

Background: HBO1 is a crucial enzyme needed for DNA replication, yet mechanisms for its disposal are unknown.
Results: A ubiquitin E3 ligase subunit, Fbxw15, mediates HBO1 degradation in concert with Mek1 kinase after endotoxin exposure.
Conclusion: Fbxw15 targets HBO1 for its site-specific ubiquitination and degradation leading to reduced histone acetylation and cellular proliferation.
Significance: The molecular control of HBO1 protein levels may impact fundamental cellular processes during inflammation.
Keywords: Proteolysis, Acetylation, Histones
Abstract
Histone acetyltransferase binding to origin recognition complex (HBO1) plays a crucial role in DNA replication licensing and cell proliferation, yet its molecular regulation in cells is relatively unknown. Here an uncharacterized protein, Fbxw15, directly interacts with HBO1, a labile protein (t½ = ∼3 h), to mediate its ubiquitination (Lys338) and degradation in the cytoplasm. Fbxw15-mediated HBO1 depletion required mitogen-activated protein kinase 1 (Mek1), which was sufficient to trigger HBO1 phosphorylation and degradation in cells. Mek1 ability to produce HBO1 degradation was blocked by Fbxw15 silencing. Lipopolysaccharide induced HBO1 degradation, an effect abrogated by Fbxw15 or Mek1 cellular depletion. Modulation of Fbxw15 levels was able to differentially regulate histone H3K14 acetylation and cellular proliferation by altering HBO1 levels. These studies authenticate Fbxw15 as a ubiquitin E3 ligase subunit that mediates endotoxin-induced HBO1 depletion in cells, thereby controlling cell replicative capacity.
Introduction
HBO1 (histone acetyltransferase binding to origin recognition complex) is a member of the histone acetyltransferase family that associates with distinct protein complexes to modulate cell cycle progression and proliferation through epigenetic mechanisms (1–3). HBO1 was first identified to bind Cdt1 in the origin recognition complex at the G1 and S phase boundary of the cell cycle (1). During the late G1 phase, Mcm2–7 helicase, in concert with Cdt1 and HBO1, loads onto chromatin to trigger DNA replication (4, 5). HBO1 initiates DNA replication licensing by acetylating both histone H3 and H4 (6–8). In the S phase, DNA replication is terminated by the degradation of Cdt1 and the recruitment of geminin (5). HBO1 also recruits the Ing (inhibitor of growth) family of tumor suppressors to regulate cell proliferation (9). The physiologic and molecular factors that control the availability of HBO1 in cells are less studied. Phosphorylation of HBO1 by Polo-like kinase 1 at Ser57 appears to be important in replication licensing because mutation of this residue leads to cell cycle arrest at the G1-S phase, thereby impairing DNA replication (10). Presumably, other factors act during cell cycle progression to terminate HBO1 function, perhaps by lowering of histone acetyltransferase concentrations to ensure homeostatic control of cell replicative activity.
The ubiquitin proteasomal machinery provides a universal mechanism to terminate the function of specific proteins by mediating their degradation to regulate numerous processes (11–15). Once a protein is targeted for ubiquitin-mediated degradation, it is labeled by monoubiquitin or polyubiquitin moieties. The conjugation of ubiquitin to the targeted protein is orchestrated by enzymatic reactions using an E1 ubiquitin-activating enzyme and ubiquitin transfer to an E2 ubiquitin-conjugating enzyme. In some instances, the targeted protein for degradation is modified before binding to an E3 ubiquitin ligase, and these modifications often involve phosphorylation (16, 17). The E3 ubiquitin ligase then catalyzes an isopeptide linkage between the ϵ-amino group of a lysine residue within the targeted protein and the COOH group of the carboxyl terminus in ubiquitin. Of note, the E3 ubiquitin ligase is substrate-specific, and yet one E3 ligase may control the degradation of tens or hundreds of substrate proteins. Of the many E3 ligases, activities of several members of the Skp-Cullin 1-F box (SCF)2 superfamily have been amply documented (17–19). The SCF complex is comprised of Skp1 (20), Cullin, E2 ubiquitin conjugation enzyme (Ubc), and an F box receptor component that engages substrates. In essence, the F box protein binds Skp1 with its NH2-terminal F box domain and docks on the targeted protein via its carboxyl-terminal leucine-rich domain or WD repeat motif. One F box protein, Fbxw15 was identified during follicular development, yet its function and substrate remains unknown (21).
The virulence of LPS-containing microbial pathogens appears to be intricately linked to the ubiquitin proteasome system that can trigger robust activation of pro-inflammatory signaling cascades or control host cell proliferative capacity (22–27). LPS activation of the canonical NF-κB pathway involving the ubiquitin proteasome leading to transactivation of pro-inflammatory genes is well established (28, 29). In some cases, LPS alters histone acetylation to modify NF-κB recruitment to selected target genes (30, 31). We have shown that LPS also activates the SCF F box component β-Trcp (β-transducing repeat-containing protein) to degrade a histone lipidation enzyme and cortactin (32, 33). Interestingly, LPS administration to mice deficient for the HBO1 co-regulator, Ing4, fails to form spontaneous tumors, yet the mice are hypersensitive to LPS-induced pro-inflammatory cytokine expression (34). Hence, these latter results indicate that Ing4 may be indispensable for tumorigenesis and partake in innate immunity, but the data may also implicate HBO1 in LPS signaling. In aggregate, these observations led us to hypothesize that LPS impairs cell proliferation by ubiquitin-dependent degradation of HBO1. Here we show that LPS activates Fbxw15 that targets HBO1 for its degradation in cells by a mitogen-activated protein kinase 1 (Mek1)-dependent mechanism.
EXPERIMENTAL PROCEDURES
Cell Lines and Reagents
Murine lung epithelial (MLE-12) cells were maintained in HITES medium with 10% of FBS as previously described (32, 35). Rabbit polyclonal HBO1 antibody (catalog no. ab70183) was purchased from Abcam (Cambridge, MA). Anti-Mek1 (catalog no. 9122), HA tag (catalog no. 2367), phosphothreonine (catalog no. 9386), lamin A/C (catalog no. 4777), ubiquitin (catalog no. 3936), Skp1 (catalog no. 2156), and Cul1 (catalog no. 4995) antibodies were from Cell Signaling (Danvers, MA). Anti-phosphoserine antibody (catalog no. 05-1000X) was from EMD Millipore (Billerica, MA), V5 tag antibody (catalog no. P/N46–0705) was from Invitrogen and protein A/G-agarose beads were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-acetyl histone H3K14 antibody (catalog no. A-4023) was from Epigentec (Farmingdale, NY). Proteasome inhibitor MG132 was from Calbiotech (Spring Valley, CA). Protease inhibitor mixture tablets were purchased from Roche Applied Science. Protein synthesis inhibitor cycloheximide and lysosome inhibitor leupeptin were from Sigma-Aldrich. The chemical reagents in the highest grades used in the experiments are commercially available.
Nucleofection
The cells were nucleofected with plasmids as previously described (32). Briefly, 1 × 106 MLE cells in their exponential growth stage were suspended in 100 μl of nucleofection buffer (20 mm of Hepes in PBS buffer) and well mixed with 3 μg of plasmid DNA in an electroporation cuvette. Electroporation was performed with a preset program T-013 in the NucleofectionTM II system (Amaxa Biosystems, Gaithersburg, MD), and the cells were cultured in 2 ml of complete HITES medium for 48 h. shRNA plasmids were also delivered into cells by using nucleofection with the same protocol (36).
Immunoblotting and Immunoprecipitation
Immunoblotting was conducted as previously described (32, 36). For immunoprecipitation, MLE cells were harvested and lysed with lysis buffer (1× PBS, 0.3% Tween 20, 1:1000 protease inhibitor mixture). The cells in the lysis buffer were lysed by sonication at 25% amplitude for 12 s followed by centrifugation at 10,000 rpm for 10 min. The cleared cell lysates (containing 1 mg of protein) were incubated with 2 μg of HBO1 antibody and rotated at room temperature for 30 min. Protein A/G-agarose beads of 30 μl were added to the mixture and rotated for another 2 h. To collect the immunoprecipitates, the mixtures were spun down at 3,000 rpm for 2 min and washed three times with 1 ml of cell lysis buffer under the same condition. The washed beads were used in immunoblotting with indicated antibodies. The ubiquitin immunoprecipitation experiments are carried out in the presence of DUB inhibitor ubiquitin-aldehyde. The immunoprecipitates were mixed with 50 μl of SDS loading dye (containing 1% SDS and 10 mm DTT) and heated at 95 °C for 5 min. The samples were separated by regular SDS-PAGE using Tris-glycine gel (Bio-Rad).
Extraction of Subcellular Proteins
MLE cells in their exponential growth stage were transfected with pcDNA3.1/Fbxw15-V5 plasmids and grown for 48 h. The cells were then treated with cycloheximide at a concentration of 20 μm for a various times. Subcellular protein extraction was performed as previously described (32). Briefly, the cells were washed with PBS buffer twice and detached by scraping. Harvested cells were collected by centrifuging at 1,500 rpm for 10 min. The pelleted cells were suspended in 1 ml of cytoplasmic membrane lysis buffer (10 mm Hepes, pH 8.0, 1.5 mm MgCl2, 10 mm KCl, 1 mm DTT, 0.1% Igepal CA-630, and 1:1000 protease inhibitor mixture). The cell suspension was centrifuged at 2,000 rpm for 5 min, and the supernatant was collected as the cytosolic fraction and condensed by Amicon Ultra filters. The pellet was lysed with 150 μl of nuclear envelope lysis buffer (20 mm Hepes, pH 8.0, 1.5 mm MgCl2, 420 mm NaCl, 1 mm DTT, 0.2 mm EDTA, 25% (v/v) glycerol, and 1:1000 protease inhibitor mixture) and cleared at 13,000 rpm for 10 min, and this supernatant was collected as the nuclear fraction. The fractions were subjected to immunoblot analysis.
Plasmid Cloning and Site-directed Mutagenesis
The human HBO1 cDNA was from Origene (Rockville, MD), and the open reading frames were amplified by PCR using the following primers: 5′-caccatgccgcgaaggaagagg-3′ (forward) and 5′-agtgcccttgggaggggtccat-3′ (reverse). The PCR products were inserted into pcDNA3.1/His-V5 plasmids (Invitrogen). The pcDNA3.1/Fbxw15/His-V5 plasmids were constructed with the same method by employing the primers of 5′-caccatggcgatccatttaccttg-3′ (forward) and 5′-agagcagatgttcaaggcatatg-3′ (reverse). The HBO1 deletion mutants were constructed using a similar approach with the following primers: 55F, 5′-caccatggattccagtcctgttcg-3′; 101F, 5′-caccatgggttcagaaactgagcaag-3′; 151F, 5′-caccatggagagcattgccaaggacatg-3′; 198F, 5′-caccatgggaaaacatgagagacatttct-3′; 252F, 5′-caccatggcaccaacggagaggcag-3′; 550R, 5′-cacagccgtctcctgactgattt-3′; 500R, 5′-gactttggaaagcaaataactg-3′; 450R, 5′-gtccgcctctgtcataacatag-3′; 400R, 5′-gcgatatatctcatcaccaggtgg-3′; 350R, 5′-ccaggtatcaagctcatagcgg-3′; and 300R, 5′-gttttctaaaagaggttcccgtgtg-3′. Site-directed mutagenesis of lysine 323 and lysine 338 within HBO1 were obtained as previously described (32) using the following primers, K323RF, 5′-cagaggatttggagaGgttaaggctgcaagg-3′; K323RR, 5′-ccttgcagccttaacctctccaaatc ctctg-3′; K338RF, 5′-gaagcaacatgattaGaacaattgcttttgg-3′; and K338RR, 5′-ccaaaagcaattgttctaatcatgttgcttc-3′.
Mek1 in Vitro Kinase Assays
HBO1 was immunoprecipitated from MLE cells by using HBO1 antibody. The washed immunoprecipitates from the cell lysates (containing 1 mg of protein) were mixed with 50 μl of kinase assay buffer (50 mm Tris, 10 mm MgCl2, 1 mm EDTA, 2 mm DTT, 200 μm ATP, and 0.01% Triton X-100) and equally allocated into five reactions. For the reaction, 1 μCi of [γ-32P]ATP (PerkinElmer Life Sciences) and 0.2 μg of Mek1 protein with kinase activity (Invitrogen) were mixed with the immunoprecipitates and incubated at 30 °C for 10 min. As system controls, one reaction was without Mek1 kinase and another without the addition of [γ-32P]ATP. As a negative control, the Mek1 protein was heat-inactivated at 95 °C for 10 min. The reaction products were mixed with SDS-PAGE loading dye and separated by SDS-PAGE. Phosphorylated HBO1 with radioactive signals was detected from the transferred nitrocellulose membrane with a phosphorus imager (Bio-Rad).
In Vitro Ubiquitin Conjugation Assays
HBO1 protein was isolated using immunoprecipitation. SCF box complex components Skp1, Cullin1, Rbx1, and substrate-specific E3 ubiquitin ligase subunit, Fbxw15, were synthesized using the cell-free reticulocyte lysate in vitro transcription/translation system with pcDNA3.1/His/V5/Lpcat1 as the template (Promega, Madison, WI). The ubiquitination assay was performed in a total volume of 25 μl containing 50 mm Tris (pH 7.6), 5 mm MgCl2, 0.6 mm DTT, 2 mm ATP, 1.5 ng/μl E1 (Boston Biochem, Cambridge, MA), 10 ng/μl Ubc5, 10 ng/μl Ubc7, 1 μg/μl ubiquitin (Calbiochem, Spring Valley, CA), 1 μm ubiquitin aldehyde, and 2 μl of each Cullin1, Skp1, Rbx1, and Fbxw15, at room temperature for 30 min. Reaction mixtures were then resolved using SDS-PAGE, and HBO1 ubiquitination was analyzed by immunoblotting.
Quantitative RT-PCR
MLE cells transfected with Fbxw15 plasmid or knockdown plasmid were treated with 20 μm of cycloheximide for various times. The collected cells were lysed with 1 ml of Tri reagents (Invitrogen), and total RNA were isolated as previously described (35). The cDNA was synthesized from isolated total RNA with an iScript cDNA synthesis kit (Bio-Rad) following the directions of the manufacturer. The primers encoding a DNA fragment of ∼120 bp in length were designed based on the mouse Fbxw15 gene sequence in the NCBI gene bank. The forward primer was 5′-ctacagtttgctacagg-3′, and the reverse primer was 5′-atgtctctttgccctgg-3′. Quantitative PCR was conducted with the CFXTM-96 thermocycle system (Bio-Rad).
Fluorescence-activated Cell Sorting
FACS analysis of the cells was conducted by using BD PharmingenTM BrdU flow kits (BD Biosciences, San Jose, CA) following the instructions of the manufacturer. Briefly, MLE cells at a concentration of 106 cells/ml were transfected with HBO1 plasmid or shRNA constructs by way of electroporation. The cells were inoculated into 6-well plates for 48 h and then incubated with 10 μm of BrdU for 40 min. The cells were harvested and washed with cold PBS and fixed with 100 μl of Cytofix buffer for 30 min. The fixed cells were treated with 100 μl of permeabilization buffer for 10 min on ice and with 100 μl of Cytofix buffer for 10 min. The cells were then digested with DNase (30 μg/106 cells) for 1 h at 37 °C. The cells were stained with FITC-conjugated anti-BrdU antibody (v/v 50:1) for 20 min. The cell nuclei were stained with 7-aminoactinomycin D before cell cycle analysis. Cell sorting was conducted with an Accuri C6 system (Bio-Rad), and the results were analyzed with FCS3 version 3 analysis software (De Novo Software).
Cell Growth Analysis
MLE cells were lentivirally transduced to overexpress or knockdown Fbxw15. The cells were seeded at 3 × 104 cells/ml in 6-well plates and allowed to grow in a standard cell culture incubator. For each cell line, three independent wells were harvested after 48 h postseeding. The cells were counted using a T10 automated cell counter (Bio-Rad). Cells at the same density were grown for 24 h, and the cells were then treated with a various concentrations of LPS in the presence of 0.1% FBS overnight. The cells were harvested and counted as described above.
Statistical Analysis
Statistical analysis was carried out by two-way analysis of variance. The data were collected from three independent experiments and presented as the means ± S.D.
RESULTS
HBO1 Is Degraded by the Proteasome
MLE cells were treated with cycloheximide to inhibit protein synthesis, and the endogenous HBO1 protein levels were then analyzed by immunoblotting. The results demonstrate that HBO1 is a short-lived protein with a predicted t½ of ∼3 h (Fig. 1A). To determine the pathway for HBO1 disposal, the cells were exposed to the proteasomal inhibitor MG132, or lysosomal inhibitor, leupeptin in a time course analysis prior to harvesting for immunoblotting. Protein levels remained unchanged after leupeptin exposure (Fig. 1B). However, endogenous HBO1 accumulates with time as a function of duration with MG132 treatment (Fig. 1C). Densitometric quantitation of immunoblots indicated that HBO1 is processed for its elimination via a proteasomal but not lysosomal pathway (Fig. 1D).
FIGURE 1.

HBO1 is degraded by the proteasome. A–C, MLE cells were treated with cycloheximide (CHX, 20 μm, A) with either leupeptin (20 μm, B) or MG132 (20 μm, C) for various time points prior to immunoblotting. D, densitometry was performed on the above immunoblots, and results were plotted. The relative immunoblotting density was adjusted by the corresponding levels of β-actin. The data are representative of three separate experiments.
HBO1 Is Selectively Targeted by Fbxw15 for Degradation
Because HBO1 is regulated by its phosphorylation status and SCF ubiquitin E3 ligase complexes target phosphorylated substrates (10, 37), we randomly analyzed several F box proteins by their ability to mediate HBO1 ubiquitination and degradation. By overexpression of plasmids encoding various F box proteins in cells, we found that HBO1 was specifically degraded by ectopic expression of Fbxw15, an unstudied member of the SCF W family (Fig. 2A). Unlike Fbxw14, overexpressed Fbxw15 plasmid was sufficient to mediate degradation of HBO1 using increasing amounts of plasmid transfected in cells (Fig. 2B). To assess specificity of Fbxw15 activity further, we co-overexpressed and knocked down Fbxw15 simultaneously. Fbxw15 mRNA was detectable by quantitative PCR, and cellular depletion of Fbxw15 effectively reduced Fbxw15 mRNA and stabilized HBO1 (Fig. 2C). Overexpressed V5-Fbxw15 plasmid in cells led to accelerated degradation of HBO1 in the presence of cycloheximide (Fig. 2D, left panel). As a negative control, we overexpressed F box plasmid encoding Fbxl17 in cells that did not alter the rate of decay of levels of immunoreactive HBO1 with cycloheximide (Fig. 2D, right panel). These observations demonstrate that Fbxw15 specifically targets HBO1 to mediate its degradation in cells.
FIGURE 2.

Fbxw15 mediates HBO1 degradation. A, several SCF V5-tagged F box subunit plasmids were overexpressed in MLE cells for 48 h, and the cells were harvested prior to immunoblotting with HBO1, V5, and β-actin antibodies. B, different amounts of pcDNA3.1/Fbxw15/V5 or pcDNA3.1/Fbxw14/V5 plasmids were overexpressed in cells for 48 h. The samples were cells harvested prior to immunoblotting with HBO1, V5, and β-actin immunoblotting. C, scrambled RNA or Fbxw15 shRNA plasmids (4 μg) was transfected into cells. The cells were then treated with 20 μm of cycloheximide (CHX) for various time points prior to immunoblotting with HBO1 and β-actin antibodies. The bar graph represents steady-state levels of Fbxw15 mRNA. D, a pcDNA3.1/Fbxw15/V5 plasmid or pcDNA3.1/Fbxw17/V5 plasmid (4 μg) was transfected into cells. The transfected cells were then treated with 20 μm of CHX for various time points. The samples were subjected to HBO1, V5, and β-actin immunoblotting. In B–D, the results of densitometric analysis of immunoblots are represented graphically on the right. The data are representative of three separate experiments.
Fbxw15 Binds and Ubiquitinates HBO1
To explore whether Fbxw15 binds HBO1, we overexpressed V5-tagged Fbxw15 plasmid in cells and immunoprecipitated Fbxw15 using V5 antibody in the presence of MG132. Analysis of the immunoprecipitates by HBO1 immunoblotting demonstrated that HBO1 binds Fbxw15 (Fig. 3A). Binding of an E3 ubiquitin ligase subunit to the target protein initiates substrate ubiquitination. To determine whether HBO1 is polyubiquitinated, we co-transfected HBO1 and HA-tagged ubiquitin plasmids in cells, and the cell lysates were subjected to HBO1 or HA immunoprecipitation, followed by HBO1 or HA immunoblotting. In these co-immunoprecipitation (co-i.p.) studies, ubiquitin was bound to HBO1 in immunoprecipitates in a pattern consistent with polyubiquitinated substrate (Fig. 3B). In reciprocal studies when antibodies were reversed, immunoblotting of the HA immunoprecipitates with HBO1 antibody confirmed polyubiquitinated HBO1 (Fig. 3C). To confirm whether HBO1 ubiquitination is Fbxw15-dependent, we knocked down Fbxw15 using shRNA and subjected these cell lysates to HBO1 co-i.p. Here, HBO1 bound to immunoprecipitated HA-ubiquitin was markedly reduced after Fbxw15 depletion in cells (Fig. 3D). The SCF E3 ubiquitin ligase machinery requires several components whereby the F box directly binds to Skp1. Fbxw15 interacted with Skp1 in co-i.p. experiments where cell lysates were subjected to V5 immunoprecipitation after cellular transfection with V5-tagged Fbxw15 plasmid (Fig. 3E). To test whether Fbxw15 catalyzes HBO1 ubiquitination, we performed in vitro ubiquitination assays in the presence or absence of Fbxw15, using Fbxw14 as a control. In the presence of SCF components Cul1, Skp1, ubiquitin-conjugating E2 enzyme, and Fbxw15, HBO1 protein was polyubiquitinated, and levels of modified HBO1 were dependent on the ubiquitin concentration in the reaction mixture. Fbxw14 did not polyubiquitinate HBO1 (Fig. 3F). These data demonstrate that Fbxw15 interacts with and mediates HBO1 ubiquitination, authenticating Fbxw15 as a bona fide E3 ubiquitin ligase component.
FIGURE 3.

Fbxw15 interacts with and ubiquitinates HBO1. A, pcDNA3.1/Fbxw15/V5 plasmid (4 μg) were transfected into MLE cells for 48 h. Cell lysates were subjected to V5 immunoprecipitation (IP). The immunoprecipitates were subjected to HBO1 immunoblotting (IB). B, pcDNA3.1/ubiquitin/HA plasmid (4 μg) and pcDNA3.1/HBO1/V5 plasmid (4 μg) were co-transfected into cells. Cell lysates (1 mg of protein) were subjected to HBO1 immunoprecipitation followed by HA immunoblotting. C, in reciprocal studies, cell lysates in B were immunoprecipitated with HA antibody, and the immunoprecipitates were analyzed by HBO1 immunoblotting. D, pcDNA3.1/ubiquitin/HA plasmids (4 μg) and pcDNA3.1/HBO1/V5 plasmids (4 μg) were co-transfected with Fbxw15 shRNA plasmids into cells for 48 h. Cell lysates (1 mg of protein) were subjected to HA immunoprecipitation followed by HBO1 immunoblotting. E, Fbxw15-V5 plasmid was overexpressed in cells, and lysates were subjected to V5 immunoprecipitation followed by Skp1 immunoblotting. F, HBO1 in vitro ubiquitination assays were conducted in the presence of Fbxw15 or a control, Fbxw14, and the full complement of ubiquitin reaction components. The samples were analyzed by HBO1 immunoblotting. The data are representative of three separate experiments.
HBO1K338 Is a Ubiquitination Acceptor Site
To identify the ubiquitin acceptor site, HBO1 deletion and point mutation plasmids were constructed and expressed in cells in the presence of MG132 (Fig. 4, A and B). We hypothesized that if an HBO1 mutant protein is devoid of a ubiquitin acceptor site, it would not be ubiquitinated, and this would be reflected in its inability to accumulate in immunoblots. By using several NH2-terminal and carboxyl-terminal HBO1 deletion mutants, all fragments showed accumulation in response to MG132 with the exception of a variant containing residues 1–300 of HBO1 (Fig. 4B). The finding that a plasmid construct encoding residues 1–350, but not the 1–300 variant, was increased after MG132 treatment suggests that a ubiquitin acceptor site resided within a span of 50 residues (amino acids 300–350). In this region, there are two lysine residues (Lys323 and Lys338) within HBO1. Site-directed mutagenesis at these sites and expression of these constructs in cells indicated that the K338R mutant displayed an extended t½ after cycloheximide treatment, whereas the K323R mutant degrades in manner comparable with wild type HBO1 (Fig. 4C). To confirm that Lys338 is a ubiquitination site within HBO1 by Fbxw15, we conducted in vitro ubiquitination assays with the K338R mutant as a substrate. The results indicate that ubiquitination of the HBO1 K338R mutant is markedly reduced in the presence of Fbxw15 as compared with the wild type HBO1 (Figs. 4D and 3F). These data suggest that Lys338 is a ubiquitination acceptor site within the histone acetyltransferase.
FIGURE 4.
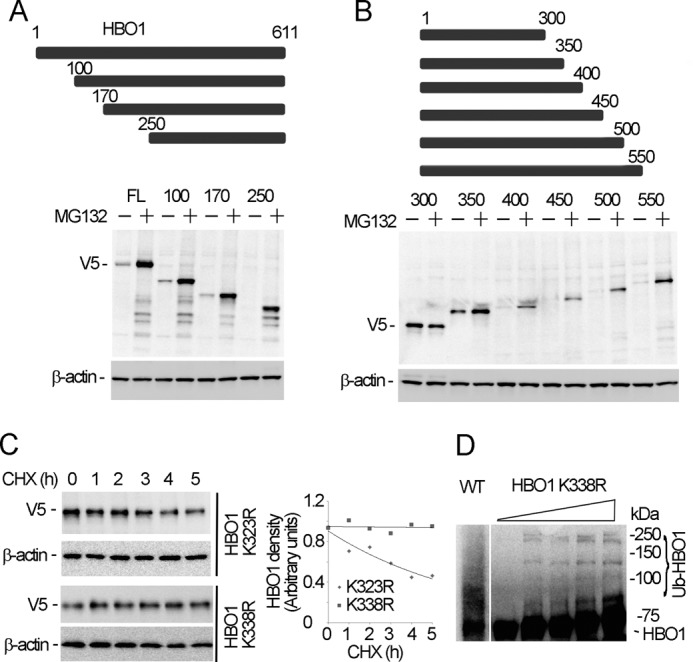
Identification of a ubiquitination acceptor site within HBO1. A and B, HBO1 deletion mutant plasmids were overexpressed in MLE cells for 48 h, followed by 8 h of MG132 (20 μm) treatment. The cell lysates were analyzed by V5 and β-actin immunoblotting. The upper panels schematically present the deletion constructs. NH2-terminal deletions are shown in A, and carboxyl-terminal deletions are shown in B. C, HBO1 mutant K323R and K338R V5-tagged plasmids were overexpressed in cells for 48 h, and the cells were treated with 20 μm of cycloheximide (CHX) for various time points. The cell lysates were analyzed with V5, and β-actin immunoblotting. The results of densitometric analysis of immunoblots are represented graphically on the right. D, in vitro ubiquitination assays were conducted (as in Fig. 3F) using K338R-mutated HBO1 as a substrate and WT (left) HBO1; the reaction products were used for HBO1 immunoblotting. The data are representative of three separate experiments.
HBO1 Is Degraded in the Cytosol
HBO1-associated histone acetyltransferase activities occur in the nucleus. To identify the site of HBO1 degradation, we isolated subcellular fractions in Fbxw15 overexpressed cells and analyzed the localization of Fbxw15 and HBO1 in cell compartments. Immunoreactive HBO1 and Fbxw15 were detected in the cytosol and the nucleus, with the majority of ectopically expressed F box protein in the cytoplasm (Fig. 5A). To determine the site of degradation, the cells were treated with cycloheximide prior to fractionation for HBO1 immunoblotting. The results show that HBO1 is exclusively degraded in the cytosol because levels were unchanged in the nuclear fraction (Fig. 5B). Densitometry of these immunoblots are presented illustrating cytosolic HBO1 degradation (Fig. 5C).
FIGURE 5.

HBO1 is cytosolically degraded. A, pcDNA3.1/Fbxw15/V5 plasmids (4 μg) were transfected into MLE cells for 48 h. The cells were harvested and subjected subcellular protein fractionation. The cytosolic (lane C) and the nuclear (lane N) fractions were analyzed by HBO1, lamin A/C, V5 and β-actin immunoblotting. Lamin A/C was used as the nuclear protein marker, and β-actin was used as the cytosolic marker. B, cells treated with 20 μm of cycloheximide (CHX) for various time points were subjected to subcellular protein fractionation. The fractions were analyzed by HBO1, lamin A/C, and β-actin immunoblotting. C, densitometric results from immunoblots in B were plotted and presented. The data are representative of three separate experiments.
Mek1 Phosphorylates HBO1 to Accelerate Its Degradation
Proteins targeted for degradation may be modified by protein kinase-directed phosphorylation. The primary sequence of HBO1 is highly enriched with serine, threonine, and tyrosine residues, comprising 22.4% of the total amino acid residues. In a preliminary screen of several candidate kinases tested, only mitogen-activated protein kinase (Mek1) was able to modify HBO1 phosphorylation (data not shown). Using in vitro kinase assays, Mek1 phosphorylated HBO1, an effect not observed using heat-inactivated Mek1 or JNK1 (Fig. 6A). Further, Mek1 overexpression augmented HBO1 degradation in the presence of cycloheximide, whereas Mek1 silencing stabilized the acetyltransferase (Fig. 6B). Further, knockdown of Fbxw15 using shRNA abolished ability of ectopically expressed Mek1 plasmid to trigger HBO1 degradation, indicating that Mek1-induced HBO1 degradation is Fbxw15-dependent (Fig. 6C). Fbxw15 shRNA transfection effectively decreased mRNA levels of Fbxw15 in cells (Fig. 6D). Further, knockdown of Mek1 with shRNA abrogated effects of ectopically expressed Fbxw15 on HBO1 degradation (Fig. 6E). Analysis of Mek1 immunoprecipitates demonstrated that HBO1 directly interacts with Mek1 (Fig. 6F, left panel). However, depletion of Mek1 abolished HBO1 and Fbxw15 interaction (Fig. 6F, right panel). Further immunoprecipitation results confirmed that HBO1 interacts with Mek1, and also Cullin1, a component of the SCF complex (Fig. 6G, left panel). Knockdown of Mek1 abrogated the HBO1-Cullin1 interaction (Fig. 6G, right panel). Mek1 silencing also diminished HBO1 ubiquitination in cells (Fig. 6H). These results suggest that Mek1 triggers HBO1 phosphorylation that leads to its proteasomal degradation mediated by Fbxw15 through polyubiquitination.
FIGURE 6.

Mek1 phosphorylates HBO1 and accelerates HBO1 degradation. A, HBO1 protein was subjected to in vitro Mek1 kinase phosphorylation assays. The lower panel shows HBO1 loading in each reaction as determined by immunoblotting (IB). B, Mek1 was overexpressed using Mek1 plasmid or knocked down by Mek1 shRNA in MLE cells for 48 h; the cells were treated with 20 μm of cycloheximide (CHX) for various time points. The cell lysates were analyzed by HBO1, Mek1, and β-actin immunoblotting. C, the cells were co-transfected with Mek1 plasmid and Fbxw15 shRNA plasmids for 48 h, and the cells were treated with 20 μm of CHX for various time points. The cell lysates were analyzed by HBO1, Mek1, and β-actin immunoblotting. D, Fbxw15 mRNA levels in the above Fbxw15 shRNA-treated cells were determined by quantitative RT-PCR. E, pcDNA3.1/Fbxw15/V5 plasmids (4 μg) were co-transfected with Mek1 shRNA plasmids into cells for 48 h. The cell lysates (1 mg of protein) were subjected to immunoblotting analysis as indicated. F, left panel, the cell lysates were subjected to Mek1 immunoprecipitation (IP) followed by HBO1 immunoblotting. Right panel, pcDNA3.1/Fbxw15/V5 plasmids (4 μg) were co-transfected with Mek1 shRNA plasmids into cells for 48 h, and V5 immunoprecipitation followed by HBO1 immunoblotting was performed. G, MLE cells were treated with MG132 (20 μm) for 1 h, and the cell lysates were subjected to HBO1 immunoprecipitation and followed by Cullin1, Mek1, and HBO1 immunoblotting (left panels). Mek1 was also knocked down with lentiviral shRNA constructs for 48 h, and the cell lysates were used for HBO1 immunoprecipitation followed by Cullin1 and HBO1 immunoblotting analysis. The cell lysates were also used for Mek1 and β-actin immunoblotting analysis (bottom panels). H, Mek1-depleted cellular immunoprecipitates were analyzed for ubiquitin and HBO1 immunoblotting after HBO1 immunoprecipitation. Bottom two panels, cell lysates were subjected to Mek1 and β-actin immunoblotting. In B, C, and E, results of densitometric analysis of immunoblots are represented graphically on the right. The data are representative of three separate experiments.
LPS Triggers HBO1 Degradation via Mek1 and Fbxw15
Under many conditions, LPS is inhibitory to epithelial growth (22–25). Hence, we evaluated whether LPS impairs cell proliferative capacity by activation of a Mek1 → Fbxw15 → HBO1 pathway in MLE cells. As predicted, HBO1 protein levels decreased in LPS-treated cells using concentrations of 5 μg/ml, an effect blocked after Fbxw15 cellular depletion (Fig. 7, A–C). In addition, LPS increased Fbxw15 mRNA levels and increased HBO1 serine phosphorylation in cells (Fig. 7, B and D). Overall, knockdown of Mek1 attenuated LPS induced HBO1 serine phosphorylation (Fig. 7E). Last, Mek1 knockdown by shRNA prior to LPS stimulation markedly attenuated LPS-induced HBO1 degradation (Fig. 7F). These data indicate that LPS induces HBO1 degradation via Mek1 phosphorylation and Fbxw15-mediated ubiquitin proteasomal degradation.
FIGURE 7.

LPS induces HBO1 degradation. A, MLE cells were treated with different concentrations of LPS for 18 h, and the cells were analyzed by HBO1 and β-actin immunoblotting (IB). B and C, scrambled RNA or Fbxw15 shRNA plasmids were transfected into cells for 48 h. The cells were treated with LPS at the indicated concentrations for 18 h. The cell lysates were subjected to HBO1 and β-actin immunoblotting analysis (C) and analysis of Fbxw15 mRNA levels by quantitative RT-PCR (B). D, the LPS-treated cells in A were immunoprecipitated (IP) with HBO1 antibody. The immunoprecipitates were probed with phosphoserine, phosphothreonine, and HBO1 antibodies. E, Mek1 knockdown cells were treated with LPS, and the cell lysates were immunoprecipitated with HBO1 antibody. The immunoprecipitates were probed with phosphoserine and HBO1 antibodies. The cell lysates were analyzed with Mek1 immunoblotting. F, cells were transfected with Mek1 shRNA plasmid or scrambled RNA for 48 h. The cells were treated with LPS at the indicated concentrations for 18 h. The cell lysates were subjected to HBO1, Mek1, and β-actin immunoblotting. In A, C, D, and F, the results of densitometric analysis of immunoblots are represented graphically on the right. The data are representative of three separate experiments.
Fbxw15-mediated HBO1 Degradation Alters Histone H3K14 Acetylation and Cell Proliferation
Because HBO1 selectively acetylates histone H3 and H4 tails, we assessed histone H3K14 acetylation by Fbxw15. First, silencing of Fbxw15 mRNA in cells resulted in increased HBO1 protein levels coupled with increased histone H3K14 acetylation (Fig. 8A). Conversely, overexpression of Fbxw15 plasmid in cells reduced histone H3K14 acetylation levels and immunoreactive HBO1 (Fig. 8B). Importantly, Fbxw15 depletion in cells increased cell proliferative capacity, whereas overexpression of the F box plasmid decreased cell proliferation (Fig. 8C). Consistent with these observations, H3K14 acetylation levels and the cellular proliferative rate of LPS-treated cells were reduced (Fig. 8D). To delineate the role of HBO1 in H3K14 acetylation and cell proliferation, we depleted HBO1 levels by shRNA in cells. HBO1 knockdown ablated H3K14 acetylation (Fig. 8E). Analysis of the cell cycle in HBO1 knockdown cells indicated that cell numbers in the S phase decreased significantly along with an increased population of apoptotic cells. No obvious change was noticed in the G1 and G2 phases (Fig. 8F). HBO1 depletion also significantly reduced cell proliferation rates, and this reduction was rescued by overexpression of wild type or a K338R HBO1 mutant plasmid (Fig. 8G). Indeed, LPS-mediated inhibitory effects on proliferation were also rescued by overexpression of wild type HBO1 or K338R mutant plasmid (Fig. 8G). Interestingly, either overexpression or knockdown of Fbxw15 in cells did not alter expression of several transcripts encoding pro-inflammatory products (data not shown). Thus, LPS-induced HBO1 degradation appears to be growth inhibitory and mediated by Fbxw15 modulation of histone H3K14 acetylation.
FIGURE 8.

Fbxw15 regulates histone H3K14 acetylation and cell proliferation. A and B, MLE cells were separately transfected with pcDNA3.1/Fbxw15/V5 (B) plasmids and Fbxw15 shRNA plasmids (A) for 48 h. The cell lysates were analyzed by H3K14 acetylation, histone H3, HBO1, and β-actin immunoblotting. C, treated cells from A and B were counted in cell proliferation assays. *, p < 0.05 versus control. D, cells were treated with various concentrations of LPS overnight, and the samples were subjected to anti-H3K14 acetylation immunoblot analysis (upper panels) and analyzed for cell proliferation (lower panel). The results of densitometric analysis of immunoblots are represented graphically on the right. E, HBO1 knockdown was conducted in MLE cells. The cell lysates were immunoblotted with the indicated antibodies. F, MLE cells were transfected with HBO1 plasmid or HBO1 was silenced in cells. The cells were subjected to FACS analysis. S phase and apoptotic cells were presented for different groups (lower right panel). G, cell proliferation was determined after HBO1 plasmid overexpression or silencing in cells. In HBO1 overexpressed cells, wild type HBO1 and K338R mutants were separately assayed. One set of the cells was treated with LPS (5 μg/ml) overnight. *, p < 0.05 versus vector control; **, p < 0.01 versus HBO1 shRNA. The data are representative of three separate experiments.
DISCUSSION
HBO1, by promoting histone H3 and H4 acetylation, is crucial for DNA replication, but little is known regarding factors that control its cellular abundance. Here, we demonstrate that a previously unrecognized component of the SCF family, Fbxw15, selectively mediates HBO1 disposal during LPS exposure. Specifically, we show that (i) ectopically expressed Fbxw15 targets HBO1 for its site-specific ubiquitination and degradation leading to reduced histone acetylation and cellular proliferation; (ii) actions of Fbxw15 are mediated by Mek1, which was both sufficient and required for HBO1 degradation; (iii) Mek1 phosphorylates HBO1, thereby potentially providing a phosphodegron molecular signal for Fbxw15 recruitment to HBO1; and (iv) endotoxin inhibition of HBO1 stability is mediated by Mek1 and Fbxw15. In essence, these findings suggest that endotoxin impairs cell replicative capacity by targeting abundance of a critical epigenetic modifier using the Mek1-SCFFbxw15 apparatus. The results open the possibility of devising Fbxw15 small molecule antagonists that oppose growth inhibitory actions of endotoxin during pro-inflammatory stress.
Some histone modification enzymes are subject to degradation, but the pathophysiologic context whereby the ubiquitin-proteasomal machinery, specifically the SCF components, controls epigenetic programs is more limited. For example the Gcn5 acetylase is degraded by the Cullin4-RING E3 ubiquitin ligase (CRL4) complex because ablation of CRL4 leads to its stabilization in U2OS cells (38). The demethylase KDM4A is also targeted for ubiquitin-mediated proteasomal degradation by SCFFbxo22 (39). Shima et al. (40) demonstrated that a nuclear protein PML specifically binds to SCFFbx3 to prevent degradation of the p300 acetyltransferase. These studies perhaps underscore the importance of SCF E3 ligase components because they may relate to transcriptional programs and tumorigenesis. Another E3 ligase component, β-Trcp, mediates disposal of the histone lipidation enzyme, lysophosphatidylcholine acyltransferase 1, through glycogen synthase kinase-3β activation; these data support an emerging role for SCF components in modulating levels of nuclear proteins involved in epigenetic processes linked to innate immunity (32). Our data here suggest that endotoxin-associated microbial virulence might involve activation of SCFFbxw15 to reduce cytosolic HBO1 concentrations to impair transcriptional networks involved in cellular proliferation. However, one limitation of our studies is that we showed actions of ectopically expressed F box subunit on HBO1 ubiquitination, but given the lack of existing Fbxw15 antibodies, we were not able to confirm interaction of endogenous proteins. Nevertheless, depletion of endogenous Fbxw15 stabilized HBO1 by reducing its ubiquitination, increased immunoreactive HBO1 levels to up-regulate histone H3 acetylation in response to endotoxin, and increased cell proliferation. Hence, the ability of SCFFbxw15 to modulate epigenetic processes may be especially important during cellular repair after inflammatory tissue injury.
Although HBO1 and SCF-based E3 ligase components are detected in the nucleus, the turnover of HBO1 occurs in the cytoplasm. Compartmentalization of HBO1 with Mek1 and SCFFbxw15 within the cytosol would facilitate efficient phosphorylation-mediated ubiquitination of HBO1 for its rapid elimination. HBO1 has a relative short life span, resembling the t½ of other acetyltransferases which are immediate-early response proteins (41). The sensing mechanism that relays signals reflecting nuclear HBO1 concentrations to the cytoplasm requires further study. Presumably, HBO1 concentrations are tightly regulated within the soluble compartment by SCFFbxw15 as part of an exquisite feedback control system to provide an appropriate supply of histone acetyltransferase molecules based on nuclear content. The localization of HBO1 in the cytosol may also confer additional roles for the enzyme distinct from histone acetylation. In this regard, histone modification enzymes regulate behavior of cytosolic proteins involved in diverse processes (42). Finally, others have shown that Fbxw15 is restricted to ovarian tissues during perinatal development (21); however, our data raise a unique role for reactivation of the Fbxw15 gene within the mature lung and specifically within distal lung epithelia to control histone modification.
Protein phosphorylation is an important molecular signal for recruitment of SCF E3 ligases to their substrates. An unexpected finding from our work was the identification of a potentially new Mek1 substrate. Mek1 primarily targets Erk for its phosphorylation to promote transduction of surface signals to the nucleus. The only other phosphorylation targets identified include MyoD, STAT5, and possibly peroxisome proliferator-activated receptor γ (43–45). Here, Mek1 interacts with HBO1 to mediate a robust increase in its phosphorylation in vitro and overexpression of Mek1 accelerated HBO1 degradation. Our finding that Mek1 depletion impairs Fbxw15 interaction with HBO1 is indicative of an important role of the kinase in providing a signal for assembly of the SCFFbxw15 apparatus with its substrate. LPS also activates Mek1 and increases HBO1 phosphorylation providing further evidence of a role for Mek1 within this pathway. The primary sequence of HBO1 contains numerous candidate phosphorylation sites (67 Ser, 36 Thr, and 28 Tyr) with approximately one-third of these sites residing within a span of the first 60 residues of the NH2 terminus. This region is highly phosphorylated (46). Despite several attempts, we were not able to conclusively identify the phosphorylation site(s) within HBO1 using a reductionist approach. It is likely, given the multitude of candidate phosphoacceptors within HBO1, that there is redundancy for Mek1 targeting in vivo. Whether Mek1 antagonizes actions of Polo-like kinase 1 with regard to HBO1 phosphorylation at the NH2 terminus to coordinately regulate replication licensing and cell cycle progression requires further study.
This work was supported, in whole or in part, by National Institutes of Health Grants HL096376, HL097376, and HL098174 (to R. K. M.), HL116472 (to B. B. C.), and HL01916 (to Y. Z.). This work was also supported in part by the United States Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development, a Merit Review Award from the United States Department of Veterans Affairs, and American Heart Association Award 12SDG12040330 (to C. Z.).
- SCF
- Skp-Cullin 1-F box
- MLE
- murine lung epithelial.
REFERENCES
- 1. Iizuka M., Stillman B. (1999) Histone acetyltransferase HBO1 interacts with the ORC1 subunit of the human initiator protein. J. Biol. Chem. 274, 23027–23034 [DOI] [PubMed] [Google Scholar]
- 2. Iizuka M., Matsui T., Takisawa H., Smith M. M. (2006) Regulation of replication licensing by acetyltransferase Hbo1. Mol. Cell. Biol. 26, 1098–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miotto B., Struhl K. (2011) JNK1 phosphorylation of Cdt1 inhibits recruitment of HBO1 histone acetylase and blocks replication licensing in response to stress. Mol. Cell 44, 62–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Miotto B., Struhl K. (2010) HBO1 histone acetylase activity is essential for DNA replication licensing and inhibited by Geminin. Mol. Cell 37, 57–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wong P. G., Glozak M. A., Cao T. V., Vaziri C., Seto E., Alexandrow M. (2010) Chromatin unfolding by Cdt1 regulates MCM loading via opposing functions of HBO1 and HDAC11-geminin. Cell Cycle 9, 4351–4363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Miotto B., Struhl K. (2008) HBO1 histone acetylase is a coactivator of the replication licensing factor Cdt1. Genes Dev. 22, 2633–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saksouk N., Avvakumov N., Champagne K. S., Hung T., Doyon Y., Cayrou C., Paquet E., Ullah M., Landry A. J., Côté V., Yang X. J., Gozani O., Kutateladze T. G., Côté J. (2009) HBO1 HAT complexes target chromatin throughout gene coding regions via multiple PHD finger interactions with histone H3 tail. Mol. Cell 33, 257–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chadha G. S., Blow J. J. (2010) Histone acetylation by HBO1 tightens replication licensing. Mol. Cell 37, 5–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Avvakumov N., Lalonde M. E., Saksouk N., Paquet E., Glass K. C., Landry A. J., Doyon Y., Cayrou C., Robitaille G. A., Richard D. E., Yang X. J., Kutateladze T. G., Côté J. (2012) Conserved molecular interactions within the HBO1 acetyltransferase complexes regulate cell proliferation. Mol. Cell. Biol. 32, 689–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu Z. Q., Liu X. (2008) Role for Plk1 phosphorylation of Hbo1 in regulation of replication licensing. Proc. Natl. Acad. Sci. U.S.A. 105, 1919–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Silverman J. S., Skaar J. R., Pagano M. (2012) SCF ubiquitin ligases in the maintenance of genome stability. Trends Biochem. Sci 37, 66–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Starostina N. G., Kipreos E. T. (2012) Multiple degradation pathways regulate versatile CIP/KIP CDK inhibitors. Trends Cell Biol. 22, 33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Karabinova P., Kubelka M., Susor A. (2011) Proteasomal degradation of ubiquitinated proteins in oocyte meiosis and fertilization in mammals. Cell Tissue Res. 346, 1–9 [DOI] [PubMed] [Google Scholar]
- 14. Lau A. W., Fukushima H., Wei W. (2012) The Fbw7 and βTRCP E3 ubiquitin ligases and their roles in tumorigenesis. Front. Biosci. 17, 2197–2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nakayama K. I., Nakayama K. (2006) Ubiquitin ligases. Cell-cycle control and cancer. Nat. Rev. Cancer 6, 369–381 [DOI] [PubMed] [Google Scholar]
- 16. Xu C., Kim N. G., Gumbiner B. M. (2009) Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle 8, 4032–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frescas D., Pagano M. (2008) Deregulated proteolysis by the F-box proteins SKP2 and β-TrCP. Tipping the scales of cancer. Nat. Rev. Cancer 8, 438–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deshaies R. J. (1999) SCF and Cullin/Ring H2-based ubiquitin ligases. Annu. Rev. Cell Dev. Biol. 15, 435–467 [DOI] [PubMed] [Google Scholar]
- 19. Ho M. S., Ou C., Chan Y. R., Chien C. T., Pi H. (2008) The utility F-box for protein destruction. Cell Mol. Life Sci. 65, 1977–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bai C., Sen P., Hofmann K., Ma L., Goebl M., Harper J. W., Elledge S. J. (1996) SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell 86, 263–274 [DOI] [PubMed] [Google Scholar]
- 21. De La Chesnaye E., Kerr B., Paredes A., Merchant-Larios H., Méndez J. P., Ojeda S. R. (2008) Fbxw15/Fbxo12J is an F-box protein-encoding gene selectively expressed in oocytes of the mouse ovary. Biol. Reprod. 78, 714–725 [DOI] [PubMed] [Google Scholar]
- 22. Potoka D. A., Upperman J. S., Zhang X. R., Kaplan J. R., Corey S. J., Grishin A., Zamora R., Ford H. R. (2003) Peroxynitrite inhibits enterocyte proliferation and modulates Src kinase activity in vitro. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G861–G869 [DOI] [PubMed] [Google Scholar]
- 23. Kim C. O., Huh A. J., Han S. H., Kim J. M. (2012) Analysis of cellular senescence induced by lipopolysaccharide in pulmonary alveolar epithelial cells. Arch. Gerontol. Geriatr. 54, e35–e41 [DOI] [PubMed] [Google Scholar]
- 24. Ruemmele F. M., Beaulieu J. F., Dionne S., Levy E., Seidman E. G., Cerf-Bensussan N., Lentze M. J. (2002) Lipopolysaccharide modulation of normal enterocyte turnover by toll-like receptors is mediated by endogenously produced tumour necrosis factor α. Gut 51, 842–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang L., Wang J., Fan Y., Chen S., Wang L., Ma J. (2011) Effect of 1,25(OH)2D3 on rat peritoneal mesothelial cells treated with high glucose plus lipopolysaccharide. Cell Immunol. 271, 173–179 [DOI] [PubMed] [Google Scholar]
- 26. Hamilton J. A., Vairo G., Cocks B. G. (1992) Inhibition of S-phase progression in macrophages is linked to G1/S-phase suppression of DNA synthesis genes. J. Immunol. 148, 4028–4035 [PubMed] [Google Scholar]
- 27. Takada Y., Andreeff M., Aggarwal B. B. (2005) Indole-3-carbinol suppresses NF-κB and IκBα kinase activation, causing inhibition of expression of NF-κB-regulated antiapoptotic and metastatic gene products and enhancement of apoptosis in myeloid and leukemia cells. Blood 106, 641–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carter A. B., Monick M. M., Hunninghake G. W. (1998) Am. J. Respir. Cell Mol. Biol. 18, 384–391 [DOI] [PubMed] [Google Scholar]
- 29. Rothe M., Sarma V., Dixit V. M., Goeddel D. V. (1995) TRAF2-mediated activation of NF-κB by TNF receptor 2 and CD40. Science 269, 1424–1427 [DOI] [PubMed] [Google Scholar]
- 30. Haller D., Holt L., Kim S. C., Schwabe R. F., Sartor R. B., Jobin C. (2003) Transforming growth factor-beta 1 inhibits non-pathogenic Gram negative bacteria-induced NF-κB recruitment to the interleukin-6 gene promoter in intestinal epithelial cells through modulation of histone acetylation. J. Biol. Chem. 278, 23851–23860 [DOI] [PubMed] [Google Scholar]
- 31. Saccani S., Pantano S., Natoli G. (2001) Two waves of nuclear factor κB recruitment to target promoters. J. Exp. Med. 193, 1351–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zou C., Butler P. L., Coon T. A., Smith R. M., Hammen G., Zhao Y., Chen B. B., Mallampalli R. K. (2011) LPS impairs phospholipid synthesis by triggering β-transducin repeat-containing protein (β-TrCP)-mediated polyubiquitination and degradation of the surfactant enzyme acyl-CoA:lysophosphatidylcholine acyltransferase I (LPCAT1). J. Biol. Chem. 286, 2719–2727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao J., Wei J., Mialki R., Zou C., Mallampalli R. K., Zhao Y. (2012) Extracellular signal-regulated kinase (ERK) regulates cortactin ubiquitination and degradation in lung epithelial cells. J. Biol. Chem. 287, 19105–19114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Coles A. H., Gannon H., Cerny A., Kurt-Jones E., Jones S. N. (2010) Inhibitor of growth-4 promotes IκB promoter activation to suppress NF-κB signaling and innate immunity. Proc. Natl. Acad. Sci. U.S.A. 107, 11423–11428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zou C., Ellis B. M., Smith R. M., Chen B. B., Zhao Y., Mallampalli R. K. (2011) Acyl-CoA:lysophosphatidylcholine acyltransferase I (Lpcat1) catalyzes histone protein O-palmitoylation to regulate mRNA synthesis. J. Biol. Chem. 286, 28019–28025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen B. B., Coon T. A., Glasser J. R., Mallampalli R. K. (2011) Calmodulin antagonizes a calcium-activated SCF ubiquitin E3 ligase subunit, FBXL2, to regulate surfactant homeostasis. Mol. Cell. Biol. 31, 1905–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kipreos E. T., Pagano M. (2000) The F-box protein family. Genome Biol. 1, REVIEWS3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li Y., Jaramillo-Lambert A., Hao J., Yang Y., Zhu W. (2011) The stability of histone acetyltransferase general control non-derepressible (Gcn) 5 is regulated by Cullin4-RING E3 ubiquitin ligase. J. Biol. Chem. 286, 41344–41352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tan M. K., Lim H. J., Harper J. W. (2011) SCF(FBXO22) regulates histone H3 lysine 9 and 36 methylation levels by targeting histone demethylase KDM4A for ubiquitin-mediated proteasomal degradation. Mol. Cell. Biol. 31, 3687–3699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shima Y., Shima T., Chiba T., Irimura T., Pandolfi P. P., Kitabayashi I. (2008) PML activates transcription by protecting HIPK2 and p300 from SCFFbx3-mediated degradation. Mol. Cell. Biol. 28, 7126–7138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jain S., Wei J., Mitrani L. R., Bishopric N. H. (2012) Auto-acetylation stabilizes p300 in cardiac myocytes during acute oxidative stress, promoting STAT3 accumulation and cell survival. Breast Cancer Res. Treat 135, 103–114 [DOI] [PubMed] [Google Scholar]
- 42. Inuzuka H., Gao D., Finley L. W., Yang W., Wan L., Fukushima H., Chin Y. R., Zhai B., Shaik S., Lau A. W., Wang Z., Gygi S. P., Nakayama K., Teruya-Feldstein J., Toker A., Haigis M. C., Pandolfi P. P., Wei W. (2012) Acetylation-dependent regulation of Skp2 function. Cell 150, 179–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jo C., Cho S. J., Jo S. A. (2011) Mitogen-activated protein kinase kinase 1 (MEK1) stabilizes MyoD through direct phosphorylation at tyrosine 156 during myogenic differentiation. J. Biol. Chem. 286, 18903–18913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maki K., Ikuta K. (2008) MEK1/2 induces STAT5-mediated germline transcription of the TCRgamma locus in response to IL-7R signaling. J. Immunol. 181, 494–502 [DOI] [PubMed] [Google Scholar]
- 45. Burgermeister E., Chuderland D., Hanoch T., Meyer M., Liscovitch M., Seger R. (2007) Interaction with MEK causes nuclear export and downregulation of peroxisome proliferator-activated receptor γ. Mol. Cell. Biol. 27, 803–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dephoure N., Zhou C., Villén J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., Gygi S. P. (2008) A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]