

Background: Malonyl-CoA reductase of the CO2 assimilating 3-hydroxypropionate/4-hydroxybutyrate cycle reduces malonyl-CoA to malonic semialdehyde.
Results: Malonyl-CoA reductase complexed with CoA and NADP+ was structurally characterized.
Conclusion: The protein acts as rigid template to press CoA and NADP+ into similar S-shaped, superimposable forms.
Significance: The data indicate how to construct a bispecific cofactor binding site and to engineer a malonyl- into methyl-malonyl-CoA reductase for polyester building block production.
Keywords: Biotechnology, Coenzyme A, Crystal Structure, Enzyme Mechanisms, Nicotinamide
Abstract
Autotrophic members of the Sulfolobales (crenarchaeota) use the 3-hydroxypropionate/4-hydroxybutyrate cycle to assimilate CO2 into cell material. The product of the initial acetyl-CoA carboxylation with CO2, malonyl-CoA, is further reduced to malonic semialdehyde by an NADPH-dependent malonyl-CoA reductase (MCR); the enzyme also catalyzes the reduction of succinyl-CoA to succinic semialdehyde onwards in the cycle. Here, we present the crystal structure of Sulfolobus tokodaii malonyl-CoA reductase in the substrate-free state and in complex with NADP+ and CoA. Structural analysis revealed an unexpected reaction cycle in which NADP+ and CoA successively occupy identical binding sites. Both coenzymes are pressed into an S-shaped, nearly superimposable structure imposed by a fixed and preformed binding site. The template-governed cofactor shaping implicates the same binding site for the 3′- and 2′-ribose phosphate group of CoA and NADP+, respectively, but a different one for the common ADP part: the β-phosphate of CoA aligns with the α-phosphate of NADP+. Evolution from an NADP+ to a bispecific NADP+ and CoA binding site involves many amino acid exchanges within a complex process by which constraints of the CoA structure also influence NADP+ binding. Based on the paralogous aspartate-β-semialdehyde dehydrogenase structurally characterized with a covalent Cys-aspartyl adduct, a malonyl/succinyl group can be reliably modeled into MCR and discussed regarding its binding mode, the malonyl/succinyl specificity, and the catalyzed reaction. The modified polypeptide surrounding around the absent ammonium group in malonate/succinate compared with aspartate provides the structural basis for engineering a methylmalonyl-CoA reductase applied for biotechnical polyester building block synthesis.
Introduction
Autotrophic members of the archaeal order Sulfolobales (Crenarchaeota) use acetyl-CoA/propionyl-CoA carboxylase as the primary CO2 fixing enzyme in the 3-hydroxypropionate/4-hydroxybutyrate cycle (1–6). This cycle starts with acetyl-CoA carboxylation to malonyl-CoA, which is further reduced by malonyl-CoA reductase (MCR)3 catalyzing the following: malonyl-CoA + NADPH + H+ → malonic semialdehyde + NADP+ + CoA (see Fig. 1A) (7). The enzyme is unrelated to the bifunctional malonyl-CoA reductase in some bacteria that catalyzes, in addition, the reduction of malonic semialdehyde to 3-hydroxypropionate (8, 9). Yet, the archaeal enzyme catalyzes also the reduction of succinyl-CoA onwards in the autotrophic cycle (10), which is required for regeneration of the starting molecule acetyl-CoA: succinyl-CoA + NADPH + H+ → succinic semialdehyde + NADP+ + CoA (see Fig. 1A).
FIGURE 1.
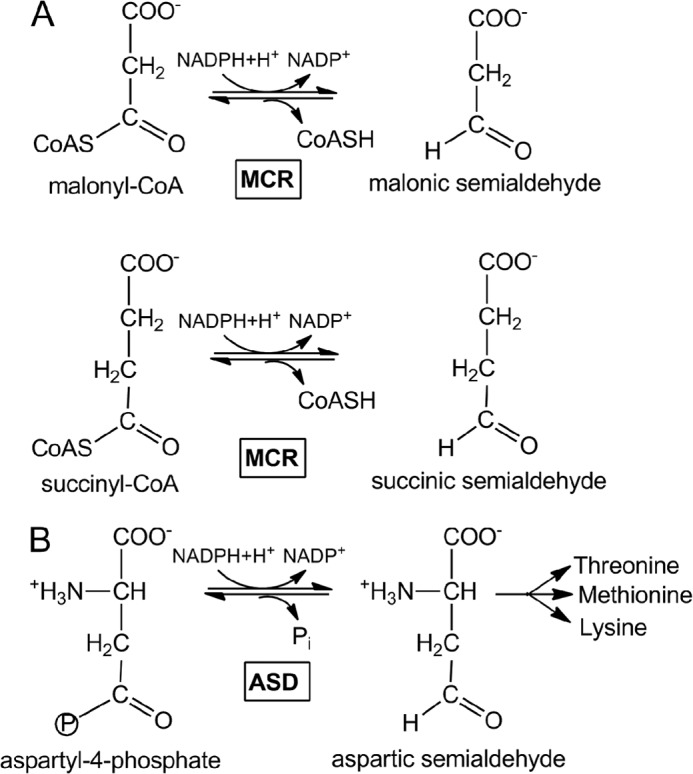
Reactions catalyzed by MCR (A) and ASD (B).
Sequence comparison suggests that the archaeal malonyl-CoA reductase gene (mcr) has evolved from the duplication of a common ancestral aspartate-β-semialdehyde dehydrogenase (ASD) gene (asd) and has since diverged from the parent copy by mutation and selection to achieve its new function. Aspartate-semialdehyde dehydrogenase catalyzes as follows: aspartyl-4-phosphate + NADPH + H+ → aspartic β-semialdehyde + NADP+ + inorganic phosphate (Fig. 1B). The two paralogous MCR and ASD enzymes show ∼40% amino acid sequence identity in the same organism, and their genes mcr and asd have different chromosomal locations. The mcr gene occurs only in autotrophic Sulfolobales underlining its role in the carbon fixation cycle and its neighbor genes are generally not conserved among different species. The asd gene clusters together with threonine synthase, aspartokinase, and ornithine carbamoyltransferase, among other genes, which is in line with its role in the biosynthesis of amino acids of the aspartate family.
MCR is a homotetramer. The monomer (Mr of 39 kDa) is topologically classified as member of the GADPH family. Activity is stimulated by thiols and inactivated by thiol-blocking agents suggesting an essential role of a cysteine residue in catalysis (7). In fact, one conserved cysteine and histidine are crucial for catalysis of this enzyme class. Interestingly, recombinant malonyl-CoA reductase binds small RNA of 60 to 180 nucleotides, which could be a hint for nonspecific cleaving of RNA (7). RNA cleavage has been shown for other metabolic enzymes, e.g. glyceraldehyde-3-phosphate dehydrogenase; however, the physiological role is not known (11, 12).
An x-ray structure was so far not reported for MCR but for homologous enzymes, with the most related being the well characterized aspartate-β-semialdehyde dehydrogenase (ASD) (13). However, MCR catalyzes a different reaction, which might implicate significant differences in the active site architecture and the enzymatic mechanism. In addition, MCR is also of biotechnological interest for the production of polyester building blocks. It may be used either for production of 3-hydroxypropionate (precursor of acrylate) from malonyl-CoA, or for production of 3-hydroxyisobutyrate (precursor of methacrylate) from methylmalonyl-CoA using an engineered enzyme.
For a more profound understanding of substrate binding and the catalyzed reaction and for a rational design of a productive methylmalonyl-CoA reductase detailed structural information is indispensable, which prompted us to initiate an x-ray crystallographic analysis of MCR. The determined structures of substrate-free MCR and of MCR in complex with NADP+ (MCRNADP) and CoA (MCRCoA) reveal insights into the geometry and evolution of a bispecific cofactor binding site.
EXPERIMENTAL PROCEDURES
Heterologous production of the Sulfolobus tokodaii MCR in Escherichia coli
Materials used, strains and culture conditions, preparation of cell extract, enzyme assay, heterologous expression of the malonyl-CoA reductase (mcr) gene from S. tokodaii, production of the protein in E. coli, and purification of heterologously expressed malonyl-CoA reductase have been described (7, 10).
In brief, S. tokodaii (Deutsche Sammlung von Mikroorganismen und Zellkulturen 16993) (14) was grown aerobically and heterotrophically at 75 °C on a chemically defined medium (pH 3.0) with 1 g per liter of glucose (generation time, 6 h). Cells were stored in liquid nitrogen until use. E. coli strain Rosetta 2 (Merck) was grown at 37 °C in Luria-Bertani (LB) medium. Antibiotics were added to E. coli cultures to the following final concentration: ampicillin, 100 μg ml−1; chloramphenicol, 34 μg ml−1.
Harvested cells were resuspended in a 2-fold volume of 50 mm Tris/HCl, pH 7.8, containing 5 mm MgCl2 and 0.1 mg ml−1 DNase I. The cell suspension was passed through a French pressure cell at 137 megapascal and ultracentrifuged (100,000 × g) at 4 °C for 1 h. The cell extract was used immediately or kept frozen at −70 °C. The malonyl-CoA-dependent oxidation of NADPH was followed spectrophotometrically at 365 nm (ϵ365 nm NADPH = 3,400 m−1 cm−1). Similarly, the reductions of succinyl-CoA and of methylmalonyl-CoA were followed. Chromosomal DNA from S. tokodaii was isolated using standard techniques. Two synthetic oligonucleotides were designed to amplify the complete mcr gene using chromosomal DNA from S. tokodaii as a template (for genome, see Ref. 15), and the sequence of the insert was checked. The plasmid pTrc99A was digested with NcoI and BamHI, and the plasmid pTrc99A-Mcr was generated after ligation of the fragment with the mcr gene. A plasmid-derived lac promotor in front of mcr allows expression of the gene after induction of isopropyl thiogalactopyranoside. Competent E. coli Rosetta 2 cells were transformed with pTrc99A-Mcr, grown in a 200-liter fermenter at 37 °C in LB broth containing 100 μg of ampicillin ml−1, and induced at an optical density of 0.6 (1-cm light path) with 0.5 mm isopropyl thiogalactopyranoside. After additional growth for 3 h, the cells were harvested and stored in liquid nitrogen until use.
Purification of the Recombinant Enzyme
The recombinant enzyme was purified from the supernatant of the cell extract after centrifugation at 100,000 × g (starting normally from 10 g (wet weight) of cells of E. coli) in three steps: 1) heat precipitation at 85 °C and concentration by ultrafiltration (Amicon YM30 membrane, Millipore); 2) gel filtration chromatography using a Superdex 200 HR 26/60 gel filtration column (Pharmacia; diameter, 2.6 cm; volume, 320 ml); and 3) Resource Phenyl chromatography using a Resource Phenyl column (1 ml of Resource PHE, Amersham Biosciences). According to SDS-PAGE, the enzyme was pure.
Crystallization and X-ray Structure Determination
MCR from S. tokodaii was concentrated to 15 mg/ml in 10 mm Hepes, pH 7.0, 5 mm MgCl2 (only in some cases used) and 100 mm NaCl and crystallized at a temperature of 18 °C using the sitting drop method. Successful crystallization experiments were undertaken with MCR and MCR supplemented with NADP+ or malonyl/succinyl-CoA. The detailed conditions are listed in Table 1. Data were collected at the SLS-PXII beamline (Villigen) and processed with XDS (16). The structure of MCRsubstrate-free from S. tokodaii was determined at 2.05 Å resolution by the molecular replacement method (17) using the ASD model of Methanococcus jannaschii (18). Model building and refinement were performed with COOT (19) and REFMAC (20), respectively. The obtained MCR model was subsequently applied to solve MCRNADP and MCRCoA structures. Refinement was performed using mostly “medium/loose” non-crystallographic symmetry restraints for the main and side chains of MCRsubstrate-free, MCRNADP, and MCRCoA. Translation, libration, and screw-motion parameters were also refined using one monomer as group. The root mean square deviations between MCRsubstrate-free and MCRNADP, MCRsubstrate-free, and MCRCoA, and MCRNADP and MCRCoA structures are 0.3 Å, 0.4 Å, and 0.3 Å, respectively, using Cα atoms for calculations. Crystallographic data are summarized in Table 1. Figs. 2–4 were generated with PyMOL (Schrodinger, LLC). Atomic coordinates and structure factors of MCRsubstrate-free, MCRNADP, and MCRCoA have been deposited in the Protein Data Bank under codes 4DPK, 4DPL, and 4DPM.
TABLE 1.
X-ray crystallographic statistics
| Crystallization and data collection | MCRsubstrate-free | MCRNADP | MCRCoA |
|---|---|---|---|
| Protein solution | 10 mm HEPES, pH 7.0, 0.1 m NaCl | 10 mm HEPES, pH 7.0, 0.1 m NaCl, 5 mm NADP+ | 10 mm HEPES, pH 7.0, 0.1 m NaCl, 5 mm NADP+, 5 mm succinyl-CoA |
| Conditionsa | 34% MPD, 0.1 m Hepes pH 7.5, 100 mm CaCl2 | 33% PEP 426, 0.1 m NaOAc, pH 4.6, 0.15 m (NH4)2SO4 | 11% PEG 6000, 10 mm MgCl2 |
| Space group | C2 | P212121 | C2221 |
| Unit cell parameter (Å) | 167.1, 81.9, 124.6 | 94.7, 128.8, 140.5 | 111.6, 137.6, 363.0 |
| 105.0° | |||
| Nr. mol/asymmetrical unit | 1 tetramer | 1 tetramer | 1.5 tetramer |
| Detector | MARCCD 225 mm | PILATUS 6 m | MARCCD 225 mm |
| Wavelength (Å) | 0.9764 | 1.0 | 0.9999 |
| Resolution (Å) (highest shell) | 50.0-2.05 (2.1-2.05) | 50.0-1.9 (2.0-1.9) | 50.0-2.4 (2.5-2.4) |
| Rsym (%) | 5.8 (25.8) | 5.5 (45.6) | 6.4 (41.4) |
| I/σ(I) | 15.0 (4.5) | 15.5 (3.2) | 20.5 (3.9) |
| Completeness (%) | 95.8 (74.2) | 99.5 (99.2) | 92.5 (65.4) |
| Redundancy | 4.1 (2.8) | 3.8 (3.8) | 7.0 (4.4) |
| Refinement | |||
| Resolution (Å) (highest shell) | 20.0-2.05 (2.10-2.05) | 20.0-1.9 (1.95-1.9) | 30.0-2.4 (2.46-2.4) |
| Rcryst/Rfree (%) (highest shell) | 18.5/23.1 (23.1/26.3) | 16.3/19.4 (25.2/29.2) | 20.2/24.5 (34.8/38.2) |
| No. of residues | 4 × 360 | 4 × 354 | 6 × 355 |
| No. of solvent molecules | 500 | 891 | 557 |
| r.m.s.d. bond lengths (Å) | 0.017 | 0.021 | 0.019 |
| r.m.s.d. bond angles | 1.58° | 1.82° | 1.72° |
| Average B (Å2) Protein/ligand/solvent | 44.2/NA/39.0b | 34.7/53.3/38.9 | 35.5/43.9/48.7c |
| PDB accession code | 4DPK | 4DPL | 4DPM |
a Crystals were originally found in the JBScreen Classic kit (Jena Bioscience) and optimized by own solutions.
b NA, not available; r.m.s.d., root mean square deviation.
c The values are only related to the polypeptide and CoA of chains A and B.
FIGURE 2.

Structure of MCR from S. todokaii. The MCR homotetramer is arranged as a dimer of dimers (colored in magenta/blue and gray/green). Each monomer is built up of a dinucleotide binding (dark green) and a dimerization domain (light green). The active site cleft lies between them and is occupied in the MCRCoA structure by CoA (drawn as a stick model). Two dimers form an extended interface that includes both cover loops (black arrow) being involved in substrate binding.
FIGURE 3.

Cofactor binding to MCR. A, NADP+ binding in stereo. NADP+ binds in a characteristic S-shaped conformation and is sandwiched between the loops of the C-terminal end of the β-sheet (light gray) and the cover loop (dark gray). Only residues forming hydrogen bonds to the polypeptide are shown. NADP+ is firmly anchored to the polypeptide at the 2′-ribose phosphate binding site. Proton donors comprise Lys41 NH, Gly42 NH, Ser43 NH, Ser43 OγH, and Thr16 Oγ1H as well as via a solvent molecule Ala39 O, Thr16 NH, and Ala15 NH. The 2Fobs − Fcalc electron density (cyan) is drawn at a contour level of 1.3σ. B, CoA binding in stereo. CoA is arranged in an S-shaped, rather compressed conformation (distance between α-phosphate O2A and β-cysteamine N4P, 8.7 Å) and embedded into the same binding site as NADP+. A metal ion seems to be positioned between the α- and β-phosphate and the Tyr187 carbonyl oxygens. The 2Fobs − Fcalc electron density (cyan) is drawn at a contour level of 1.5σ. C, superposition of NADP+ and CoA. The carbons of NADP+ are colored in yellow, and CoA carbons are in green. The distances between the β-phosphate of CoA and the α-phosphate of NADP+ and between their 3′- and 2′-ribose phosphates are ∼1.4 Å.
FIGURE 4.

Binding of the enzyme-malonyl thioacyl adduct. The Cys153-malonyl adduct (malonyl carbons in green) of MCR is modeled on the basis of the corresponding cysteine-aspartyl template of ASD. Although the thioacyl and the carboxylate surroundings of malonyl and aspartyl are highly conserved, the absence of the ammonium group tracks with an exchange of a glutamate and an asparagine in ASD with a tyrosine (Tyr206) and a leucine (Leu152) in MCR, respectively.
RESULTS AND DISCUSSION
Overall Architecture of MCR
The structure of recombinantly produced MCR of S. tokodaii was characterized at 2.05 Å resolution in a substrate-free state (Table 1). The enzyme shows, as expected, a GAPDH-like fold (Fig. 2), the most related family members are ASD, N-acetyl-γ-glutamyl phosphate reductase, and GAPDH with root mean square deviations between 1.4 and 2.6 Å for the corresponding M. jannaschii (Protein Data Bank code 1YS4), Salmonella typhimurium (Protein Data Bank code 2G17), and Bacillus thermophilus (Protein Data Bank code 1GD1) enzymes. The structures of MCR and the most related ASD from M. jannaschii deviate only in a few loop segments in the order of maximally 3 Å except for the loop after helix 204:219 (deviation > 20 Å).
In agreement with previous solution data (7), MCR was found in the crystal structure as a homotetramer that is organized as a dimer of two dimers (Fig. 2). Each monomer is composed of a classical dinucleotide binding domain and a dimerization domain consisting of a mixed six-stranded β-sheet primarily flanked by three α-helices on one side and the dimerization domain of the counter subunit on the other side. The two central β-sheets of the dimerization domains face each other, thereby forming an extended monomer-monomer contact area (Fig. 2). The two dimers of MCR forming the tetramer are stacked on each other at right angles and possess an interface at their centers (Fig. 2) that is conserved in GAPDH-like family members. However, tetramerization is not uniformly maintained (i.e. ASD is a dimer). No electron density was visible for RNA that strongly binds in substantial amounts to recombinant MCR (7) suggesting a flexible binding at the protein surface (if bound at all in the crystalline state).
The substrate binding site of MCR (and the entire GAPDH-like family) is accommodated in a mostly shallow cleft with a size of ∼20 × 10 × 7 Å3 located between the dinucleotide binding and dimerization domains of each monomer (Fig. 2). The cleft extends toward the protein interior forming the active site region with the catalytic residues Cys153 and His248 (MCR of S. todokaii nomenclature) both strictly conserved in the GADPH-like family and reminiscent to thiol proteases (21). The molecular basis for substrate binding has been established by solving the structure of MCRNADP at 1.9 Å resolution and of MCRCoA at 2.4 Å resolution (Table 1). Comparisons between MCRsubstrate-free, MCRNADP, and MCRCoA structures at the substrate binding cleft resulted in Cα deviations of maximally 0.9 Å. They are mostly below 0.3 Å.
NADP+ Binding
The structure of MCRNADP revealed a highly occupied NADP+ molecule in an S-shaped conformation (Fig. 3A), which resembles that found in the ASD-NADP+ and GAPDH-NADP+-structures (22, 23). NADP+ is positioned along the C-terminal side of the central β-sheet of the dinucleotide binding domain partly capped by the protruding cover loop that bridges strand 171:180 and 199:202 of the dimerization domain (Fig. 2). The 2′-phospho-ADP moiety is primarily attached to the dinucleotide recognition loop 14GxxGxxG20. The 2′-phospho group of ribose is accommodated into a well designed pocket formed besides the dinucleotide recognition loop by the expanded hairpin-shaped segment linking strands 37:40 and 82:89. It is anchored to the polypeptide by multiple proton donors partly via a solvent molecule (Fig. 3A). This strong fixation of the phosphate group also provides the molecular basis for the specificity of NADPH against NADH and thus confirms the kinetic data (7). In comparison, the phospho group of the most related M. jannaschii ASD is more exposed and the number of hydrogen bond contacts with the polypeptide significantly reduced compared with MCR. This difference rationalizes its dual specificity for NADPH and NADH as electron donor, albeit still with a clear preference for NADPH (18). As expected, the diphosphate group is sandwiched between the dinucleotide recognition loop and the conserved cover loop region 183SGAGY187; hydrogen bonds are formed between the α-phosphate oxygens and residues Leu18 NH and Tyr187 NH (Fig. 3A). A minor displacement of the cover loop upon NADP+ binding shrinks the binding site of the phenol group of Tyr187, which is rotated about 90° toward the bulk solvent. A crucial function in diphosphate binding is attributed to the second dimer of tetrameric MCR, as mutual interactions between the two cover loops (i.e. Arg153 and Asp194) rigidify their conformations relative to each other (Fig. 2). A similar contact was found in GADPH (23).
The conformation of the catalytically competent ribose-nicotinamide moiety is related to that of other members of the GADPH family. Notably, Asn335, Ser183 (part of the SGxG motif conserved in MCR and ASD), and the β-phosphate are hydrogen-bonded with the amide group of the nicotinamide group and fixes the latter in an optimal position for hydride transfer (Fig. 3A). The hydride transferring C4 atom of NADP+ (and presumably of NADPH) and the thioacyl group of the Cys153 malonyl model (see below) are in van der Waals distance, which supports the functional relevance of the obtained nicotinamide conformation.
CoA Binding
Co-crystallization experiments between MCR and malonyl-CoA or succinyl-CoA resulted in a structure of MCR in complex with CoA (MCRCoA), suggesting that its malonyl/succinyl head group was hydrolyzed during the crystallization process (Fig. 3B). The MCRCoA structure revealed an identical binding site for CoA and NADP+. Similar to NADP+, CoA is present in an S-shaped, rather compressed conformation that is reflected in several intramolecular contacts (Fig. 3B).
When superimposing the MCRNADP and MCRCoA structures, NADP+ and CoA globally align fairly well but not necessarily their analogous parts (Fig. 3C). Accordingly, the binding positions of the adenine rings are identical albeit mutually rotated and the 3′-ribose phosphate group of CoA approximately occupies the site of the 2′-ribose phosphate of NADP+ (Fig. 3B). In addition, the diphosphate and the ribose of CoA are shifted ∼2 Å away from the active site implicating that the β-phosphate of CoA occupies the site of the α-phosphate of NADP+. The pantoate part of CoA approximately occupies the position of the β-phosphate of NADP+, the β-alanine that of ribose, and the β-cysteamine that of the nicotinamide moiety (Fig. 3C).
CoA is primarily anchored to the polypeptide via its 3′-ribose phosphate using the same binding pocket as the 2′-ribose phosphate group of NADP+ and its β-phosphate, which is strongly fixed between the dinucleotide binding and the cover loop (Fig. 3B). The latter anchor site is occupied by a phosphate (sulfate) in the MCRsubstrate-free structure documenting its high phosphate affinity. The residual components of CoA, also the α-phosphate, are essentially connected by unspecific van der Waals contacts. The more flexible pantetheine part is placed in an oversized binding site, and the number of contacts to the polypeptide is small (Fig. 3B). Solely, the terminal β-cysteamine sulfur forms polar interactions to Asn335 and Cys153. The distance between the sulfurs (∼3.1 Å) does not argue for a disulfide bond but rather for a hydrogen bond, similar to the interaction between the Cys153 thiolate and the (protonated) thiol group of CoA. However, in two subunits of the asymmetric unit, the β-cysteamine is largely disordered, and a highly flexible Cys153 adduct appears to be formed. We interpret the electron density as a mixture between Cys153 and CoA in a reduced and in an oxidized disulfide state. A covalent Cys153 CoA adduct has also been found in acetaldehyde dehydrogenase (24). We assume that the binding of malonyl/succinyl-CoA (not only CoA) with the strongly bound malonyl/succinyl (see below) rigidifies the β-cysteamine.
One Binding Site for Both NADP+ and CoA
Enzymatic ping pong reactions are characterized either by covalent polypeptide (coenzyme)-ligand intermediates (25) or by prosthetic groups such as FAD that can store electrons (26). The consecutive binding of two substrates to the corresponding enzyme in principle allow them to share one binding site. Overlapping binding sites for NAD and CoA have been reported on the basis of hydrogen-deuterium exchange and mass spectrometric experiments for acetaldehyde dehydrogenase, another member of the GAPDH family (27, 28). The MCRNADP and MCRCoA structures, presented here, provide detailed insights into the architecture of a bispecific binding site that can host two large and structurally different cofactors. From the viewpoint of molecular engineering, this challenge might be responded by constructing an equivalent and high affinity binding site for the common ADP part of NADP+ and CoA and flexible polypeptide segments that become selectively rearranged and fixed upon NADP and CoA binding, respectively. ADP as major component for NADP+ binding has also been experimentally verified by ASD of M. jannaschii structurally related to MCR (18). Interestingly, nature adopts another strategy by essentially utilizing the protein as a rigid template that presses the conformationally variable coenzymes into highly similar S-shaped conformations (Fig. 3C). CoA and NADP+ are surprisingly well superimposable, implicating that the β-phosphate of CoA is positioned on the α-phosphate in NADP+ and the 3′- on the 2′-ribose phosphate of CoA and NADP+, respectively. This shape/size-controlled cofactor binding forces the common ADP parts into different binding modes but smartly considers the increased length of pantetheine compared with the more voluminous ribose-nicotinamide part as well as the displacement of 3′- and 2′-ribose phosphate groups in CoA and NADP+, respectively (Fig. 3C). In addition, the adjusted conformation allows both coenzymes to use the same tailor made ribose phosphate binding pocket as major fixation point (Fig. 3, A and B), which ensures, in parallel, the specificity for NADPH relative to NADH (7). Although remarkably superimposable, the slightly different surface profile of NADP+ and CoA is accounted by tiny side chain rearrangements of contacting residues below 1.0 Å and by evading into the free space of the adjacent solvent.
The described template-governed cofactor shaping requires a strongly fixed and rigid substrate binding cleft that is presumably provided by the compact globular domains and the multiple domain-domain and oligomeric interactions (Fig. 2). A crucial factor might also be the stabilization of the cover loop by tetramerization.
Evolution of the Bispecific Binding Site
MCR has evolved from ASD and, consequently, the bispecific binding site for NADP+ and CoA originate from an NADP+ binding site. The direction of evolution is weakly reflected on the atomic scale by the increased and more equally distributed enzyme-NADP+ compared with the enzyme-CoA interactions and the unusually compressed conformation of CoA that is related to that of NADP+ (Fig. 3C). CoA is normally found in a more elongated conformation even in those CoA-dependent enzymes that use, similar to MCR a dinucleotide binding domain for CoA binding (29, 30).
Comparison between ASD-NADP+ and MCR-NADP+ structures should provide information about how an NADP+ is converted into an NADP+ and CoA binding site. However, analysis is intricate, as the additional CoA specificity is accomplished by many amino acid exchanges at and far away from the cofactor binding site; three pronounced features are described in more detail. First, the shape/size of the ribose-phosphate binding pocket is modified by the conformationally changed hairpin segment between strands 37:40 and 82:89, in particular, due to the exchange of two residues in ASD by one residue (Gly42) in MCR. This change provides space at the ribose phosphate binding site for the 3′-phosphate of CoA (Fig. 3B). For forming optimal polypeptide-phosphate interactions, the 2′-ribose phosphate of NADP+ is moved toward 3′-ribose phosphate of CoA compared with that of the ASD-NADP+ complexes. As a consequence, the adenine ring has to be rotated, which is compensated by the exchange of a small residue at position 41 in ASD by lysine in MCR and of a medium-sized residue such as leucine or threonine at position 91 by an alanine. This finding indicates an adaptation of the NADP+ position to chemical restraints imposed by the CoA structure. Second, the interactions between the β-/α-phosphate of CoA/NADP+ and Tyr187 implicates a small rearrangement of this region primarily caused by the mentioned altered hairpin segment and a hydrogen bond to Asp194 of the partner dimer (not present in dimeric ASD). Thus, tetramerization might also be used as a tool to optimize the common binding site for the β-/α-phosphate of CoA/NADP+. Third, the ribose-nicotinamide binding site is geometrically conserved but appears to be more unpolar in MCR than in ASD reflecting the increased hydrophobicity of the pantetheine part of CoA. Thus, residues similar to alanine and serine in positions 18, 86, and 111 are replaced by a leucine and two prolines. In addition, the polar amide group of Asn94 is turned away in MCR but directed to the nicotinamide binding site in ASD. Asn94 becomes hydrogen bonded to Tyr23 in MCR that is substituted in ASD by an unpolar residue, mostly a methionine.
Substrate Binding and Catalyzed Reaction
A reaction cycle for the MCR reaction was already postulated (7), consisting of a nucleophilic attack of the Cys153-thiolate onto the malonyl/succinyl-CoA thioester bond forming a covalent enzyme-malonyl/succinyl adduct. Its relatively stable thioacyl bond is subsequently reduced to malonic/succinic semialdehyde by NADPH (Fig. 1). The presented structural data support this proposal and revealed a remarkable relationship between the active sites of MCR and ASD that allows a direct application of the wealth of structural and mechanistic data available for ASD (13). This high degree of conservation, despite substantial differences of the substrates used, becomes comprehensible when considering similarities between the aspartate and malonate/succinate and the CoA and NADP+ structures as well as between the catalyzed reactions. In either case, an activated ester is reduced to an aldehyde with NADPH as electron donor (Fig. 1). Accordingly, the catalytic key players of MCR and ASD, the nucleophile Cys153, the acid/base catalyst His248, and most of the residues binding malonyl/succinyl or aspartyl, are found in similar positions. Therefore, a covalent MCR-malonyl/succinyl adduct can be transferred from the superimposed ASD-aspartyl template (31) without any interference with the surrounding protein matrix of MCR (Fig. 4). In addition, the distance of ca. 3.5 Å between the thioacyl carbon and the hydride donating C4 atom of the experimentally determined NADP+ is reasonable for a hydride transfer and justifies the modeling operation. Consequently, the experimentally determined CoA and the deduced malonyl/succinyl binding positions (18) allow a reliable modeling of malonyl-CoA and, together with the found NADP+ position, the postulation of a structure-based enzymatic mechanism for the MCR reaction (Fig. 5).
FIGURE 5.

Proposed enzymatic mechanism. A, malonyl-CoA is bound and subsequently attacked by the Cys153 thiolate forming a tetrahedral intermediate. The negative charge of the oxyanion is stabilized by basic residues and by the positively charged N-terminal end of helix 152:168. B, the tetrahedral intermediate is converted to a thioacyl adduct, CoA is released, and NADPH is bound. CoA and NADPH use an identical binding site. C, NADPH transfers a hydride from the B-side to the thioacyl carbon forming a hemithioacetal intermediate, which is converted to the product malonic semialdehyde, thereby restoring Cys153 (D).
In complete agreement with the structure of the ASD-aspartyl thioacyl adduct (22, 31), the malonyl group is mainly anchored to the polypeptide via its carboxylate group by forming hydrogen bonds with the strictly conserved residues Gln180, Arg241, and His248 (Fig. 4). In addition, Arg241 appears to be the key amino acid to provide the promiscuity of MCR. The conformational variability of its side chain and of the loop following strand 233:241 (Fig. 4) allows accommodation of both malonyl- and succinyl-CoA that are distinguished by one methylene group (Fig. 1) (10).
The thioacyl oxygen of the modeled enzyme malonyl adduct most likely interacts with Arg114 Nη1/Nη2 and Lys209 Nζ (after rigidification upon substrate binding) via a modeled water molecule and with Cys153 NH and/or Thr154 NH, both located at the positively charged N-terminal end of helix 152:168 (Fig. 4). This positively charged oxygen surrounding is well suited for stabilizing the tetrahedral oxoanion intermediate generated during catalysis (Fig. 5), either directly by charge compensation, or by activating the water molecule that may act as proton donor/acceptor. Oxoanion stabilization might also be the reason for the stimulation of the catalyzed reaction with Mg2+ ions (7).
Malonyl/succinyl is mainly distinguished from aspartyl by the absent positively charged ammonium group (Fig. 1), which interacts in ASD with invariant glutamate and asparagine side chains (18). In MCR, these residues are replaced by a tyrosine (Tyr206) and a leucine (Leu152); both strictly conserved residues contact the methylene group of malonyl/succinyl and are interpreted as the response of the more apolar character of malonyl/succinyl compared with aspartyl. Tyr206, in contact to Leu152, Thr154 (Ser in ASD), Gln180, and Leu202, maintains on one hand the hydrogen bond network important for malonyl carboxylate binding and catalysis and on the other hand maintains the position/conformation of Leu152 in a highly polar environment (Fig. 4). Its introduction required an unpredictable rearrangement of the N-terminal side of helix 203:220 compared with ASD (i.e. Ile → Gly203, Lys → Asp204). The resulting outward shift of Tyr206 prevents an interference of its long side chain with malonyl binding. In contrast, the stretch containing Leu152 is nearly identical in MCR and ASD reflecting its importance for catalysis (next residue, Cys153). Leu152 would not only interfere with the ammonium group of aspartyl-4-phosphate but also with its phosphate group. A possible ASD activity is thus suppressed in agreement with kinetic data, indicating that NADP+ is hardly reduced by malonic semialdehyde and phosphate (7).
Engineered Enzyme
A planned enzyme-based methacrylate synthesis pathway involves the reduction of 2-methylmalonyl-CoA to the corresponding semialdehyde, which requires the transformation of MCR to a productive methylmalonyl-CoA reductase. The structure of MCR is essential for substrate engineering as the exact surrounding of the new methyl group differs from that of ASD in an unpredictable manner. Broad mutational experiments are promising especially as attractive side chains exist and no significant large-scale conformational changes are expected upon substrate binding. The increased hydrophobicity around the methylene group of malonyl/succinyl in MCR compared with the ammonium group in ASD is, in principle, favorable; however, Leu152 would interfere with a methyl substituent, suggesting its exchange to a smaller residue such as valine, threonine, or alanine. Other side chains in the environment of the methyl group are Tyr206 (∼3 Å), Lys209 (4 Å), and Pro111 (6 Å). Pro111 is especially interesting because of its more indirect participation in substrate binding and its position in a loop that might be variably designed.
Acknowledgments
We thank Hartmut Michel for continuous support and the staff of the beamline PXII at the Swiss Light Source (Villigen) for help during data collection.
This work was supported by Deutsche Forschungsgemeinschaft (Fu 118/15-4) and Evonik Industries AG.
The atomic coordinates and structure factors (codes 4DPK, 4DPL, and 4DPM) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- MCR
- malonyl (succinyl)-CoA reductase
- ASD
- aspartate-β-semialdehyde dehydrogenase
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- CoA
- coenzyme A.
REFERENCES
- 1. Burton N. P., Williams T. D., Norris P. R. (1999) Carboxylase genes of Sulfolobus metallicus. Arch. Microbiol. 172, 349–353 [DOI] [PubMed] [Google Scholar]
- 2. Chuakrut S., Arai H., Ishii M., Igarashi Y. (2003) Characterization of a bifunctional archaeal acyl coenzyme A carboxylase. J. Bacteriol. 185, 938–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Norris P., Nixon A., Hart A. (1989) Acidophilic, mineral-oxidizing bacteria: the utilization of carbon dioxide with particular reference to autotrophy in Sulfolobus in Microbiology of Extreme Environments and Its Potential for Biotechnology (Da Costa M. S., Duarte J. C., Williams R. A. D., eds), pp. 24–39, Elsevier, London, United Kingdom: ) Elsevier, London, United Kingdom [Google Scholar]
- 4. Menendez C., Bauer Z., Huber H., Gad'on N., Stetter K. O., Fuchs G. (1999) Presence of acetyl coenzyme A (CoA) carboxylase and propionyl-CoA carboxylase in autotrophic Crenarchaeota and indication for operation of a 3-hydroxypropionate cycle in autotrophic carbon fixation. J. Bacteriol. 181, 1088–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ishii M., Miyake T., Satoh T., Sugiyama H., Oshima Y., Kodama T., Igarashi Y. (1996) Autotrophic carbon dioxide fixation in Acidianus brierleyi. Arch. Microbiol. 166, 368–371 [DOI] [PubMed] [Google Scholar]
- 6. Berg I. A., Kockelkorn D., Buckel W., Fuchs G. (2007) A 3-hydroxypropionate/4-hydroxybutyrate autotrophic carbon dioxide assimilation pathway in Archaea. Science 318, 1782–1786 [DOI] [PubMed] [Google Scholar]
- 7. Alber B., Olinger M., Rieder A., Kockelkorn D., Jobst B., Hügler M., Fuchs G. (2006) Malonyl-coenzyme A reductase in the modified 3-hydroxypropionate cycle for autotrophic carbon fixation in archaeal Metallosphaera and Sulfolobus spp. J. Bacteriol. 188, 8551–8559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hügler M., Menendez C., Schägger H., Fuchs G. (2002) Malonyl-coenzyme A reductase from Chloroflexus aurantiacus, a key enzyme of the 3-hydroxypropionate cycle for autotrophic CO(2) fixation. J. Bacteriol. 184, 2404–2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zarzycki J., Fuchs G. (2011) Coassimilation of organic substrates via the autotrophic 3-hydroxypropionate bi-cycle in Chloroflexus aurantiacus. Appl. Environ. Microbiol. 77, 6181–6188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kockelkorn D., Fuchs G. (2009) Malonic semialdehyde reductase, succinic semialdehyde reductase, and succinyl-coenzyme A reductase from Metallosphaera sedula: enzymes of the autotrophic 3-hydroxypropionate/4-hydroxybutyrate cycle in Sulfolobales. J. Bacteriol. 191, 6352–6362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Evguenieva-Hackenberg E., Schiltz E., Klug G. (2002) Dehydrogenases from all three domains of life cleave RNA. J. Biol. Chem. 277, 46145–46150 [DOI] [PubMed] [Google Scholar]
- 12. Cieśla J. (2006) Metabolic enzymes that bind RNA: yet another level of cellular regulatory network? Acta Biochim. Pol. 53, 11–32 [PubMed] [Google Scholar]
- 13. Viola R. E., Faehnle C. R., Blanco J., Moore R. A., Xuying L., Arachea B. T., Pavlovsky A. G. (2011) The Catalytic Machinery of a Key Enzyme in Amino Acid Biosynthesis. Amino Acids 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Suzuki T., Iwasaki T., Uzawa T., Hara K., Nemoto N., Kon T., Ueki T., Yamagishi A., Oshima T. (2002) Sulfolobus tokodaii sp. nov. (f. Sulfolobus sp. strain 7), a new member of the genus Sulfolobus isolated from Beppu Hot Springs, Japan. Extremophiles 6, 39–44 [DOI] [PubMed] [Google Scholar]
- 15. Kawarabayasi Y., Hino Y., Horikawa H., Jin-no K., Takahashi M., Sekine M., Baba S., Ankai A., Kosugi H., Hosoyama A., Fukui S., Nagai Y., Nishijima K., Otsuka R., Nakazawa H., Takamiya M., Kato Y., Yoshizawa T., Tanaka T., Kudoh Y., Yamazaki J., Kushida N., Oguchi A., Aoki K., Masuda S., Yanagii M., Nishimura M., Yamagishi A., Oshima T., Kikuchi H. (2001) Complete genome sequence of an aerobic thermoacidophilic crenarchaeon, Sulfolobus tokodaii strain7. DNA Res. 8, 123–140 [DOI] [PubMed] [Google Scholar]
- 16. Kabsch W. (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 26, 795–800 [Google Scholar]
- 17. Kissinger C. R., Gehlhaar D. K., Fogel D. B. (1999) Rapid automated molecular replacement by evolutionary search. Acta Crystallogr. D Biol. Crystallogr. 55, 484–491 [DOI] [PubMed] [Google Scholar]
- 18. Faehnle C. R., Ohren J. F., Viola R. E. (2005) A new branch in the family: structure of aspartate-β-semialdehyde dehydrogenase from Methanococcus jannaschii. J. Mol. Biol. 353, 1055–1068 [DOI] [PubMed] [Google Scholar]
- 19. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 20. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 21. Storer A. C., Ménard R. (1994) Catalytic mechanism in papain family of cysteine peptidases. Methods Enzymol. 244, 486–500 [DOI] [PubMed] [Google Scholar]
- 22. Faehnle C. R., Le Coq J., Liu X., Viola R. E. (2006) Examination of key intermediates in the catalytic cycle of aspartate-β-semialdehyde dehydrogenase from a gram-positive infectious bacteria. J. Biol. Chem. 281, 31031–31040 [DOI] [PubMed] [Google Scholar]
- 23. Skarzyński T., Moody P. C., Wonacott A. J. (1987) Structure of holo-glyceraldehyde-3-phosphate dehydrogenase from Bacillus stearothermophilus at 1.8 A resolution. J. Mol. Biol. 193, 171–187 [DOI] [PubMed] [Google Scholar]
- 24. Smith L. T., Kaplan N. O. (1980) Purification, properties, and kinetic mechanism of coenzyme A-linked aldehyde dehydrogenase from Clostridium kluyveri. Arch. Biochem. Biophys. 203, 663–675 [DOI] [PubMed] [Google Scholar]
- 25. Ferrer J. L., Jez J. M., Bowman M. E., Dixon R. A., Noel J. P. (1999) Structure of chalcone synthase and the molecular basis of plant polyketide biosynthesis. Nat. Struct. Biol. 6, 775–784 [DOI] [PubMed] [Google Scholar]
- 26. Pejchal R., Sargeant R., Ludwig M. L. (2005) Structures of NADH and CH3-H4folate complexes of Escherichia coli methylenetetrahydrofolate reductase reveal a spartan strategy for a ping-pong reaction. Biochemistry 44, 11447–11457 [DOI] [PubMed] [Google Scholar]
- 27. Lei Y., Pawelek P. D., Powlowski J. (2008) A shared binding site for NAD+ and coenzyme A in an acetaldehyde dehydrogenase involved in bacterial degradation of aromatic compounds. Biochemistry 47, 6870–6882 [DOI] [PubMed] [Google Scholar]
- 28. Manjasetty B. A., Powlowski J., Vrielink A. (2003) Crystal structure of a bifunctional aldolase-dehydrogenase: sequestering a reactive and volatile intermediate. Proc. Natl. Acad. Sci. U.S.A. 100, 6992–6997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wolodko W. T., Fraser M. E., James M. N., Bridger W. A. (1994) The crystal structure of succinyl-CoA synthetase from Escherichia coli at 2.5-A resolution. J. Biol. Chem. 269, 10883–10890 [DOI] [PubMed] [Google Scholar]
- 30. Ricagno S., Jonsson S., Richards N., Lindqvist Y. (2003) Formyl-CoA transferase encloses the CoA binding site at the interface of an interlocked dimer. EMBO J. 22, 3210–3219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blanco J., Moore R. A., Viola R. E. (2003) Capture of an intermediate in the catalytic cycle of l-aspartate-β-semialdehyde dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 100, 12613–12617 [DOI] [PMC free article] [PubMed] [Google Scholar]