

Background: VWD mutations in the A2 domain increase cleavage by ADAMTS13.
Results: Three VWD mutations decreased thermal stability and altered single molecule force resistance.
Conclusion: VWD mutations destabilize A2 and the R1597W mutation slows refolding by disrupting calcium stabilization.
Significance: Three mutations highlight structural features important in A2 function as a force sensor.
Keywords: Immunology, Protein Folding, Single Molecule Biophysics, Structural Biology, Von Willebrand Factor, A2 Domain, VWD
Abstract
von Willebrand Factor (VWF) is an ultralong, concatameric, and adhesive glycoprotein. On short time scales, adhesiveness for platelets is activated by elongation of VWF by altered hydrodynamics at sites of hemostasis. Over longer time scales, the length of VWF is regulated by ADAMTS13 (a disintegrin and metalloprotease with a thrombospondin type 1 motif, member 13), cleavage by which in the VWF A2 domain is dependent on elongational force. Patients with von Willebrand disease type 2A present with increased bleeding due to mutations within the VWF A2 domain that enhance cleavage. We tested using temperature and force the hypothesis that von Willebrand disease mutations disrupt A2 force sensing by destabilizing the folded state. Mutations R1597W, M1528V, and E1638K reduced A2 thermal stability by 10–18 °C. M1528V and E1638K showed a marked further decrease in stability upon calcium removal. In contrast, R1597W, which resides within the A2 calcium-binding loop, exhibited similar stability in the presence and absence of calcium. Using single molecule optical tweezers and R1597W, we measured the force dependence of unfolding and refolding kinetics. In the presence of calcium, the R1597W mutation slowed the rate of refolding but had no effect on unfolding. The three mutations highlight the calcium-binding loop (R1597W), the hydrophobic core around the vicinal disulfide (M1528V), and hydrogen bonds to the α4-less loop (E1638K), as structural features critically important to the function of A2 as a force sensor in regulating thrombogenic activity in the vasculature.
Introduction
von Willebrand factor (VWF)3 multimers mediate hemostasis in the arteriolar circulation by cross-linking platelets and damaged vessel walls. Endothelial cells and platelets secrete VWF at sites of vascular injury as ultralong, disulfide-bonded concatamers reaching >12 MDa in mass and >15 μm in length (1–3). VWF functional activity correlates with its length.
VWF concatamer length and hence thrombogenic activity is regulated through cleavage in the VWF A2 domain by ADAMTS13 (a disintegrin and metalloprotease with a thrombospondin type 1 motif, member 13). ADAMTS13 circulates as a constitutively active protease. Cleavage is regulated by hydrodynamic force applied to its substrate, VWF. The ADAMTS13 cleavage site is buried within a central β4-strand that is inaccessible in the native state (Fig. 1A). Hydrodynamic forces on VWF concatamers in the vasculature result in elongational forces exerted throughout the length of VWF. These forces are experienced in the A2 domain as tensile forces exerted on its N and C termini where it connects to the neighboring A1 and A3 domains. Single molecule studies have demonstrated that ADAMTS13 cleavage requires unfolding of the A2 domain to expose the scissile bond (4).
FIGURE 1.

A2 structure. Windings 1 and 2 are shown in beige and orange, respectively. Tyr-1605 and Met-1606 at the scissile bond have black side chains. The Ca2+ ion is a silver sphere. Residues mutated in VWD have side chains with green carbons. Other residues important in the Ca2+ site or in the local environment of mutations are shown with side chains or backbones with white carbons. Metal coordination and hydrogen bonds are dashed red and black lines, respectively. Important water molecules are small red spheres. A, the A2 domain. B, Ca2+-binding site in the α3β4 loop. C, Glu-1638 in the α5-helix interacts with the α4-less loop and is mutated to Lys in VWD. D, Met-1528 at the end of the α1-helix interacts with the end of the α6-helix and is mutated to Val in VWD. E, Arg-1597 coordinates Ca2+ through its carbonyl and its side chain has multiple Van der Waals and hydrogen bond interactions with residues in two loops and near the A2 N terminus. Arg-1597 is mutated to Trp or Gln in VWD. The structure shown is a chimera containing residues 1495–1578 and 1602–1672 of Protein Data Bank code 3GXB (4) and residues 1579–1601 of Protein Data Bank code 3PPV (6). The figure was made using PyMOL software.
VWF A2 is comprised of two topologically distinct windings (Fig. 1A) (5–7). Unfolding is predicted to begin in winding 2 at the C terminus of the α6-helix, which resides unprotected on the exterior of the domain, whereas the β1-strand at the N terminus in winding 1 is highly protected at the center of the fold and as a middle β-strand. Several unique features distinguish A2 from other VWA domains and may regulate folding, unfolding, and sensitivity of A2 to force (5). Compared with neighboring A1 and A3 domains, A2 lacks a long range disulfide bond to protect the domain against unfolding. An “α4-less” loop in place of an α4-helix is thought to slow refolding to promote cleavage site accessibility (5). At the junction between winding 1 and winding 2, A2 features a unique calcium-binding site demonstrated to protect VWF against ADAMTS13 cleavage (6, 7) and enhance refolding (8).
Mutations within the VWF gene cause von Willebrand disease (VWD), the most common inherited bleeding disorder in humans. Patients with type 2A VWD present with increased bleeding due to the decreased thrombogenicity of smaller VWF multimers (9). Certain mutations have been described to impair VWF biosynthesis (group 1) (10), whereas other mutations increase VWF proteolysis despite normal VWF multimer assembly and secretion (group 2) (11, 12). Group 2A mutations cluster within the A2 domain and are thought to enhance cleavage by disrupting the stability of the folded A2 structure. This hypothesis, however, has not yet been tested. Previous studies have investigated the conformational stability of VWF A2 relative to neighboring domains. Consistent with the absence of a long-range disulfide bond, A2 was found to be more susceptible to chemical denaturation compared with A1 and A3 (13).
In this study, we seek to understand the impact of VWD type 2A mutations on A2 conformational stability, and in particular, the dynamics of unfolding and refolding underlying enhanced cleavage of A2. We specifically focus on group 2 mutations to isolate effects on protein function from biosynthesis. R1597W is the most common VWD type 2A mutation described in patients (14) and resides within the α3β4 calcium-binding loop (Fig. 1C). We also examined one clinically well characterized VWD type 2A mutation in each of windings 1 and 2. M1528V (12) resides at the end of the α1-helix within winding 1 (Fig. 1, A and D). E1638K (15–17) resides within the α5-helix, C-terminal to the scissile bond and within winding 2 (Fig. 1, A and B). Mutation of Ca2+-coordinating side chains or removal of Ca2+ destabilizes A2 against cleavage, thermal denaturation, and refolding (6–8). Thus, we have also investigated how Ca2+ removal interacts with VWD mutations, and whether mutation R1597W, within a Ca2+-binding loop, acts similarly to Ca2+ removal. We characterize the impact of these mutations on conformational stability using thermodynamic denaturation. In parallel, we utilize single molecule optical tweezers to characterize the effect of VWD mutations on the force dependence of A2 conformational dynamics. The results demonstrate the impact of VWD 2A mutations on A2 stability and provide insight into structural features regulating A2 unfolding and refolding dynamics that dictate ADAMTS13 cleavage.
EXPERIMENTAL PROCEDURES
Circular Dichroism
Circular dichroism (CD) spectra were obtained on an Aviv 62 DS spectropolarimeter equipped with a Peltier temperature control unit (Aviv Associates, Lakewood, NJ). A2 protein was monitored within a 1.0-cm path length quartz cuvette with a magnetic stirrer to allow uniform thermal mixing. Spectra shown are corrected for background contribution of buffer (either 20 mm Tris or phosphate, pH 7.5, and 30 mm NaCl), and were obtained at 2 μm protein concentrations to yield signals at least 10-fold greater than buffer absorbance alone. Base-line spectra of the folded and unfolded states were obtained at 4 and 90 °C, respectively, with five accumulations scanned from 260 to 195 nm at 1-nm intervals. Thermal unfolding curves were derived from linear fits to thermal denaturation signal before and after the unfolding transition. Single wavelength CD temperature melts were fit to a Gibbs-Helmholtz equation as described by Greenfield (18).
Protein Expression and Purification
VWF A2 was prepared as described previously (4). VWD mutations were introduced using overlap PCR.
Single Molecule Force Experiments
Single molecule constructs (Fig. 2) and optical tweezers experiments were prepared as described previously (8).
FIGURE 2.

Biochemical coupling of R1597W A2 to DNA handles. 802-bp DNA handles were PCR-amplified using 5′-thiol forward primers and 5′-biotin or -digoxigenin reverse primers. DNA handles were then activated using 2,2′-dithiodipyridine and reacted with freshly reduced R1597W A2 to disulfide link the handles to A2, as described previously (8). Disulfide coupling was verified by subjecting reaction products to 4–20% polyacrylamide Tris borate-EDTA gel electrophoresis with or without reduction with 1 mm DTT and staining with ethidium bromide. Shifts in migration upon coupling or binding to an antibody to C-terminal region of A2 (R&D Systems) report successful coupling of DNA handles to R1597W A2. A, DNA handles only; B, DNA handles + R1597W; C, same as B + A2 antibody. *, linkage to DNA was abolished under reducing conditions. DNA markers are shown on the left of each gel.
Statistical Analysis
Errors were estimated by χ2 minimized fits using Origin (version 6.1, OriginLab Corp., Northampton, MA) and are shown as S.E. or 95% confidence intervals.
RESULTS
Reversibility of A2 Thermal Denaturation
CD spectra were obtained to monitor changes in A2 secondary structure. Wavelength scans obtained in phosphate buffer highlight the strong α-helical content of the A2 domain. Double minima at 222 and 208 nm (19) were observed for both wild-type and R1597W A2 at 4 °C (Fig. 3A, filled and open squares, respectively). At 90 °C, both proteins lose their α-helical signature, reflecting thermal denaturation (Fig. 3A, circles). Thermal denaturation was reversible. Cooling denatured samples back to 4 °C completely recovered the native α-helical signatures (Fig. 3A, triangles).
FIGURE 3.
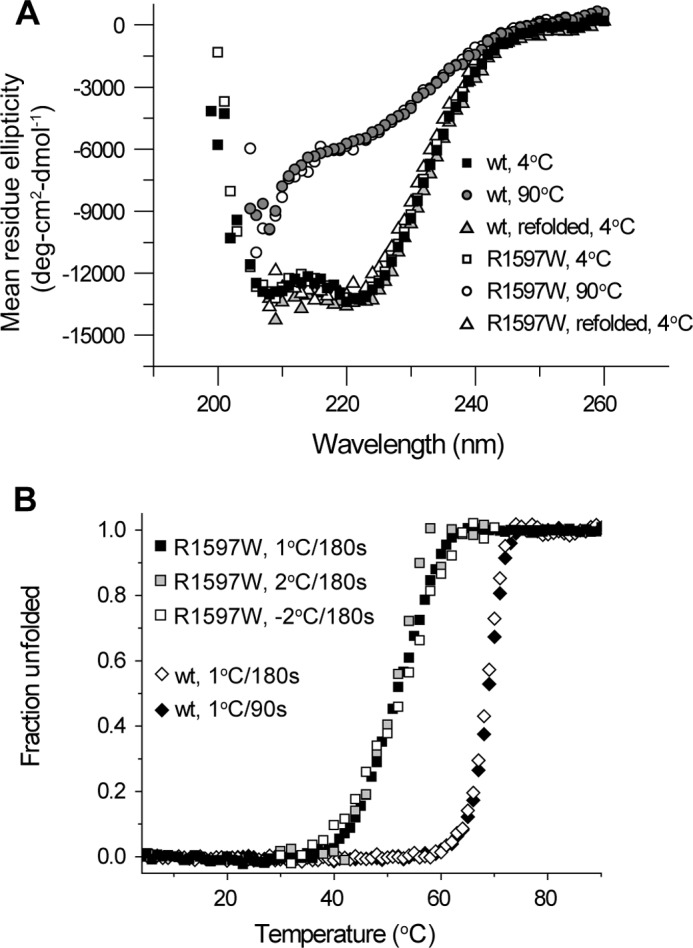
Reversible equilibrium thermal denaturation of A2. A, wavelength scan in 20 mm phosphate buffer demonstrates the α-helical signature that is lost with thermal denaturation. Upon cooling wild-type or R1597W protein back down to 4 °C, the same α-helical spectra are recovered. B, thermal denaturation profiles are independent of thermal scan rates and are reversible. The key in the figure gives the step size for heating (1 or 2 °C) or cooling (−2 °C) and the total length of each step (90 or 180 s). Similar profiles were obtained with different heating rates. Furthermore, the same thermal profile was recovered with stepwise cooling from 70 to 25 °C.
Thermal denaturation was quantitated at 222 nm, the wavelength of greatest contrast between native and denatured states (Fig. 3A), over a stepwise increase in temperature. Spectra remained stable until at least 30 °C, above which α-helical signature was lost in a transition region until again becoming stable in a thermally denatured state at 90 °C (Fig. 3B). Temperature steps of 1 °C per 180 s, 2 °C per 180 s, or 1 °C per 90 s all yielded similar denaturation profiles. These findings demonstrated that measurements obtained with 1 °C steps per 180 s, used below in Fig. 4, were sufficiently slow to reach equilibrium.
FIGURE 4.

CD spectra of VWD A2. Under physiologic calcium concentrations, all VWD mutants exhibited similar native spectra compared with wild-type at 4 °C (black). At 90 °C (white), the spectra lost α-helical signature. Upon removal of calcium with 1 mm EDTA at 4 °C (gray), wild-type, R1597W, and M1528V retained and E1638K lost α-helical content.
Furthermore, stepwise cooling of the denatured sample from 70 to 25 °C yielded refolding along the same thermal profile as for unfolding, as demonstrated with the R1597W mutant (Fig. 3B, white squares). The lack of distinction between heat-dependent denaturation and renaturation definitively demonstrated reversibility and that our data were obtained at equilibrium.
Thermal Stability of VWD Mutants in Physiologic Calcium
VWF A2 binds calcium with a Kd of 3.8 μm (8), and thus the calcium binding site is predicted to be fully saturated at physiologic concentrations of 1.1–1.3 mm ionized calcium in healthy human blood. Because Ca2+ complexes with phosphate, experiments described below were conducted in Tris buffer, which we found had no effect on A2 CD spectra >205 nm. To quantitate the effect of calcium in stabilizing VWD mutants, equilibrium spectra were obtained with either 1 mm EDTA or 1.25 mm CaCl2 at 1 °C steps of 180-s each. In the presence of calcium, all three VWD mutants and wild-type A2 exhibited similar spectra at 4 °C (Fig. 4, A–D, black symbols). Under physiologic 1.25 mm Ca2+, all three VWD mutants were less stable against thermal denaturation compared with wild-type (Fig. 5A). Whereas the half-point melting temperature Tm of wild-type A2 was 67.7 °C, M1528V, R1597W, and E1638K exhibited decreased Tm temperatures of 57.9, 53.9, and 50.2 °C, respectively (Table 1).
FIGURE 5.

VWD A2 thermal denaturation. Thermal denaturation monitored at 222 nm over a stepwise increase in temperature at equilibrium (3 min per °C). A, under physiologic calcium concentrations (1.25 mm Ca2+), all three mutants unfolded at lower temperatures compared with wild-type. B–D, effect of Ca2+ removal by EDTA on thermal denaturation of wild-type and mutant A2 domains.
TABLE 1.
Tm values determined using CD
| WT | R1597W (Tm) | M1528V | E1638K | |
|---|---|---|---|---|
| °C | ||||
| 1.25 mm Ca | 67.7 ± 0.1 | 53.9 ± 0.1 | 57.9 ± 0.1 | 50.2 ± 0.1 |
| 1 mm EDTA | 56.7 ± 0.1 | 51.5 ± 0.1 | 42.6 ± 0.2 |
Calcium Stabilization in VWD Mutants
To monitor the effect of calcium on stabilizing A2 mutants, thermal denaturation was similarly monitored in the presence and absence of calcium (Fig. 5, B–D, Table 1). Wild-type A2 exhibited an 11 °C decrease in Tm in 1 mm EDTA compared with 1.25 mm Ca2+ (Fig. 5B) (8). The Tm of M1528V also exhibited a large decrease of 15 °C upon removal of calcium (Fig. 5C, Table 1). E1638K, featuring the lowest Tm in the presence of calcium (Fig. 5A), exhibited a sharp change in spectrum upon calcium removal at 4 °C. The E1638K 4 °C spectrum in 1 mm EDTA (gray symbols, Fig. 4D) differed little from the 90 °C spectrum obtained in the presence of calcium and remained in this altered state throughout the stepwise temperature increase to 90 °C (data not shown). The large shift in Tm observed in M1528V and loss of secondary structure observed in E1638K at 4 °C suggest that similar to its effect on wild-type A2, calcium plays an important role in stabilizing the structures of M1528V and E1638K mutants. In contrast, the R1597W mutant exhibited only a 2 °C decrease in Tm in the absence of calcium (Fig. 5C and Table 1). The marked loss of calcium dependence strongly suggests that the R1597W mutation destabilizes the calcium-binding loop in which it resides.
Force-induced Unfolding of R1597W
The effect of R1597W on force-dependent A2 unfolding and refolding was investigated using single molecule optical tweezers. VWF A2 was covalently linked through its N and C termini to DNA handles and suspended between two polystyrene beads, with one anchored to the tip of a micropipette and another trapped within the infrared dual beam laser focus of the optical tweezers (Fig. 6A). Changes in light momentum reflected excursions of the bead from the center of the trap, dx (Fig. 6A), and reported the force experienced by the protein-DNA tether with pN force sensitivity. Force extension profiles reported the stiffness of the single molecule construct, and A2 unfolding manifested as abrupt decreases in force (dashed circles, Fig. 6B). Refolding was monitored by the presence or absence of unfolding in the subsequent pull (Fig. 6B). Clamping at a low force between pulls provided time for A2 refolding.
FIGURE 6.

Monitoring protein unfolding using optical tweezers. A, experimental setup. Single A2 domains suspended between two polystyrene beads by DNA handles are subjected to force by movement of the laser trap. Excursions of the bead from the center of the trap, dx, report the force experienced by the single molecule construct. Trap force on the bead is symbolized with a spring. B, representative unfolding events observed under 40 nm/s pulling and relaxation rates (corresponding to 0.5–2 pN/s force application rates) between 2 and 20 pN with a 10-s pause at 2 pN to allow refolding. Force extension discontinuities are highlighted. C, worm-like chain (WLC) model fit to R1597W unfolding in the presence of no added Ca2+, 5 mm Ca2+, and 1 mm EDTA. Global fit for R1597W (solid black line) is compared with wild-type worm-like chain fit (dashed gray line) determined previously (8). Error bars show S.E. The inset shows the distribution of extensions for one bin of unfolding force.
Over a range of pulling rates, R1597W unfolding was similar to wild-type in displaying excellent agreement with full unfolding of VWF A2 (Fig. 6C). Unfolding extension lengths depended on the force at which unfolding occurred (symbols, Fig. 6C) and were fit to the worm-like chain model of polymer elasticity (solid black line, Fig. 6C). The fit yielded a persistence length, Lp, of 0.49 ± 0.02 nm. The contour length, or length of maximal extension, Lc, derived from the fit was 70.5 ± 1.18 nm. These results agree with previous measurements of wild-type A2 unfolding (dotted gray line, Fig. 6C; Lp, 0.41 nm; Lc, 75.6 nm) (8) and suggest full unfolding of R1597W A2 under all conditions.
M1528V and E1638K coupling to DNA handles was verified via biochemical assay. However, repeated attempts were unsuccessful in finding multiple cycles of unfolding and refolding using many single molecule tethers with these mutant domains. Whether this reflects technical problems or inefficient refolding during the time allotted for refolding before the next pull is not known.
Calcium Mechanically Stabilizes R1597W against Unfolding
Unfolding kinetics were determined by examining the forces at which A2 unfolding was observed. R1597W and wild-type A2 both exhibited a logarithmic dependence of unfolding force on loading rate (Fig. 7), consistent with biophysical models of protein unfolding (20). In presence of calcium, the force dependence of unfolding was similar for wild-type A2 and R1597W. In absence of calcium, wild-type A2 exhibited similar unfolding kinetics as in the presence of calcium (within 20%, p < 0.05; Fig. 7B) (8). In contrast, R1597W unfolded more readily with increasing force in 1 mm EDTA (Fig. 7A, dashed line and open symbols) compared with calcium (Fig. 7A, black line and grey symbols). This change in force sensitivity is reflected in the force sensitivity parameter σ, where k = ku0exp(σF/kbT), and σ is given by the slope of the lines in Fig. 7A. Thus, mechanical force destabilizes R1597W more in the absence compared with the presence of Ca2+. However, the extrapolated off-rate in the absence of force (ku0) was similar for R1597W in the presence and absence of Ca2+. Furthermore, the ku0 values for the R1597W mutant and wild-type in all conditions were indistinguishable (see ku0 values in Fig. 7). Therefore, unfolding kinetics cannot account for the impact of R597W in destabilizing A2 to thermal denaturation, which was measured in the absence of force.
FIGURE 7.

R1597W unfolding. R1597W unfolding kinetics determined from the distribution of unfolding forces using the Dudko-Hummer-Szabo equation (25). A, R1597W exhibited greater force resistance to unfolding (σ) in the presence of calcium (black) compared with EDTA (dotted line). B, for comparison, published wild-type data (8). Means are shown ± S.E., with 95% confidence intervals in brackets.
R1597W Refolding
R1597W refolding kinetics were also measured over a range of forces. Compared with wild-type A2, R1597W exhibited significantly decreased refolding in the presence of calcium (Fig. 8A). In contrast to wild-type A2 refolding, which was significantly decreased upon removal of calcium, R1597W refolding remained similar in the presence and absence of calcium. In both 5 mm Ca2+ and 1 mm EDTA, R1597W refolding was similar to calcium-free wild-type A2 (Fig. 8A).
FIGURE 8.

R1597W refolding. A, binary refolding statistics collected for 10-s pauses at 1 pN. In the presence of calcium, R1597W significantly decreased the probability of refolding compared with wild-type. B, unfolded state lifetimes were estimated by maximum likelihood at each pause force from the refolding probabilities at pause durations of 2, 5, 10, or 20 s. Lines show fits based on an exponential dependence of refolding on the square of applied force using the equation shown (4). Wild-type data shown for comparison were published previously (8).
Unfolded state lifetimes were estimated by maximum likelihood from refolding probabilities (Fig. 8B). Each data point in Fig. 8B shows the maximum likelihood estimation based on refolding data for all pause durations obtained at a given pause force. Theoretical fits (lines in Fig. 8B) are to exponential dependence on the square of applied force, f2, and the compliance of the unfolded state, κ (4). The extrapolated unfolded state lifetimes in absence of force, τ0, of R1597W were similar in presence and absence of calcium, at 7.2 and 9.2 s, respectively (Fig. 8B). In contrast, wild-type A2 exhibited enhanced refolding in presence of calcium, with τ0 of 2.5 s, compared with τ0 of 7.1 s in absence of calcium (Fig. 8B). In both the presence and absence of calcium, R1597W refolding was similar to wild-type A2 refolding with calcium removed (Fig. 8B).
DISCUSSION
In this study, we investigated the mechanisms by which mutations in VWF A2 cause VWD using bulk measurements of thermodynamic denaturation of three VWD A2 domain mutants and single molecule measurements of R1597W unfolding and refolding kinetics. Under physiologic calcium concentrations, all three VWD type 2A group 2 mutations tested, R1597W, M1528V, and E1638K, exhibited α-helical content similar to wild-type A2, suggesting that A2 structure remained largely intact. However, all three mutations exhibited decreased stability to thermal denaturation compared with wild-type A2, suggesting that these clinical mutations enhance ADAMTS13 proteolysis by destabilizing the A2 domain.
We further probed the impact of these VWD mutations on calcium-mediated stabilization of VWF A2 by monitoring thermal denaturation in the presence and absence of calcium. Wild-type A2 exhibits an 11 °C increase in stability in Ca2+ (8). M1528V and E1638K, outside of the calcium binding loop, retained stabilization by calcium binding. M1528V exhibited a 15.3 °C stabilization by Ca2+ against thermal denaturation, whereas E1638K required calcium to stabilize a folded structure at 4 °C.
In contrast, R1597W, within the calcium-binding α3β4 loop, exhibited only a 2 °C increase in Tm in calcium, demonstrating that this mutation significantly disrupts stabilization by calcium. The Arg-1597 backbone carbonyl oxygen directly coordinates calcium. Although only the side chain is altered by mutation, the Arg side chain stabilizes the conformation of the α3β4 loop and the orientation of the side chain of Asp-1498, the only residue outside of the α3β4 loop involved in A2 calcium coordination (Fig. 1B). R1597W and R1597Q are the two most frequently reported VWD 2A mutations and account for 65% of all VWD type 2A mutations described in the International Society on Thrombosis and Hemostasis database (14). The disruption of calcium-mediated stabilization in R1597W thus highlights the importance of calcium stabilization for VWF function in vivo.
Previous studies have demonstrated that calcium modulates wild-type A2 stability and ADAMTS13 sensitivity. VWF A2 binds calcium with μm affinity (8), and calcium binding has been shown to protect the domain against cleavage (6, 7) and enhance A2 refolding (8). The mechanism of calcium-mediated stabilization has also been studied through alanine substitutions of calcium coordinating residues (6, 7). Consistent with loss of calcium binding, D1596A and N1602A mutations abolished the ability of calcium to shift thermal stability of A2. Thermal stability of N1602A was similar to calcium-free wild-type A2. In contrast, D1596A thermal stability in absence of Ca2+ was similar to that of wild-type A2 in presence of Ca2+, presumably because Ca2+ binding eliminates electrostatic repulsion between Asp-1596 and Asp-1498 (7).
Our single molecule results are discussed in terms of a free energy landscape for A2 unfolding and refolding (Fig. 9). Assuming that wild-type and R1597W reach the same unfolded state, the increased thermal stability of wild-type A2 observed with circular dichroism predicts that R1597W destabilizes the folded state. In both the presence and absence of calcium, R1597W exhibited no effect on the unfolding rate in absence of force, ku0, compared with wild-type. Similar unfolding kinetics dictate similar energy barriers to unfolding between the folded (N) and unfolding transition states (TS) (Fig. 9). In contrast, in the presence of Ca2+, R1597W demonstrated markedly slower refolding compared with wild-type, with unfolded state lifetimes, τ0, of 7.7 and 2.5 s, respectively. Slowed R1597W refolding is reflected in the larger energy barrier to folding in the mutant protein (U to TS, Fig. 9).
FIGURE 9.

Postulated free energy landscape. Unfolding and refolding kinetics predict a free energy landscape describing the impact of R1597W on A2 unfolding. Our landscape features a destabilized mutant with similar unfolding kinetics but retarded refolding kinetics and assumes that R1597W (black) and wild-type A2 (gray) achieve the same unfolded state (U). Similar barrier heights between native (N) and rate determining transition states (TS) reflect similar unfolding kinetics, whereas the increased barrier between the unfolded state and transition states for R1597W reflects slowed A2 refolding. The finding that Ca2+-binding affects the rate of refolding for wild-type dictates an intermediate state, which has been postulated and observed in previous studies, and is shown with dashed segments (4, 8).
In both the presence and absence of Ca2+, R1597W refolding kinetics were similar to wild-type in EDTA. Thus, loss of calcium-dependent stabilization contributes to the decreased stability observed in the most frequently observed substitution in VWD type 2A, R1597W.
The force sensitivity parameter σ measures the compliance (deformability) of A2 at the transition state for unfolding. In the presence of calcium, the σ value for R1597W was similar to σ for wild-type in both the presence and absence of Ca2+. However, in the absence of Ca2+, the R1597W mutation markedly increased σ, i.e. the force dependence of unfolding. This result was highly significant, with no overlap in the 95% confidence intervals for R1597W σ in presence (2.57 to 2.97 nm) and absence (3.86 to 4.22 nm) of Ca2+. Thus, upon application of force, ku0 is exponentiated to a greater degree, resulting in markedly shorter folded state lifetimes with increasing force application.
These results demonstrate that Ca2+ and the Arg-1597 side chain make distinct, synergistic contributions to the stiffness of A2. A plausible structural basis for the mechanical role of Arg-1597 is suggested by its structural environment. The Arg-1597 side chain is largely buried by Val-1496 and Ala-1600, and its planar guanido group stacks closely on the aromatic side chain of His-1536 (Fig. 1E). The three Arg-1597 side chain nitrogens make a total of four hydrogen bonds, two to Asp-1498, and two to the carbonyl oxygen of Ser-1534. All of these hydrogen bonds are nearly ideal, with close apposition of the O and N heavy atoms (2.7 to 2.9 Å) and alignment of the nitrogen hydrogens with the trigonal oxygen electron orbitals (Fig. 1E). Arg-1597 thus links through its backbone to the Ca2+-binding α3β4 loop and through its side chain to residues near the N terminus at which force is applied (Val-1496 and Asp-1498) and to the α1β2 loop (Ser-1534 and His-1536). The increased σ suggests that in the combined absence of Ca2+ and the Arg-1597 side chain, force distorts this region and lowers the transition state for unfolding more than it does in presence of either or both of these elements.
Our single molecule estimates of rate constants and force sensitivity also predict differences in A2 stability at force equilibrium (Fig 10). The parameters ku0 and σ, and kf0 and κ, were used to calculate ku and kf, respectively, over a range of forces. At any given force, [unfolded A2]/[folded A2] = ku/kf. Fig. 10 plots the formula: [unfolded A2]/[total A2]. In physiologic Ca2+, 5 pN is required to unfold almost all wild-type A2, whereas 3 pN is sufficient to unfold almost all R1597W A2. Thus, in the vasculature, R1597W will unfold more readily, increasing the population of unfolded VWF substrate for cleavage by ADAMTS13, resulting in the smaller and less thrombogenic VWF concatamers characterized in VWD patients.
FIGURE 10.

Fraction of unfolded A2 under applied force. The population of unfolded A2 under equilibrium predicted from the ratio of unfolding to refolding kinetics. Kinetic (ku0 and kf0) and mechanical (σ and κ) constants determined here were used to estimate the fraction of unfolded A2 over a range of constant forces, in the presence or absence of calcium.
The three VWD mutations studied here highlight specialized features of the A2 domain that enable force-dependent size regulation of VWF. Loss of calcium stabilization accounts for the destabilization observed in the most commonly described site of VWD type 2A mutations, Arg-1597. Under physiologic calcium conditions, the R1597W mutation exhibited little effect on unfolding kinetics but significantly slowed refolding. This is in agreement with the effect of Ca2+ depletion on wild-type A2, which slows force-dependent refolding with no effect on unfolding kinetics (8).
In addition to stabilizing Ca2+ binding, Arg-1597 in the α3β4 loop highlights additional features important to A2 force sensitivity. As described above, the partially buried guanido group of Arg-1597 knits together two loops, α1β2 and α3β4, and the N terminus of A2 to which elongational hydrodynamic force exerted throughout the length of VWF is applied (Fig. 1E). Thus, Arg-1597 stabilizes A2 against unfolding by mechanical force.
The E1638K VWD mutation in winding two highlights the importance of Glu-1638 in the A2 α5-helix to stabilizing the α4-less loop (Fig. 1C). The Glu-1638 side chain hydrogen bonds to the backbone of residue 1612 in the α4-less loop and also to a remarkable network of five largely buried water molecules that hydrogen bond to the backbones of residues 1610 and 1614 in the α4-less loop. The low stability of E1638K, especially upon removal of calcium, highlights the sensitivity of the α4-less loop to structural perturbation.
The M1528V VWD mutation in winding 1 highlights the importance of precise packing of the hydrophobic core of A2 (Fig. 1D). In this mutation, the long, unbranched, hydrophobic Met-1528 side chain is replaced the smaller, β-branched, hydrophobic Val side chain. Met-1528 at the end of the α1-helix is buried by the critical vicinal disulfide bond between Cys residues 1669 and 1670 (21) and by Leu-1666. These three residues are at the end of the α6-helix and critically stabilize the C-terminal end of VWF A2 against applied elongational force.
VWF A2 is flanked on both its N and C termini by O-glycosylated linkers that connect it to the A1 and A3 domains within the VWF protein. Although our study is limited in characterizing conformational dynamics of isolated A2, the relevance of A2 unfolding has been affirmed by studies demonstrating that force-induced A2 unfolding is required for cleavage of the A1-A2-A3 tridomain as well (22, 23). Furthermore, CD measurements of urea-induced unfolding of the tridomain unit show no significant differences from the sum of the unfolding transitions of the individual domains (13). Moreover, EM studies at physiologic pH show separation between the neighboring A1, A2, and A3 domains and variable orientations among them (24). These results are consistent with the presence of O-glycosylated linker regions between A1 and A2 and between A2 and A3. Thus, the force sensitivity of A2 is likely to be minimally impacted by interactions with neighboring domains.
The impacts of these three VWD type 2A mutations on A2 stability highlight structural features, including calcium binding, the α4-less loop, and the hydrophobic environment around the vicinal disulfide bond, that are unique to A2 and help to specialize it as a sensor within VWF concatamers for hydrodynamic forces experienced in the vasculature. Kinetic measurements of R1597W unfolding and refolding demonstrate how such structural perturbations result in altered mechanoregulation. Specifically, our measurements predict that VWD mutations result in a greater population of unfolded A2, thus increasing the concentration of the substrate available for cleavage by ADAMTS13. The end result is the smaller, less thrombogenic VWF fragments that characterize clinical disease.
Acknowledgments
We thank Dr. S. C. Blacklow for CD equipment and advice and Dr. W. P. Wong for helpful discussion about statistical analysis.
This work was supported by National Institutes of Health Grant HL-48675 (to T. A. S.) and Molecular Biophysics and General Medical Sciences Training Grants T32 GM008313 and GM007753 (from NIGMS; to A. J. X.).
- VWF
- von Willebrand factor
- VWD
- von Willebrand disease
- ADAMTS13 (a disintegrin and metalloprotease with a thrombospondin type 1 motif
- member 13).
REFERENCES
- 1. Hoyer L. W., Shainoff J. R. (1980) Factor VIII-related protein circulates in normal human plasma as high molecular weight multimers. Blood 55, 1056–1059 [PubMed] [Google Scholar]
- 2. Schneider S. W., Nuschele S., Wixforth A., Gorzelanny C., Alexander-Katz A., Netz R. R., Schneider M. F. (2007) Shear-induced unfolding triggers adhesion of von Willebrand factor fibers. Proc. Natl. Acad. Sci. U.S.A. 104, 7899–7903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Springer T. A. (2011) Biology and physics of von Willebrand factor concatamers. J. Thromb. Haemost. 9, 130–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang X., Halvorsen K., Zhang C. Z., Wong W. P., Springer T. A. (2009) Mechanoenzymatic cleavage of the ultralarge vascular protein von Willebrand factor. Science 324, 1330–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang Q., Zhou Y. F., Zhang C. Z., Zhang X., Lu C., Springer T. A. (2009) Structural specializations of A2, a force-sensing domain in the ultralarge vascular protein von Willebrand factor. Proc. Natl. Acad. Sci. U.S.A. 106, 9226–9231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou M., Dong X., Baldauf C., Chen H., Zhou Y., Springer T. A., Luo X., Zhong C., Gräter F., Ding J. (2011) A novel calcium-binding site of von Willebrand factor A2 domain regulates its cleavage by ADAMTS13. Blood 117, 4623–4631 [DOI] [PubMed] [Google Scholar]
- 7. Jakobi A. J., Mashaghi A., Tans S. J., Huizinga E. G. (2011) Calcium modulates force sensing by the von Willebrand factor A2 domain. Nat. Commun. 2, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xu A. J., Springer T. A. (2012) Calcium stabilizes the von Willebrand factor A2 domain by promoting refolding. Proc. Natl. Acad. Sci. U.S.A. 109, 3742–3747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sadler J. E. (2005) New concepts in von Willebrand disease. Ann Rev. Med. 56, 173–191 [DOI] [PubMed] [Google Scholar]
- 10. Lyons S. E., Bruck M. E., Bowie E. J., Ginsburg D. (1992) Impaired intracellular transport produced by a subset of type IIA von Willebrand disease mutations. J. Biol. Chem. 267, 4424–4430 [PubMed] [Google Scholar]
- 11. Sadler J. E., Budde U., Eikenboom J. C., Favaloro E. J., Hill F. G., Holmberg L., Ingerslev J., Lee C. A., Lillicrap D., Mannucci P. M., Mazurier C., Meyer D., Nichols W. L., Nishino M., Peake I. R., Rodeghiero F., Schneppenheim R., Ruggeri Z. M., Srivastava A., Montgomery R. R., Federici A. B. (2006) Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J. Thromb. Haemost. 4, 2103–2114 [DOI] [PubMed] [Google Scholar]
- 12. Hassenpflug W. A., Budde U., Obser T., Angerhaus D., Drewke E., Schneppenheim S., Schneppenheim R. (2006) Impact of mutations in the von Willebrand factor A2 domain on ADAMTS13-dependent proteolysis. Blood 107, 2339–2345 [DOI] [PubMed] [Google Scholar]
- 13. Auton M., Cruz M. A., Moake J. (2007) Conformational stability and domain unfolding of the Von Willebrand factor A domains. J. Mol. Biol. 366, 986–1000 [DOI] [PubMed] [Google Scholar]
- 14. Hampshire D. J., Goodeve A. C. (2011) The international society on thrombosis and haematosis von Willebrand disease database: an update. Semin. Thromb. Hemost. 37, 470–479 [DOI] [PubMed] [Google Scholar]
- 15. Lavergne J. M., Laurian Y., Dudilleux A., Larrieu M. J., Bahnak B. R., Meyer D. (1991) Carrier detection and prenatal diagnosis in 98 families of haemophilia A by linkage analysis and direct detection of mutations. Blood Coagul. Fibrinolysis 2, 293–301 [DOI] [PubMed] [Google Scholar]
- 16. Christophe O., Ribba A. S., Baruch D., Obert B., Rouault C., Niinomi K., Piétu G., Meyer D., Girma J. P. (1994) Influence of mutations and size of multimers in type II von Willebrand disease upon the function of von Willebrand factor. Blood 83, 3553–3561 [PubMed] [Google Scholar]
- 17. Ribba A. S., Voorberg J., Meyer D., Pannekoek H., Pietu G. (1992) Characterization of recombinant von Willebrand factor corresponding to mutations in type IIA and type IIB von Willebrand disease. J. Biol. Chem. 267, 23209–23215 [PubMed] [Google Scholar]
- 18. Greenfield N. J. (2006) Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions. Nat. Protoc. 1, 2527–2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Toumadje A., Alcorn S. W., Johnson W. C., Jr. (1992) Extending CD spectra of proteins to 168 nm improves the analysis for secondary structures. Anal. Biochem. 200, 321–331 [DOI] [PubMed] [Google Scholar]
- 20. Evans E., Ritchie K. (1997) Dynamic strength of molecular adhesion bonds. Biophys. J. 72, 1541–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luken B. M., Winn L. Y., Emsley J., Lane D. A., Crawley J. T. (2010) The importance of vicinal cysteines, C1669 and C1670, for von Willebrand factor A2 domain function. Blood 115, 4910–4913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu T., Lin J., Cruz M. A., Dong J. F., Zhu C. (2010) Force-induced cleavage of single VWFA1A2A3 tridomains by ADAMTS-13. Blood 115, 370–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ying J., Ling Y., Westfield L. A., Sadler J. E., Shao J. Y. (2010) Unfolding the A2 domain of von Willebrand factor with the optical trap. Biophys. J. 98, 1685–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou Y. F., Eng E. T., Nishida N., Lu C., Walz T., Springer T. A. (2011) A pH-regulated dimeric bouquet in the structure of von Willebrand factor. EMBO J. 30, 4098–4111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dudko O. K., Hummer G., Szabo A. (2008) Theory, analysis, and interpretation of single-molecule force spectroscopy experiments. Proc. Natl. Acad. Sci. U.S.A. 105, 15755–15760 [DOI] [PMC free article] [PubMed] [Google Scholar]