 5). Furthermore, the mutations had no effect on K+ conductance and on Ca2+ selectivity (PCa/PK, in the presence of 10 mm SR luminal Ca2+) compared with WT (Table 5), indicating that these mutations did not affect channel conductance and stability and supporting the model that the side chain of Gln-4947 faces away from the pore. However, channel gating was significantly different from WT, with reduced channel open probability, decreased durations of open times, and increased durations of closed times, indicating stabilization of the closed state (Fig. 4, D and E, and Table 4), in accordance with the model.
TABLE 5.
Properties of S6 mutants
The values denote means ± S.E.
| RyR1 mutant | Cells with caffeine response | Bmax of [3H]ryanodine bindinga | γb | PCa/PKc |
|---|---|---|---|---|
| % WT | % WT | pS | ||
| Q4947N | 69.7 ± 19.6 (6)d | 28.3 ± 16.1 (5)d | 788 ± 8 (16) | 6.3 ± 0.2 (4) |
| Q4947T | 67.7 ± 10.1 (6)d | 47.9 ± 5.4 (4)d | 783 ± 6 (11) | 6.4 ± 0.2 (5) |
| Q4947S | 59.8 ± 7.3 (5)d | 54.8 ± 22.0 (4)d | 784 ± 7 (12) | 6.9 ± 0.3 (4) |
a Maximum binding capacity.
b Conductivity.
c Selectivity of Ca2+ over K+.
d p < 0.05 compared with WT.
DISCUSSION
Our models for the open and closed states of RyR1 provide the structural basis for altered gating behavior of 45L and S6 mutants. The V4830W mutation in the open state features increased contacts between the N-terminal end of 45L and the S6 helix from the same monomer and the S5 helix from the adjacent monomer due to the larger side chain of tryptophan compared with valine (Fig. 5A). The increased contacts lead to the stabilization of the open state of the mutant compared with the closed state because the closed state features no additional contacts with S6 due to the mutation. Experimentally, V4830W features a 356% increase in the Po and a 545% increase in the duration of the open state compared with WT RyR1. We observed a similar effect in the structural models and experiments for the T4840W mutant (Fig. 6A). Thus, the mutations that stabilize the open state according to our calculations increase the channel Po.
FIGURE 5.

Structural models of 45L and S6 mutations in open and closed states. A, the structural models of V4830W in the open and closed states depict relative stabilization of the mutation in the open state due to formation of new van der Waals contacts. Residues forming contacts with residue 4830 are shown with surface representation, with the largest such interaction observed for V4830W in the open state. B, the structural models of G4834W in the open and closed states depict destabilization of the open state due to clashes between residue 4834 and residues in the S6 helix (shown as red dotted lines) and no such clashes in the closed state. C, the structural models of Q4947N show loss of van der Waals contacts between S6 and 45L due to a shortened side chain in the open state but no such loss in the closed state. The residues proximal to residue 4947 that can form contacts are shown with surface representation, and the atoms belonging to the side chain at residue 4947 are shown as spheres. All three mutants are compared with WT.
FIGURE 6.

Structural models of linker mutations in open and closed states. A, the structural models of T4840W in the open and closed states depict relative stabilization due to the mutation in the open state caused by formation of new van der Waals contacts. Residues forming contacts with residue 4840 are shown with surface representation, with the largest such interaction observed for T4840W in the open state. B, the structural models of H4832W in the open and closed states depict destabilization of the open state due to clashes between residue 4832 and residues in the S5 helix from the adjacent monomer (shown as red dotted lines) and no such clashes in the closed state. Both mutants are compared with WT.
The G4834W mutation in our structural model leads to unfavorable steric clashes between the tryptophan side chain and the S6 helix (Gln-4947 and Glu-4948), indicative of a destabilizing effect on the open state. However, in the closed state, due to the movement of S6 toward the C-terminal side of 45L, position 4834 is farther away from S6, and the G4834W mutation results in slight stabilization (Fig. 5B). Experimentally, the Po of G4834W is only 37% of that of WT RyR1. Similarly, the H4832W mutation also leads to increased clashes in the open state but no such destabilization of the closed state, with the net effect being the relative stabilization of the closed state (Fig. 6B). Thus, mutations that stabilize the closed state according to the structural model of the mutants decrease the channel Po (G4834W and H4832W).
On S6, we focused on Gln-4947, which faces away from the pore (Fig. 7, A and B) and is part of the S6 interface with 45L. From the structural models of the three S6 mutants (Q4947N, Q4947T, and Q4947S), we observed that the open state is destabilized due to reduced contacts of S6 with 45L in the open state, as the glutamine side chain is replaced with smaller side chain amino acids (Fig. 5C and Fig. 7, C and D). There is no such destabilization in the closed state because Gln-4947 is away from the 45L interface in the closed state (Figs. 5C and 7); thus, we observed the relative stabilization of the closed state due to these mutations. In accordance with the model, the three single S6 mutations Q4947N, Q4947T, and Q4947S have a reduced Po compared with WT (Table 4).
FIGURE 7.

Orientation of S6 residues and models of S6 mutations in open and closed states. A, a helical wheel representation of the C-terminal part of the S6 helix depicts the face of the helix that forms the interface with 45L (shown in blue) and the face of S6 that lines the pore (shown in red). B, the position of Gln-4947 in the structural model when observed from the cytoplasmic side indicates that the residue faces away from the pore (marked in the center). The Gln-4947 side chain is shown as sticks, whereas the structural model of the open state is shown by ribbon representation. C and D, the structural models of Q4947T and Q4947S, respectively, show loss of van der Waals contacts due to shortened side chains in the open state but no such contacts in the closed state. The residues proximal to residue 4947 that can form contacts are shown with surface representation, and the side chain at residue 4947 is shown as spheres. Both mutants are compared with WT.
To directly compare the computational predictions with experimental data, we plotted ΔΔG from Po measurements (left side of Equation 2) against ΔΔG from Medusa calculations (right side of Equation 2). We observed a high correlation (r = 0.88) (Fig. 8), which is statistically significant (p value of correlation = 0.008), clearly indicating the predictive power of our structural models.
FIGURE 8.
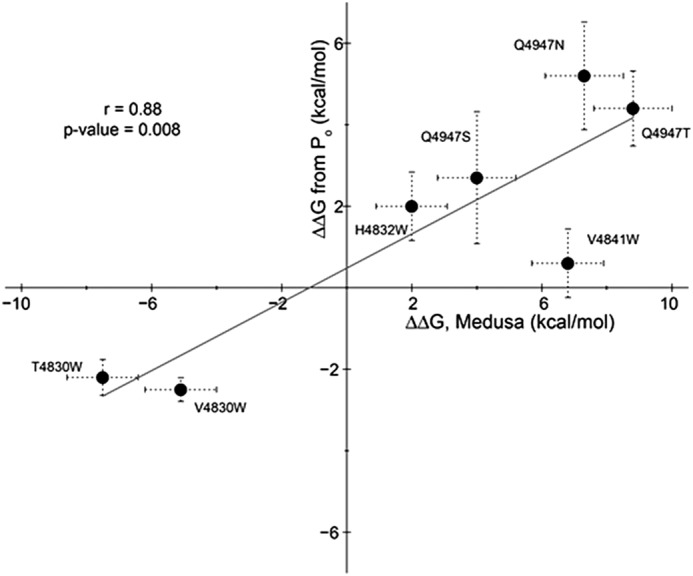
Correlation between computational predictions and experimental observations. The ΔΔG calculated from Po measurements (left side of Equation 2) is plotted against the ΔΔG from Medusa calculations (right side of Equation 2). The least-squares fit between computational and experimental data is indicated by the line.
Overall, our mutational data combined with evolutionary and structural analysis point to a 45L-S6 interface that controls Ca2+-dependent gating of RyR1. Significantly, our structural model paves the way for the characterization of the functional contribution of residues outside the predicted pore region of RyR1. Even in the context of the pore, our data point to residues in S6 that are important for gating but not for selectivity and conductance. In contrast to earlier studies, we were able to design both activating and deactivating mutants based on molecular interactions that lead to such changes in gating behavior. The importance of 45L residues in Ca2+-dependent gating of RyR1 is highlighted by the decoupling of channel gating from Ca2+ binding in the cytosolic side that is observed in the V4830W and T4840W mutants. The validation of 45L mutants indicates the structural conservation of the 6-TM topology between RyR1 and voltage-gated K+ and Na+ channels, which leads to the intriguing possibility of a common origin for the ligand-gated RyR and voltage-gated cation channels.
Acknowledgments
We thank Daniel Pasek for expressing and purifying WT and mutant RyR1 and Jazmyne Hefflefinger for performing part of the Ca2+ imaging experiments.
This work was supported, in whole or in part, by National Institutes of Health Grant AR018687 (to G. M. and N. V. D.). This work was also supported by American Heart Association Predoctoral Fellowship 09PRE2090068 (to S. R.) and Virginia Commonwealth University startup funds (to M. S.).
- RyR1
- ryanodine receptor type 1
- 6-TMD
- six-transmembrane domain
- SR
- sarcoplasmic reticulum
- pS
- picosiemens
- TM
- transmembrane.
REFERENCES
- 1. Du G. G., Sandhu B., Khanna V. K., Guo X. H., MacLennan D. H. (2002) Topology of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum (RyR1). Proc. Natl. Acad. Sci. U.S.A. 99, 16725–16730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Radermacher M., Rao V., Grassucci R., Frank J., Timerman A. P., Fleischer S., Wagenknecht T. (1994) Cryo-electron microscopy and three-dimensional reconstruction of the calcium release channel/ryanodine receptor from skeletal muscle. J. Cell Biol. 127, 411–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Samsó M., Feng W., Pessah I. N., Allen P. D. (2009) Coordinated movement of cytoplasmic and transmembrane domains of RyR1 upon gating. PLoS Biol. 7, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lanner J. T., Georgiou D. K., Joshi A. D., Hamilton S. L. (2010) Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2, a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Franzini-Armstrong C., Protasi F. (1997) Ryanodine receptors of striated muscles: a complex channel capable of multiple interactions. Physiol. Rev. 77, 699–729 [DOI] [PubMed] [Google Scholar]
- 6. Ríos E., Pizarro G. (1991) Voltage sensor of excitation-contraction coupling in skeletal muscle. Physiol. Rev. 71, 849–908 [DOI] [PubMed] [Google Scholar]
- 7. Williams A. J. (1992) Ion conduction and discrimination in the sarcoplasmic reticulum ryanodine receptor/calcium-release channel. J. Muscle Res. Cell Motil. 13, 7–26 [DOI] [PubMed] [Google Scholar]
- 8. Meissner G. (2004) Molecular regulation of cardiac ryanodine receptor ion channel. Cell Calcium 35, 621–628 [DOI] [PubMed] [Google Scholar]
- 9. Balshaw D., Gao L., Meissner G. (1999) Luminal loop of the ryanodine receptor: a pore-forming segment? Proc. Natl. Acad. Sci. U.S.A. 96, 3345–3347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao M., Li P., Li X., Zhang L., Winkfein R. J., Chen S. R. (1999) Molecular identification of the ryanodine receptor pore-forming segment. J. Biol. Chem. 274, 25971–25974 [DOI] [PubMed] [Google Scholar]
- 11. Gao L., Balshaw D., Xu L., Tripathy A., Xin C., Meissner G. (2000) Evidence for a role of the lumenal M3-M4 loop in skeletal muscle Ca2+ release channel (ryanodine receptor) activity and conductance. Biophys. J. 79, 828–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu L., Wang Y., Gillespie D., Meissner G. (2006) Two rings of negative charges in the cytosolic vestibule of type-1 ryanodine receptor modulate ion fluxes. Biophys. J. 90, 443–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiang Y., Lee A., Chen J., Cadene M., Chait B. T., MacKinnon R. (2002) The open pore conformation of potassium channels. Nature 417, 523–526 [DOI] [PubMed] [Google Scholar]
- 14. Alam A., Jiang Y. (2009) High-resolution structure of open NaK channel. Nat. Struct. Mol. Biol. 16, 30–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Long S. B., Campbell E. B., MacKinnon R. (2005) Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309, 897–903 [DOI] [PubMed] [Google Scholar]
- 16. Doyle D. A., Morais Cabral J., Pfuetzner R. A., Kuo A., Gulbis J. M., Cohen S. L., Chait B. T., MacKinnon R. (1998) The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280, 69–77 [DOI] [PubMed] [Google Scholar]
- 17. Jiang Y., Lee A., Chen J., Cadene M., Chait B. T., MacKinnon R. (2002) Crystal structure and mechanism of a calcium-gated potassium channel. Nature 417, 515–522 [DOI] [PubMed] [Google Scholar]
- 18. Ludtke S. J., Serysheva I.I., Hamilton S. L., Chiu W. (2005) The pore structure of the closed RyR1 channel. Structure 13, 1203–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Punta M., Coggill P. C., Eberhardt R. Y., Mistry J., Tate J., Boursnell C., Pang N., Forslund K., Ceric G., Clements J., Heger A., Holm L., Sonnhammer E. L., Eddy S. R., Bateman A., Finn R. D. (2012) The Pfam protein families database. Nucleic Acids Res. 40, D290–D301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Altschul S. F., Wootton J. C., Gertz E. M., Agarwala R., Morgulis A., Schäffer A. A., Yu Y. K. (2005) Protein database searches using compositionally adjusted substitution matrices. FEBS J. 272, 5101–5109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schäffer A. A., Aravind L., Madden T. L., Shavirin S., Spouge J. L., Wolf Y. I., Koonin E. V., Altschul S. F. (2001) Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 29, 2994–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Clayton G. M., Altieri S., Heginbotham L., Unger V. M., Morais-Cabral J. H. (2008) Structure of the transmembrane regions of a bacterial cyclic nucleotide-regulated channel. Proc. Natl. Acad. Sci. U.S.A. 105, 1511–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Serohijos A. W. R., Hegedus T., Aleksandrov A. A., He L., Cui L., Dokholyan N. V., Riordan J. R. (2008) Phenylalanine 508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc. Natl. Acad. Sci. U.S.A. 105, 3256–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bernsel A., Viklund H., Falk J., Lindahl E., von Heijne G., Elofsson A. (2008) Prediction of membrane-protein topology from first principles. Proc. Natl. Acad. Sci. U.S.A. 105, 7177–7181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mitaku S., Hirokawa T., Tsuji T. (2002) Amphiphilicity index of polar amino acids as an aid in the characterization of amino acid preference at membrane-water interfaces. Bioinformatics 18, 608–616 [DOI] [PubMed] [Google Scholar]
- 27. Nugent T., Jones D. T. (2009) Transmembrane protein topology prediction using support vector machines. BMC Bioinformatics 10, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Viklund H., Elofsson A. (2004) Best α-helical transmembrane protein topology predictions are achieved using hidden Markov models and evolutionary information. Protein Sci. 13, 1908–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Viklund H., Elofsson A. (2008) OCTOPUS: improving topology prediction by two-track ANN-based preference scores and an extended topological grammar. Bioinformatics 24, 1662–1668 [DOI] [PubMed] [Google Scholar]
- 30. von Heijne G. (1992) Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J. Mol. Biol. 225, 487–494 [DOI] [PubMed] [Google Scholar]
- 31. Cuff J. A., Barton G. J. (2000) Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins 40, 502–511 [DOI] [PubMed] [Google Scholar]
- 32. Ding F., Tsao D., Nie H., Dokholyan N. V. (2008) Ab initio folding of proteins with all-atom discrete molecular dynamics. Structure 16, 1010–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dokholyan N. V., Buldyrev S. V., Stanley H. E., Shakhnovich E. I. (1998) Discrete molecular dynamics studies of the folding of a protein-like model. Fold. Des. 3, 577–587 [DOI] [PubMed] [Google Scholar]
- 34. Shirvanyants D., Ding F., Tsao D., Ramachandran S., Dokholyan N. V. (2012) Discrete molecular dynamics: an efficient and versatile simulation method for fine protein characterization. J. Phys. Chem. B 116, 8375–8382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ding F., Dokholyan N. V. (2006) Emergence of protein fold families through rational design. PLoS Comput. Biol. 2, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yin S., Ding F., Dokholyan N. V. (2007) Modeling backbone flexibility improves protein stability estimation. Structure 15, 1567–1576 [DOI] [PubMed] [Google Scholar]
- 37. Yin S., Ding F., Dokholyan N. V. (2007) Eris: an automated estimator of protein stability. Nat. Methods 4, 466–467 [DOI] [PubMed] [Google Scholar]
- 38. Ramachandran S., Kota P., Ding F., Dokholyan N. V. (2011) Automated minimization of steric clashes in protein structures. Proteins 79, 261–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kota P., Ding F., Ramachandran S., Dokholyan N. V. (2011) Gaia: automated quality assessment of protein structure models. Bioinformatics 27, 2209–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yin S., Biedermannova L., Vondrasek J., Dokholyan N. V. (2008) MedusaScore: an accurate force-field based scoring function for virtual drug screening. J. Chem. Inf. Model. 48, 1656–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chacón P., Wriggers W. (2002) Multi-resolution contour-based fitting of macromolecular structures. J. Mol. Biol. 317, 375–384 [DOI] [PubMed] [Google Scholar]
- 42. Gao L., Tripathy A., Lu X., Meissner G. (1997) Evidence for a role of C-terminal amino acid residues in skeletal muscle Ca2+ release channel (ryanodine receptor) function. FEBS Lett. 412, 223–226 [DOI] [PubMed] [Google Scholar]
- 43. Lee H. B., Xu L., Meissner G. (1994) Reconstitution of the skeletal muscle ryanodine receptor-Ca2+ release channel protein complex into proteoliposomes. J. Biol. Chem. 269, 13305–13312 [PubMed] [Google Scholar]
- 44. Wang Y., Xu L., Duan H., Pasek D. A., Eu J. P., Meissner G. (2006) Knocking down type 2 but not type 1 calsequestrin reduces calcium sequestration and release in C2C12 skeletal muscle myotubes. J. Biol. Chem. 281, 15572–15581 [DOI] [PubMed] [Google Scholar]
- 45. Sutko J. L., Airey J. A., Welch W., Ruest L. (1997) The pharmacology of ryanodine and related compounds. Pharmacol. Rev. 49, 53–98 [PubMed] [Google Scholar]
- 46. Schoenmakers T. J., Visser G. J., Flik G., Theuvenet A. P. (1992) CHELATOR: an improved method for computing metal ion concentrations in physiological solutions. BioTechniques 12, 870–879 [PubMed] [Google Scholar]
- 47. Callaway C., Seryshev A., Wang J. P., Slavik K. J., Needleman D. H., Cantu C., 3rd, Wu Y., Jayaraman T., Marks A. R., Hamilton S. L. (1994) Localization of the high and low affinity [3H]ryanodine binding sites on the skeletal muscle Ca2+ release channel. J. Biol. Chem. 269, 15876–15884 [PubMed] [Google Scholar]
- 48. Du G. G., Avila G., Sharma P., Khanna V. K., Dirksen R. T., MacLennan D. H. (2004) Role of the sequence surrounding predicted transmembrane helix M4 in membrane association and function of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum (ryanodine receptor isoform 1). J. Biol. Chem. 279, 37566–37574 [DOI] [PubMed] [Google Scholar]
- 49. Payandeh J., Scheuer T., Zheng N., Catterall W. A. (2011) The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Caprini M., Fava M., Valente P., Fernandez-Ballester G., Rapisarda C., Ferroni S., Ferrer-Montiel A. (2005) Molecular compatibility of the channel gate and the N terminus of S5 segment for voltage-gated channel activity. J. Biol. Chem. 280, 18253–18264 [DOI] [PubMed] [Google Scholar]
- 51. Decher N., Chen J., Sanguinetti M. C. (2004) Voltage-dependent gating of hyperpolarization-activated, cyclic nucleotide-gated pacemaker channels. Molecular coupling between the S4-S5 and C-linkers. J. Biol. Chem. 279, 13859–13865 [DOI] [PubMed] [Google Scholar]
- 52. Ferrer T., Rupp J., Piper D. R., Tristani-Firouzi M. (2006) The S4-S5 linker directly couples voltage sensor movement to the activation gate in the human ether-á-go-go-related gene (hERG) K+ channel. J. Biol. Chem. 281, 12858–12864 [DOI] [PubMed] [Google Scholar]
- 53. Long S. B., Campbell E. B., Mackinnon R. (2005) Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science 309, 903–908 [DOI] [PubMed] [Google Scholar]
- 54. Lu Z., Klem A. M., Ramu Y. (2002) Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J. Gen. Physiol. 120, 663–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tristani-Firouzi M., Chen J., Sanguinetti M. C. (2002) Interactions between S4-S5 linker and S6 transmembrane domain modulate gating of HERG K+ channels. J. Biol. Chem. 277, 18994–19000 [DOI] [PubMed] [Google Scholar]
- 56. Murayama T., Kurebayashi N., Oba T., Oyamada H., Oguchi K., Sakurai T., Ogawa Y. (2011) Role of amino-terminal half of the S4-S5 linker in type 1 ryanodine receptor (RyR1) channel gating. J. Biol. Chem. 286, 35571–35577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yarov-Yarovoy V., DeCaen P. G., Westenbroek R. E., Pan C. Y., Scheuer T., Baker D., Catterall W. A. (2012) Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc. Natl. Acad. Sci. U.S.A. 109, E93–E102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Seo M. D., Velamakanni S., Ishiyama N., Stathopulos P. B., Rossi A. M., Khan S. A., Dale P., Li C., Ames J. B., Ikura M., Taylor C. W. (2012) Structural and functional conservation of key domains in InsP3 and ryanodine receptors. Nature 483, 108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Klein A., Lillis S., Munteanu I., Scoto M., Zhou H., Quinlivan R., Straub V., Manzur A. Y., Roper H., Jeannet P. Y., Rakowicz W., Jones D. H., Jensen U. B., Wraige E., Trump N., Schara U., Lochmuller H., Sarkozy A., Kingston H., Norwood F., Damian M., Kirschner J., Longman C., Roberts M., Auer-Grumbach M., Hughes I., Bushby K., Sewry C., Robb S., Abbs S., Jungbluth H., Muntoni F. (2012) Clinical and genetic findings in a large cohort of patients with ryanodine receptor 1 gene-associated myopathies. Hum. Mutat. 33, 981–988 [DOI] [PubMed] [Google Scholar]