

Background: Increasing HDL-c through ApoA-I expression is hypothesized to reduce cardiovascular deaths significantly.
Results: Genes that regulate hepatocyte ApoA-I secretion were identified using 21,789 siRNAs.
Conclusion: Forty genes of interest were confirmed as regulators of ApoA-I production by hepatocytes.
Significance: This study provides functional genomics-based data for exploring new mechanisms by which ApoA-I levels may be regulated.
Keywords: Apolipoproteins, Hepatocyte, High Density Lipoprotein (HDL), High Throughput Screening (HTS), siRNA, Coronary Artery Disease, Farnesyltransferase
Abstract
Control of plasma cholesterol levels is a major therapeutic strategy for management of coronary artery disease (CAD). Although reducing LDL cholesterol (LDL-c) levels decreases morbidity and mortality, this therapeutic intervention only translates into a 25–40% reduction in cardiovascular events. Epidemiological studies have shown that a high LDL-c level is not the only risk factor for CAD; low HDL cholesterol (HDL-c) is an independent risk factor for CAD. Apolipoprotein A-I (ApoA-I) is the major protein component of HDL-c that mediates reverse cholesterol transport from tissues to the liver for excretion. Therefore, increasing ApoA-I levels is an attractive strategy for HDL-c elevation. Using genome-wide siRNA screening, targets that regulate hepatocyte ApoA-I secretion were identified through transfection of 21,789 siRNAs into hepatocytes whereby cell supernatants were assayed for ApoA-I. Approximately 800 genes were identified and triaged using a convergence of information, including genetic associations with HDL-c levels, tissue-specific gene expression, druggability assessments, and pathway analysis. Fifty-nine genes were selected for reconfirmation; 40 genes were confirmed. Here we describe the siRNA screening strategy, assay implementation and validation, data triaging, and example genes of interest. The genes of interest include known and novel genes encoding secreted enzymes, proteases, G-protein-coupled receptors, metabolic enzymes, ion transporters, and proteins of unknown function. Repression of farnesyltransferase (FNTA) by siRNA and the enzyme inhibitor manumycin A caused elevation of ApoA-I secretion from hepatocytes and from transgenic mice expressing hApoA-I and cholesterol ester transfer protein transgenes. In total, this work underscores the power of functional genetic assessment to identify new therapeutic targets.
Introduction
Coronary artery disease (CAD)2 is one of the major causes of morbidity and mortality worldwide. About 800,000 new cases of myocardial infarction and 500,000 recurrent myocardial infarctions occur each year in the United States whereby dyslipidemia is a major risk factor for CAD and myocardial infarction. Although lowering LDL cholesterol (LDL-c) is a primary therapeutic approach through administration of statin drugs, effective treatment of CAD and dyslipidemia should include treatment of low HDL-c levels. Low HDL-c is associated with a substantial increase in risk of CAD (1), and several epidemiological and secondary prevention trials strongly support the hypothesis that agents that increase HDL-c and reduce triglycerides produce significant reduction in death due to cardiovascular events, including nonfatal and fatal myocardial infarction (1). The Veterans Affairs High-Density Lipoprotein Intervention Trial (2) demonstrated that gemfibrozil, a peroxisome proliferator-activated receptor α agonist, elevated HDL-c and lowered triglycerides without lowering LDL-c and provided a 22% reduction in relative risk of major coronary events in patients with coronary artery disease. Fenofibrate, another peroxisome proliferator-activated receptor α agonist, has been shown in the Diabetes Atherosclerosis Intervention Study to be effective at lowering triglycerides and LDL cholesterol and increasing HDL cholesterol in patients with non-insulin-dependent diabetes mellitus. Furthermore, treatment with fenofibrate was associated with a reduction in angiographic progression of coronary artery disease in non-insulin-dependent diabetes mellitus (3). Indeed, the National Cholesterol Education Program recommends that HDL-c be screened as an independent risk factor for CAD. At present, an optimum therapy for the treatment of low HDL-c is unavailable. The marketed weak peroxisome proliferator-activated receptor α agonists (fibrates) produce a moderate (10–15%) elevation in plasma HDL-c, and the newer class of promising cholesterol ester transfer protein (CETP) drugs is still in clinical development.
HDL-c is a small, high density circulating lipoprotein particle comprised of phospholipid, cholesterol, and triglycerides with myriad associated proteins. The major structural apolipoprotein associated with HDL-c is apolipoprotein A-I (ApoA-I). ApoA-I is synthesized and secreted by the liver and intestine, and upon secretion, lipid-poor ApoA-I and nascent HDL particles must acquire lipid to form mature HDL-c particles. Mechanistically, ApoA-I mediates the acquisition of phospholipids and unesterified cholesterol through cellular efflux of phospholipids and cholesterol via the transporter adenosine triphosphate-binding cassette protein A-1. Subsequently, lecithin-cholesterol acyltransferase is activated by ApoA-I to transfer fatty acid from phosphatidylcholine to unesterified cholesterol, producing cholesteryl ester, which drives the movement of unesterified cholesterol into HDL. This reverse cholesterol transport mechanism is believed to protect against or resolve atherosclerosis by relieving the cholesterol burden from macrophages in the vasculature and returning the cholesterol to the liver for catabolism through the bile acid synthetic route. The importance of ApoA-I in protecting vessels against cholesterol-laden atherosclerotic plaques was underscored by significant reduction in atheroma volume of patients suffering from acute coronary syndrome upon infusion of an ApoA-I variant peptide mimetic for only 5 weeks (4). Thus, discovering molecular mechanisms involved either in increased expression, stability, or secretion of ApoA-I from liver would provide potential therapeutic targets for elevating levels of circulating HDL-c.
Our approach to identify new mechanisms that are involved in ApoA-I production and secretion was to query the whole genome through siRNA knockdown technology coupled with measuring hepatocyte production of ApoA-I. Genes of interest found by elevation of ApoA-I in culture media upon mRNA knockdown were prioritized using a convergence of information, including literature-based human genetic associations with HDL-c levels, liver- and intestine-specific expression patterns, druggability assessments, and pathway analysis. Known and novel genes encoding secreted enzymes, proteases, G-protein-coupled receptors, metabolic enzymes, ion transporters, and proteins of unknown function were identified and evaluated as potential new therapeutic targets. One gene of interest, farnesyltransferase (FNTA), was further tested for its role in controlling hepatic ApoA-I secretion by using siRNA and a compound inhibitor, manumycin A, in a cell-based and preclinical in vivo model of human lipoprotein metabolism.
EXPERIMENTAL PROCEDURES
Cell Culture
The hepatocarcinoma cell line HepG2 (ATCC, Manassas, VA) was cultured in 3:1 DMEM:F-12 (Invitrogen), 10% FBS (Invitrogen), and 1× penicillin-streptomycin (Invitrogen). Confluent cells were treated with manumycin A (Sigma) at a variety of concentrations.
Transfection of siRNA Library
The screening consisted of reverse transfection of 5000 cells/well with 0.25 μl/well Dharmafect 3 and 50 nm Dharmacon SMARTpool siRNAs (four individual siRNAs; 12.5 nm each; Dharmacon, Lafayette, CO). Reverse transfection occurred on day 1 followed by a 72-h incubation with siRNA. To determine the impact of siRNA knockdown on secretion of ApoA-I, media were changed at 72 h post-transfection, and conditioned medium was analyzed 24 h later for ApoA-I by ELISA (Mabtech, Inc., Mariemont, OH) and viability by CellTiter-Glo (Promega, Madison, WI) according to the manufacturer's protocols. Culture media were diluted 1:20 for ApoA-I ELISA measurements.
ApoA-I Assay Validation
A 3-day, three plates per day statistical validation study was performed according to the Eli Lilly and Company assay optimization and validation procedures to assess ApoA-I signal separation between a non-targeting control (NTC) and both a positive (adrenergic receptor α1A (ADRA1A)) and a negative control (ApoA-I) (5). Two Z′ values were calculated from the controls on each plate with an acceptance criterion of Z′ ≥ 0.4. Acceptance criteria were also applied to individual plates during the screen as follows: ADRA1A to NTC ratio ≥1.5, ADRA1A coefficient of variation ≤25%, NTC coefficient of variation ≤25%, and Z′ for NTC and ApoA-I separation ≥0.3.
Primary Screen
In partnership with Dharmacon Screening Services (Lafayette, CO), the conditions described above were validated in full-screen scale. Subsets of the whole genome were screened sequentially starting with the G Protein-coupled Receptor (516) and Kinase (800) siRNA libraries followed by the Druggable Genome library (7317), and screening was completed with the Rest of Genome library (14,000). In total, 21,789 human genes were targeted with Dharmacon SMARTpools (four siRNAs/well) in triplicate where 80 SMARTpools were evaluated per plate, and 880 plates were used to screen the genome libraries in triplicate.
Statistical Analysis
Genes were delivered to triplicate plates in order by family, and as such, genes on a particular plate could be highly correlated. Therefore, the typical approach of estimating within-plate variability and identifying hits relative to the variability on each plate was not used. To avoid potential plate bias and make the best use of the replicates, ApoA-I levels were instead analyzed by a model that averages across replicate plates to obtain an estimate of gene-to-gene variability for each run. The model was applied to within-plate -fold changes relative to NTC to remove plate effects. The run-specific estimates of gene-to-gene variability were used to calculate Z-scores that express the -fold change of a particular gene relative to NTC in units of standard deviation. Genes with Z-score >2 or <−2 were considered hits. Cell viability was also expressed as -fold change relative to NTC, and hits with -fold change <0.6 or >1.4 were considered to have significantly altered cell viability and were therefore excluded.
FNTA Targeting and Inhibition
HepG2 cells were transfected with 30 nm siRNA: Dharmacon siRNA1, D-008807-02, 5′-GAAAGUGCAUGGAACUAUU; siRNA2, D-008807-03, 5′-GAAAAUGACUCACCAACAA; siRNA3, D-008807-04, 5′-CCAAAGAUACUUCGUUAUU; and siRNA4, D-008807-18, 5′-UGGCAUCAUAGGCGAGUAU targeting FNTA mRNA (NCBI Reference Sequence NM_00207). Cells were also transfected with ADRA1A siRNA SMARTpool M-005419-00 (5′-GCAGAAAGCAGUCUUCCAA, 5′-UGAGCGCUCUCGAGGAGUA, 5′-UGACAAGAACCAUCAAGUU, and 5′-GACCAAUCCUCCUGUACCA) targeting ADRA1A mRNA (NCBI Reference Sequence NM_000680)), ApoA-I siRNA SMARTpool M-010994-00 (5′-GUACGUGGAUGUGCUCAAA, 5′-UGAGCGCUCUCGAGGAGUA, 5′-GGGAUAACCUGGAAAAGGA, and 5′-UAAAGCUCCUUGACAACUG) targeting ApoA-I mRNA (NCBI Reference Sequence NM_000039), or non-targeting control siRNA pool number 2 (D-001206-14-05) using Dharmafect 3 transfection reagent according to the manufacturer's instructions. Media were changed at 72 h post-transfection, and conditioned medium was analyzed 24 h later for ApoA-I by ELISA (Mabtech, Inc.) and viability by CellTiter-Glo (Promega). Overexpression of FNTA protein was conducted by transient transfection of full-length human farnesyltransferase cDNA, MHS1010-74254 Human Mammalian Gene Collection Verified Full-length cDNA (IRAT) clone ID 3850453 (Thermo Fisher, Waltham, MA), for 96 h using FuGENE HD at a ratio of 4.5:1 (Promega, Madison, WI) into HepG2 cells.
Quantitative PCR Analysis
Total RNA was isolated using the RNeasy Plus kit from (Qiagen, Valencia, CA). One microgram of total RNA was converted to cDNA using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Carlsbad, CA). Quantitative PCR was performed using a standard curve method on a 7900HT instrument (Applied Biosystems). Twenty-microliter PCRs were prepared containing 1× Universal Master Mix (catalogue number 4305719, Applied Biosystems); either 1× FNTA gene expression assay (Hs00357739_m1 Applied Biosystems), ApoA-I gene expression assay (Hs00163641_m1, Applied Biosystems), or β-actin gene expression assay (Hs99999903_m1); and 4 μl of template cDNA diluted 1:100 in 10 mm Tris (pH 8.0). PCR conditions for FNTA and ACTB were as follows: 50 °C for 2 min and 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The data from FNTA were normalized to ACTB and calibrated relative to non-targeting control.
Mass Spectrometry Determination of ApoA-I and Apolipoprotein B (ApoB) Levels
Apolipoproteins from the culture media were captured by the affinity resin Liposorb as described previously (6, 7) with modifications. Briefly, 400 μl of HepG2 cell supernatants were spiked with 1.67 μg of 15N-labeled human ApoA4 as internal standard and then incubated in the presence of 4 mg of Liposorb for 30 min at 4 °C with constant shaking. Unbound proteins were removed by centrifugation of the Liposorb suspension through a 0.22-μm Captiva filter plate at 2000 rpm for 5 min followed by two washes with 100 mm ammonium bicarbonate containing 5 mm EDTA (ABCE). After resuspension of the Liposorb, proteins were denatured by incubation with 8 m urea in ABCE for 15 min at 55 °C and then digested with Trypsin Gold overnight at 37 °C with constant shaking (digestion mixture contained 2 μg of Trypsin Gold, 1.6 m urea, and 0.01% Nonidet P-40 in ABCE) after which samples were filtered through a 0.45-μm membrane filter before analysis by LC-MS/MS.
Tryptic peptides were separated by an HPLC system (Thermo Finnigan, Waltham, MA) on a C18 XBridge column (2.5 μm; 2.1 × 50 mm) using a two-solvent gradient system (solvent A, 0.1% formic acid in H2O; solvent B, 0.1% formic acid in acetonitrile) consisting of the following step gradients maintained at 50 °C: 100% A at 200 μl/min for 1 min, 10% B at 200 μl/min for 5.1 min, 15% B at 200 μl/min for 5 min, 30% B at 200 μl/min for 3 min, 35% B at 200 μl/min for 3.3 min, 80% B at 200 μl/min for 0.1 min, 80% B at 600 μl/min for 0.9 min, and 100% A at 600 μl/min for 1.5 min followed by 100% A at 200 μl/min for 0.1 min.
Two peptides from ApoA-I (DYSQFEGSALGK, ApoA1-52; AKPALEDLR, ApoA1-231), and two peptides from ApoB (IADFELPTIIVPEQTIEIPSIK, ApoB-3847; FSVPAGIVIPSFQALTAR, ApoB-3869) were simultaneously measured along with one peptide from 15N-labeled ApoA4 (LEPYADQLR, h_N15A4_135) that was used for normalization of differences in sample recovery and instrument performance.
Positive ion mass spectrometry was carried out using an LTQ ion trap mass spectrometer equipped with an electrospray ionization source (Thermo Finnigan) in multiple reaction monitoring mode. The entire effluent of the column was directed to the electrospray ionization source between 2.5 and 17.4 min of the HPLC run, whereas the rest was diverted away from the mass spectrometer. The m/z selection values for each peptide are given within parentheses as indicated (m/z value for the parent ion/isolation window → m/z values for each daughter ion/isolation window in mass units): apoA1_52 (701.26/4 → 562.29/3, 809.42/4, 1024.51/4), apoA1_231 (507.09/3 → 814.45/4, 726.38/4, 288.70/3), apoB_3847 (1234.46/4 → 1467.85/4, 1354.76/4, 1779.05/4), apoB_3869 (938.11/4 → 991.54/4, 771.95/4, 1203.69/4), and h_N15A4_135 (559.58/3 → 437.69/3, 874.37/4, 245.63/3).
For data analysis, the peak areas of ApoA-I and ApoB peptides were calculated using Xcalibur 2.1 peak integration and curve fitting software normalized to the internal standard. Normalized peak area for each peptide was converted as a percentage of that of the same peptide in the non-transfected cell control, and the average value of the two peptides for the same protein was reported along with percent difference between the two peptides.
Western Blotting
HepG2 cells were transfected as described, and protein was isolated by lysis in 1% SDS buffer containing 100 μl (1×) of fresh Halt protease inhibitor (Thermo Fisher) and 100 μl (1×) of fresh Halt phosphatase inhibitor (Thermo Fisher). Samples were heated at 95 °C for 5 min and then sonicated. Samples were quantified with the BCA protein kit (Thermo Fisher) prior to dilution and addition of 1× NuPAGE lithium dodecyl sulfate sample buffer (Invitrogen) containing 10% β-mercaptoethanol. Samples were heated at 95 °C for 5 min, centrifuged, and loaded with SeeBlue Plus2 prestained protein ladder (Invitrogen). Samples (15 μg) were run on Tris-glycine 4–20% gels (Bio-Rad) in 1× Tris-glycine/SDS running buffer (Bio-Rad). Samples were transferred for 6.5 min with the Invitrogen iBlot blotting unit with nitrocellulose. Blots were blocked in 5% milk in PBS with 0.1% Tween 20 for 30 min at room temperature. Blots were probed with FNTA (Epitomics, Burlingame, CA), GAPDH rabbit monoclonal IgG antibody (Cell Signaling Technology, Danvers, MA), or ADRA1A antibody (Epitomics) diluted at 1:1000, 1:5000, and 1:1000, respectively, overnight at 4 C in 5% milk in PBS with 0.1% Tween 20. Blots were washed four times for 10 min each with PBS with 0.1% Tween 20 and then incubated with HRP-conjugated goat anti-rabbit secondary antibody diluted 1:5000 (Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 h at room temperature. Blots were washed four times for 10 min each at room temperature in PBS with 0.1% Tween 20 and then developed with the Pierce SuperSignal West Pico kit (Thermo Fisher) and imaged on a LAS-4000 luminescent image analyzer (GE Healthcare).
Manumycin A Treatment of HepG2 Cells
HepG2 cells were seeded into 24-well plates at 200,000 cells/well and grown for 48 h before treatment. The culture media were removed, and manumycin A dissolved in DMSO was added to fresh media and applied to the cells to determine the dose response. The cells were incubated for 4 h, and ApoA-I levels in the media were determined by ELISA.
Manumycin A Treatment of Mice
All animals received deionized water and 2014 Teklad Global Diet (Harlan, Indianapolis, IN) ad libitum and were maintained in accordance with the Institutional Animal Use and Care Committee of Eli Lilly and Company and the National Institutes of Health Guide for the Use and Care of Laboratory Animals. Treatment with farnesyltransferase inhibitor manumycin A (5 mg/kg of body weight dissolved in 0.4% dimethyl sulfoxide in phosphate-buffered saline subcutaneously three times per week; Sigma) or vehicle alone was commenced at 7 weeks of age in hApoA-I × hCETP transgenic mice (Taconic, Germantown, NY). The treatment with manumycin A was continued for 14 days. At days 1 and 14, blood samples were obtained to measure serum hApoA-I levels by ELISA (Mabtech, Inc.) (n = 6 mice per group).
RESULTS
Genome-wide siRNA Screen
Several factors were considered in selecting the cell model for the functional genomics screen. Because the main sources of ApoA-I in human are the liver and small intestine, a human cell line from one of these tissue types to identify modulators of ApoA-I secretion was preferred. In addition, ease and reproducibility of culturing and expanding the cell line to create a single lot of cells for high throughput screening was considered. Lastly, the cell line needed to be amenable to reverse transfection methods that integrate well with automated high throughput screening methodologies. Given these requirements, the human hepatocarcinoma cell line HepG2 was selected for assay development. HepG2 cells secrete ApoA-I and have been used successfully in other high throughput screening applications (8).
Once an appropriate cell line was selected, optimal growth and siRNA transfection conditions were determined and validated in a 96-well format. As the first proof of concept for the assay, a SMARTpool siRNA targeting ApoA-I was tested for the ability to repress both ApoA-I mRNA and ApoA-I protein in the culture medium. The ApoA-I SMARTpool reduced ApoA-I mRNA levels by 88% and repressed secreted ApoA-I protein by 97% as compared with the neutral NTC siRNA (Fig. 1A). Two siRNAs that modulated ApoA-I secretion in HepG2 cells either positively or negatively over the control siRNA were identified. ADRA1A SMARTpool siRNA was found to modulate ApoA-I protein secretion in the positive direction and therefore served as the condition defining the “maximal” signal in each assay. When transfected into HepG2 cells, the ADRA1A SMARTpool siRNA caused a 92% reduction in ADRA1A mRNA and an 86% reduction in ADRA1A protein levels (Fig. 1B). Knockdown of ADRA1A caused a 75% increase in ApoA-I secretion as compared with the NTC siRNA. ADRA1A is a G-protein-coupled receptor that is expressed in liver, signals through the Gq/11 family of G-proteins, is activated by epinephrine and norepinephrine, and mediates calcium release upon ligand binding. The mechanism by which endogenous levels of ADRA1A expression appears to increase ApoA-I secretion is currently unknown. As shown previously, the SMARTpool for targeting ApoA-I mRNA itself decreased secretion and served as the definition of “minimal” signal in each assay, and the ADRA1A siRNA SMARTpool induced ApoA-I secretion by ≥50% in a three-plate, 3-day reproducibility study (Fig. 1C). Each assay plate in the genome-wide screen contained a specific configuration of control wells of NTC, ApoA-I, and ADRA1A SMARTpools as a measure of technical success. Secreted ApoA-I levels were normalized to the median of the NTC on each plate. Data were reported as -fold change from the NTC.
FIGURE 1.

Genome-wide screen for modulation of ApoA-I secretion from HepG2 cells. A, HepG2 cells were transiently transfected with siRNA directed toward human ApoA-I as a positive control or an NTC siRNA that served as a reference for 100% expression of ApoA-I mRNA. Cells were cultured for 72 h, and culture media were collected during the final 24 h for measurement of ApoA-I protein by ELISA. Repression of ApoA-I mRNA was confirmed by quantitative PCR. Data are presented as the mean ± S.E. (error bars). Statistical analysis using one-way ANOVA followed by comparison with NTC by Dunnett's method was performed (*, p < 0.01). B, HepG2 cells were transiently transfected with siRNA directed toward human ADRA1A as a positive control or an NTC siRNA as a reference for 100% expression of ADRA1A mRNA. Cells were cultured for 72 h, and culture media were collected during the final 24 h for measurement of ApoA-I protein by ELISA. Repression of ADRA1A mRNA was confirmed by quantitative PCR, and repression of ADRA1A protein was determined by Western blot analysis. GAPDH protein levels served as a loading control. Data are presented as the mean ± S.E. (error bars). Statistical analysis using one-way ANOVA followed by comparison with NTC by Dunnett's method was performed (*, p < 0.01). C, three-plate (Plt), 3-day replication studies were conducted using siRNA targeting either ADRA1A to serve as a positive control for ApoA-I protein secretion (maximum signal; circles) or ApoA-I as a control for reduction of ApoA-I protein secretion (minimum signal; diamonds) or the NTC to represent no change in ApoA-I protein secretion (middle signal; plus symbols). Individual data points are represented to illustrate the variance and reproducibility of the experimental conditions. D, human siRNA libraries targeting 21,789 genes were transfected into HepG2 cells to identify genes that are involved in production of ApoA-I protein. The data are represented as Z-score on the y axis, and the number of siRNA targets is represented on the x axis. E, testing scheme for selecting genes of interest from the whole genome screen illustrates that of 21,789 target genes assessed 432 genes were identified for follow-up. Gene class categories, including the number of genes, that may regulate ApoA-I secretion are listed. GPCR, G-protein-couple receptor; GNF, The Genomics Institute of the Novartis Research Foundation.
To ensure that the assay is robust with sufficient dynamic window, the overall requirement for the signals was that the raw signals had to show sufficient separation between the NTC and the ApoA-I and ADRA1A controls, and the -fold change from NTC had to be sufficiently reproducible. Using the conditions described above, significant separation of ApoA-I levels between the NTC siRNA and ApoA-I negative control siRNA was observed with Z′ values well above 0.4. Sufficient separation between the NTC siRNA and ADRA1A positive control siRNA was observed with nearly all Z′ values above 0.4. Although some plates illustrated “edge effects” and other patterns, none were >20% in magnitude. The application of the individual plate acceptance criteria resulted in rejection of just 10 plates for the entire screen of 880 plates, and in no case was more than one of the triplicate plates rejected.
Two Z-scores that measure how far the response of a particular gene is above or below the NTC relative to the variability in gene response were implemented to describe the change of ApoA-I protein levels. This approach expresses the response of a particular gene relative to the NTC in units of standard deviation so that a Z-score of 2 (−2) means the response of a particular gene is two standard deviations above (below) the NTC. Only Z-scores >2 or <−2 for the analysis were pursued. Hits that significantly altered cell viability based on the normalized CellTiter-Glo values were excluded if the values were less than 0.6 or greater than 1.4 (0.6 ≤ CellTiter-Glo ≤ 1.4).
Hit Triage
The distribution of siRNA activity was continuous whereby 756 of the targets met the criteria of Z-score ±2 where the highest Z-score was 6.8 and the lowest was −2.4 (Fig. 1D). Confirmation of the activity of these SMARTpools was conducted by a replicate analysis, and 85% of the original hits were reproducibly active. Genes of interest were further refined by eliminating low probability candidate genes based on biological criteria, such as tissue expression patterns. Informatics resources such as Proteome, The Genomics Institute of the Novartis Research Foundation Human Atlas, and Oncogenomics enabled filtering of genes that are expressed in liver and/or intestine. The number of hits with the appropriate tissue distribution was 432 (Fig. 1E and supplemental Table 1). In parallel fashion, two methods were undertaken to select a total of 100 genes of highest interest for further study (Fig. 2). One approach was to rank the target genes of interest solely on the maximum positive -fold induction of ApoA-I secretion (n = 50). The second approach was to curate a list of target genes of interest based on a composite of factors, including factors such as existence of genetic associations to HDL/ApoA-I levels in human, phenotype associations in knock-out mice, pathway associations and multiplicity of independent hits in a given pathway, literature text mining, and druggability. After collating the list of genes, additional literature-based research was conducted to determine which genes of interest were tractable from a drug development perspective. Specifically, whether compound inhibitors or knock-out mice that reduce the gene products of interest were available was determined. Based on reagent availability, 59 genes of interest were subjected to deconvolution analysis by studying four individual siRNAs from the SMARTpool reagents (Fig. 2). Of those genes tested, 40 genes of interest were confirmed regulators of ApoA-I secretion by at least two individual siRNAs. The list of the top 40 genes that were confirmed as ApoA-I secretion regulators included multiple cathepsin family members, ion transporters, proteins of unknown function, and metabolic enzymes. The results of mean -fold change, Z-score, and sequences of individual siRNA sequences for the confirmed 40 genes are shown in Table 1.
FIGURE 2.
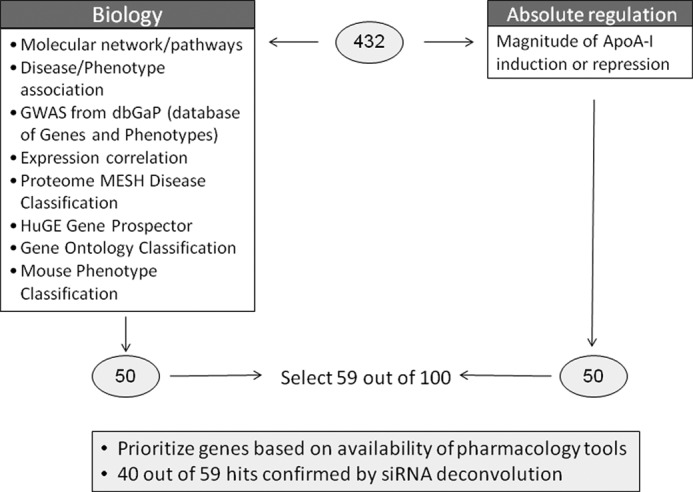
Informatics triage strategy. Two methods were undertaken to select a total of 100 genes of highest interest: ranking target genes of interest based on the maximum positive or negative -fold induction of ApoA-I secretion (n = 50) or curating genes of interest based on existence of genetic associations to HDL/ApoA-I levels in human, phenotype associations in knock-out mice, pathway associations and multiplicity of independent hits in a given pathway, literature text mining, and druggability. After collating the list of genes, additional literature-based research was conducted to determine which genes of interest were tractable from a drug development perspective (n = 59). Individual siRNAs to each gene of interest were tested for the ability to modulate ApoA-I protein, and 40 genes were confirmed as active. GWAS, genome-wide association study; MESH, medical subject headings; HuGE, human genome epidemiology.
TABLE 1.
List of genes of interest that regulate ApoA-I secretion and are verified by independent siRNA sequences
The mean -fold change in ApoA-I secretion following transfection of the Dharmacon SMARTpool siRNA for the gene of interest, the number of individual siRNAs out of four targeting the gene of interest that modulate ApoA-I secretion, and the sequences of the individual siRNAs that confirmed gene of interest involvement in ApoA-I secretion are listed.
| Entrez gene | Gene symbol | -Fold ΔApoA-I | Z-score | Confirming siRNAs | Sequence 1 | Sequence 2 | Sequence 3 | Sequence 4 |
|---|---|---|---|---|---|---|---|---|
| 13 | AADAC | 2.33 | 3.09 | 2 | GAGAAAGUCUGAAGCACUA | CAACAAAUUACGUGGCUUA | ||
| 4363 | ABCC1 | 2.16 | 2.70 | 3 | GGAUCAAGCUGAUGAACGAtt | CCGUCUACGUGACCAUUGAtt | GUUCCAAGGUGGAUGCGAAtt | |
| 31 | ACACA | 1.92 | 2.10 | 2 | GUUUCGAAAUGAACGUGCAtt | GGCUGUAUCCAUUACGUCAtt | ||
| 52 | ACP1 | 1.98 | 2.68 | 2 | GGAUGAAAGCAAUCUGAGAtt | GGGUAACAUUUGUCGAUCAtt | ||
| 51205 | ACP6 | 1.85 | 2.02 | 3 | GCACGGAUGAUCGAACAGAtt | CACUAACAUUUUUCGGAAUtt | CUGGUGGUCCGAAACCAUAtt | |
| 8424 | BBOX1 | 2.14 | 2.40 | 3 | GUAGAUGGGUUUAAUGUGU | GUCAAGGCUUCGUAUCUUA | CUACCCAGCUGUAUGGUUG | |
| 60526 | C2orf43 | 2.00 | 2.35 | 3 | GGACAGACCUCUUUCAUGA | GAGCAUUUAUGUAAGCUUA | AAACAUACCUCAUGCUUUC | |
| 1512 | CTSH | 2.89 | 4.32 | 2 | GACAUGAGCUUUGCUGAAA | GCCAGGCUUUCGAGUAUAU | ||
| 1513 | CTSK | 2.75 | 4.00 | 2 | GCAAAGGUGUGUAUUAUGA | GGAAGAGAGUUGUAUGUAC | ||
| 1520 | CTSS | 2.55 | 3.55 | 3 | GAGAAGAUGUCCUGAAAGA | GAAGUGGUGUCUACUAUGA | GUCAAACCCUAAUCGGAUA | |
| 28983 | TMPRSS11E | 1.93 | 2.11 | 2 | GCAAUGAACCUCAAGCUUA | CGCCAUAACUCCUAGAAUG | ||
| 57628 | DPP10 | 2.01 | 2.37 | 3 | CAAAUGAACCCAACAAUAA | UGAUAAAGAUCUUGGCAUA | UCACAGACUUGAAAUUGUA | |
| 78986 | DUSP26 | 1.90 | 2.46 | 2 | CUCCCUCACCAAAACAAAAtt | GGGAUACGGCAAGCAUGAAtt | ||
| 2339 | FNTA | 2.02 | 2.33 | 3 | GAAAGUGCAUGGAACUAUU | CCAAAGAUACUUCGUUAUU | UGGCAUCAUAGGCGAGUAU | |
| 148423 | C1orf52 | 3.15 | 4.24 | 3 | GAAGUCAAAUUAUGUACCA | GGACCUGACGAGCUGUUUA | GUAGGAACAAGGCGGAGAA | |
| 8111 | GPR68 | 1.88 | 1.98 | 2 | AGCUGUACCAUCGACCAUAtt | GCAUCCUCCUGUACGAGAAtt | ||
| 2939 | GSTA2 | 2.01 | 2.48 | 3 | CUUCUGCCCUUUACUCAAC | GUAACCUGCCCACAGUGAA | UAAACCAGCCAUAGAGGUC | |
| 3033 | HADH | 1.95 | 2.16 | 3 | GAAAUUAGGAGCCGGUUAC | GCAAUCAGGCUGUAUGAAC | GGACAAGUUUGCUGCUGAA | |
| 3363 | HTR7 | 1.96 | 2.15 | 2 | UGAUCAGCAUUGACAGGUAtt | GGAUGGGCUCAGAAUGUAAtt | ||
| 23415 | KCNH4 | 1.82 | 1.91 | 3 | GGAGAUCUCUCGUCUCAAUtt | UCCUAGAUAUCAUCCUGAAtt | GACCCAAACUUCGUGCUAAtt | |
| 27133 | KCNH5 | 1.84 | 2.29 | 2 | GGACGAAAUUUGCCCGAUU | CAGCUCACGCCAAUGAAUA | ||
| 23026 | MYO16 | 2.15 | 2.61 | 2 | GUGGAGAAUCUGUUCGUAA | CCCAUUAGAUUAUGCUGUA | ||
| 79815 | NIPAL2 | 2.07 | 2.51 | 4 | GAACAUAUUUACUGGUGAA | GAAACUUGGUGAUCAGUAU | CCACGAAACUCUACAAUAC | AGUAUUACCUUGUCGGAUG |
| 54681 | P4HTM | 2.08 | 2.66 | 3 | CGACAGGGAUCACUUCAUC | GAAGAGGCAAUGAGCACUA | UCAAGCCGCUGCUCUUCGA | |
| 5511 | PPP1R8 | 2.57 | 4.29 | 3 | CAAAGGAGACAAACUAAUU | AAGCGGAUUUCUACCCUUA | GCUCUAGCCUCCCGCUGUU | |
| 128674 | PROKR2 | 1.96 | 2.15 | 3 | CCCUCUACGUCUCCACCAAtt | ACAGAAACGGUCCUCUUUAtt | GUUUCACCAUCGUUCGUGAtt | |
| 6385 | SDC4 | 2.29 | 2.94 | 3 | GAUCGGCCCUGAAGUUGUC | GUGAGGAUGUGUCCAACAA | GAUCGGCCCUGAAGUUGUC | |
| 10371 | SEMA3A | 1.84 | 1.95 | 2 | GAUGUUAUGUUUAUCGGAAtt | GGUGUGAUAUUUACGGGAAtt | ||
| 462 | SERPINC1 | 2.54 | 3.50 | 4 | CAACAAAUCCUCCAAGUUA | UGAUGGAGGUAUUUAAGUU | CAACCCUUGUGUUAAGUAA | CCGAAUCACCGAUGUCAUU |
| 114789 | SLC25A25 | 4.09 | 6.11 | 4 | GCAGAGCAGUUUCCUAAGA | GCUAGACUUUGAAGAAUUU | GAAGACAGGCCAGUACUCA | GUUGGUGGCUUCACUCAGA |
| 84102 | SLC41A2 | 2.44 | 2.85 | 4 | GAAAGGACCCGGAUAGUUU | GGUCAUACUUCUUUAACUA | GCAAAUACUUGUGCCCUUU | UAUAAUAGCUGCCAAACAU |
| 7057 | THBS1 | 1.99 | 2.30 | 3 | GGACUGCGUUGGUGAUGUA | GUACAGAAACGUAGUCGUC | UCAUUAGAGUGGUGAUGUA | |
| 658 | BMPR1B | 1.50 | <1.9 | 2 | CCACAGGGCUUUACUUAUA | GACGCUAAAUCAAUGCUGA | ||
| 1600 | DAB1 | 2.51 | 3.48 | 4 | UAAGUGCUGUGACCCAAUU | UAAGUGCUGUGACCCAAUU | CAAGUUAUGUCAAGAUUCC | GCCAAUCCGUGAUCCCGAA |
| 3604 | TNFRSF9 | 1.90 | 2.10 | 3 | ACUCCUGUAUAUAUUCAAAtt | GGAUUGUAAACAAGGUCAAtt | GGUGUUUUCAGGACCAGGAtt | |
| 8303 | SNN | 1.92 | 2.28 | 3 | GGUCACAGUCAUCGUCAUC | GACCGUGCGUGGAGAGAAA | GUAGGGAGCUGUUCGUUUA | |
| 55504 | TNFRSF19 | 1.86 | 1.96 | 3 | GGAUUCAAAUAGCAGUCAAtt | CUACUGAUUUAUCUAGAUAtt | GGAUUUUAUAGGAAGACGAtt | |
| 57094 | CPA6 | 2.36 | 2.60 | 4 | GUAGCUAACUUCCUUCGAA | UCAAGGAACUCAAGGUUUC | GGCAGACGAUCACGACUCA | CAGUAUACGGGGUACGAUA |
| 90273 | CEACAM21 | 2.36 | 2.54 | 2 | GCUAAUCGCAGCAUAUGUA | CAUCAGCGCCCUUUGAAGU | ||
| 374918 | IGFL1 | 0.18 | −1.95 | 3 | GAAACUGCACCUUCAGAGU | GACCUCGGAUGACAGGCUU | CAAGAGAUGUGGGGACAAG |
Repression of Farnesyltransferase mRNA Expression and Activity Increases ApoA-I Secretion Selectively
Inhibition of FNTA by chemical inhibitors, such as manumycin A and Zarnestra, demonstrated cardiovascular benefits in preclinical models of disease (9); therefore, we were interested in determining whether ApoA-I levels could be influenced selectively as compared with other apolipoproteins by repression of FNTA mRNA and enzymatic activity in cells and in mice. As evaluated in the primary screen, HepG2 cells were transfected with four independent siRNAs and the original SMARTpool that target FNTA mRNA (Fig. 3A). FNTA mRNA levels were significantly reduced by transfection of the SMARTpool and individual siRNAs targeting FNTA mRNA. Likewise, FNTA protein levels were reduced in cells transfected with all siRNA reagents as compared with the non-targeting control protein GAPDH (Fig. 3B). Transfection of HepG2 cells with the FNTA SMARTpool increased ApoA-I secretion by 2.5-fold, whereas three of four individual siRNAs that comprise the SMARTpool caused a statistically significant increase in ApoA-I secretion, although each was to a lesser degree as compared with the pooled siRNA reagent (Fig. 3C). These data confirm that FNTA mRNA and protein reduction leads to increased ApoA-I secretion from HepG2 cells. We reasoned that changes to a cell that lead to increased secretion of ApoA-I may likewise lead to increased secretion of other apolipoproteins. ApoB is the primary apolipoprotein associated with LDL-c particles, and some evidence suggests that levels of ApoB are the most potent markers of cardiovascular events (10). To evaluate the effect of FNTA mRNA knockdown on ApoB secretion, mass spectrometry measurement of both ApoA-I and ApoB in culture media of HepG2 cells transfected with the FNTA SMARTpool and siRNA1 was conducted. In response to both siRNA treatments, ApoA-I secretion into the media was increased by greater than 2.5-fold as compared with NTC siRNA-transfected cells. By contrast, ApoB secretion was not statistically different from that of NTC siRNA-transfected cells (Fig. 3D). A profile of higher ApoA-I:ApoB ratio is indicative of improvement in metabolic disease end points, such as cardiovascular outcomes, diabetic retinopathy, and other sequelae of metabolic syndrome; therefore, a therapeutic molecule with this pharmacology may lead to improved outcomes in patients.
FIGURE 3.

In vitro confirmation of FNTA as modulator of ApoA-I secretion. A, HepG2 cells were transiently transfected with either SMARTpool or individual siRNAs directed toward human FNTA, and efficiency of mRNA knockdown was determined by quantitative PCR. Cells were cultured for 72 h prior to RNA isolation and quantitative PCR. Data are presented as the mean ± S.E. (error bars). Statistical analysis using one-way ANOVA followed by comparison with vehicle by Dunnett's method was performed (*, p < 0.01). B, HepG2 cells were transiently transfected with either SMARTpool or individual siRNAs directed toward human FNTA, and efficiency of protein knockdown was determined by Western blot analysis of FNTA and GAPDH as a protein loading control. C, specific knockdown of FNTA leads to an increase in ApoA-I secretion from HepG2 cells that were transiently transfected with siRNA directed toward human ApoA-I as a positive control or an NTC siRNA as a reference for 100% expression of ApoA-I mRNA. Cells were cultured for 72 h, and culture media were collected during the final 24 h for measurement of ApoA-I protein by ELISA. Data are presented as the mean ± S.E. (error bars). Statistical analysis using one-way ANOVA followed by comparison with vehicle by Dunnett's method was performed (*, p < 0.01). D, mass spectrometry measurement of both ApoA-I and ApoB in culture media of HepG2 cells transfected with the FNTA SMARTpool and siRNA1 was conducted. Normalized peak area for each peptide was converted as a percentage of that of the same peptide in the non-transfected cell control, and the average value of the two peptides for the same protein was reported along with percent difference between the two peptides (*, p < 0.01). E, HepG2 cells were transiently transfected with either reagent alone (Mock) or with a cDNA expression construct for human FNTA, and efficiency of overexpression was determined by Western blot analysis of FNTA and GAPDH as a protein loading control. F, HepG2 cells overexpressing human FNTA (FNTA Over Exp) have a 3-fold reduction in ApoA-I secretion.
We next determined whether overexpression of FNTA would cause a reciprocal decrease in ApoA-I secretion. Using a mammalian expression vector for human FNTA transfected into HepG2 cells, levels of FNTA were increased over mock-transfected cells, and ApoA-I secretion into the medium was decreased by 3-fold as compared with ApoA-I secretion from mock-transfected cells (Fig. 3E). These “gain-of-function” data substantiate a role for FNTA in controlling secretion of ApoA-I.
To test whether inhibition of FNTA enzymatic activity would also promote ApoA-I secretion, HepG2 cells were treated with a chemical inhibitor of farnesyltransferase, manumycin A, in a dose-response study. Manumycin A treatment dose-dependently increased ApoA-I secretion by up to 40% compared with untreated cells (Fig. 4). These results indicate that reduction in both expression and enzymatic activity of FNTA lead to increased secretion of ApoA-I from hepatocytes.
FIGURE 4.
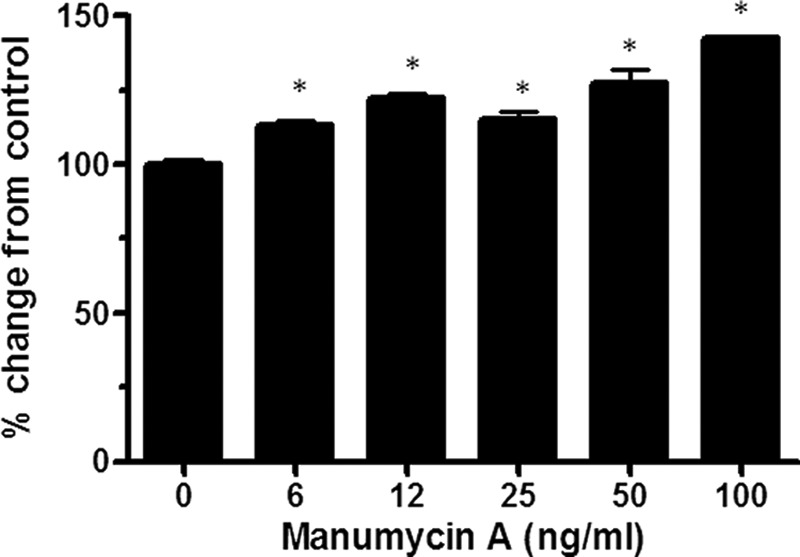
Dose-dependent inhibition of FNTA by manumycin A increases ApoA-I secretion. Confluent cultures of HepG2 cells were treated with manumycin A for 4 h prior to ApoA-I ELISA measurement from the culture media to determine the dose response. Data are represented as percent change (mean ± S.E. (error bars)) from cells treated with vehicle. Statistical analysis using one-way ANOVA followed by comparison with vehicle by Dunnett's method was performed (*, p < 0.01).
Lipoprotein profiles in mice differ from those in human in that expression of the mouse ApoA-I gene is regulated in a manner different from human, and human expresses CETP, but mice do not. Accordingly, the majority of cholesterol in mice circulates as HDL-c particles, whereas LDL-c particles predominate in human. To assess the ability of manumycin A to increase circulating levels of human ApoA-I in a preclinical model, we treated mice harboring both a human ApoA-I genomic fragment that includes a liver-specific enhancer and the entire human ApoA-I structural gene and the human CETP gene. Manumycin A was given to mice three times per week for 2 weeks, and human ApoA-I levels were measured from serum on days 1 and 14 postdose initiation (Fig. 5A). As compared with mice treated with vehicle, manumycin A caused a statistically significant 26% increase in hApoA-I levels as early as 24 h postdose. After 14 days of treatment, hApoA-I levels remained elevated 30% by manumycin A treatment as compared with vehicle-treated mice (Fig. 5B).
FIGURE 5.
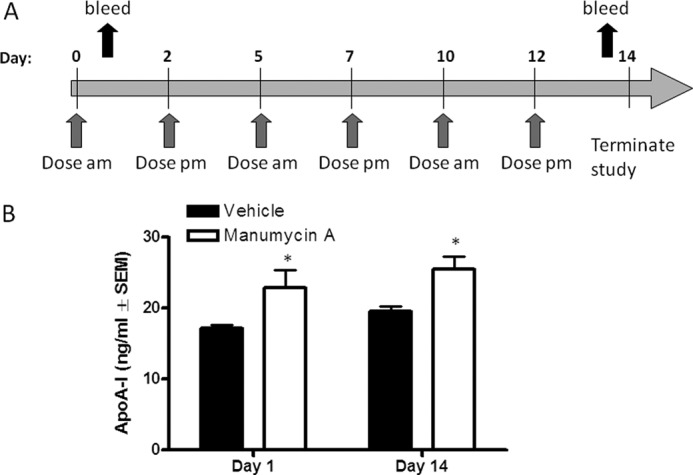
Farnesyltransferase inhibitor manumycin A regulates circulating ApoA-I in hApoA-I/hCETP mice. A, schematic representation of administration of manumycin A to hApoA-I × hCETP transgenic mice. Mice were dosed either with vehicle or manumycin A at 5 mg/kg of body weight subcutaneously 3 times per week for 2 weeks (n = 6 mice per group). At days 1 and 14, blood samples were obtained to measure serum hApoA-I levels by ELISA. Average ApoA-I levels for each group are shown (n = 6, ±S.E. (error bars)). Statistical analysis using one-way ANOVA followed by comparison with vehicle by Dunnett's method was performed. (*, p < 0.01).
DISCUSSION
The statin class of therapeutics has been successful in reducing LDL-c and cardiovascular risk. However, significant residual risk remains in patient populations; therefore, addressing low HDL-c as a causal risk factor is a promising approach to reduce the incidence of cardiovascular outcomes. HDL-c is atheroprotective because it possesses multiple properties, such as antithrombotic, anti-inflammatory, and antioxidant activities and its promotion of reverse cholesterol transport from the peripheral tissues, including atherosclerotic plaques, that improve such disease-promoting mechanisms. In this study, we leveraged a functional genomics approach to find molecular targets for the development of drugs that could elevate ApoA-I, the major protein constituent of HDL-c. The genome-wide functional siRNA screen for increased ApoA-I secretion from HepG2 cells identified numerous candidate gene targets that may be pursued as therapeutic drug targets.
Because the fields of genome-wide association study, exome sequencing, and whole genome sequencing are rapidly emerging to provide human genetic variant associations with lipid traits important to cardiovascular disease, we assessed whether our genes of interest were also identified by human genetic studies. The seminal study conducted by Teslovich et al. (11) associated common single nucleotide polymorphisms (SNPs) with lipid traits in over 100,000 individuals (11). We merged our genes of interest list with the 95 loci for blood lipids that were identified in the Teslovich et al. (11) study and rank-ordered the results based on ApoA-I secretion effect. As expected, the ApoA-I gene itself is a determinant of HDL-c levels in humans (+16.95 effect size; p = 7 × 10−240) and a determinant of ApoA-I secretion in our assay whereby siRNA targeting of the gene resulted in an 88% reduction in secreted ApoA-I. Other genes with functional congruence include MC4R, encoding melanocortin 4 receptor, whereby an MC4R SNP associates with a −0.42 effect size (p = 7 × 10−9) and MC4R siRNA causes a 1.82-fold increase in ApoA-I secretion, as well as PLTP, encoding phospholipid transfer protein, whereby a PLTP SNP associates with a −0.93 effect size (p = 2 × 10−22) and PLTP siRNA causes a 1.85-fold increase in ApoA-I secretion. None of the genes that were identified in our siRNA screen that were also identified in the genome-wide association study met the cutoff criteria of a Z-score >2 or <−2. Supplemental Table 2 lists loci associated with the HDL-c trait and siRNA targets that modulated ApoA-I secretion.
The genes of interest that garnered the most interest were those that modulated ApoA-I secretion by the highest magnitude because those genes may serve either as direct molecular targets for therapeutics or as pathway participants that may highlight druggable pathways for HDL modulation. We reasoned that the functional siRNA screen may identify gene products that could regulate transcriptional activity of the ApoA-I gene, ApoA-I translation efficiency, the rate of secretory flux, or the proteolysis of ApoA-I. Indeed, several gene products with such hypothesized activity that may also be modulated by therapeutics, such as the cathepsin family of proteases, were identified in the screen (Table 1). The hypothesis that cathepsins may directly degrade ApoA-I protein is substantiated by previous reports showing that cathepsin F and K proteolytic activity can partially degrade lipid-free ApoA-I, thereby reducing its ability to induce cholesterol efflux from macrophages. Cathepsin S was shown to completely degrade ApoA-I, leading to complete loss of ApoA-I cholesterol acceptor function (12). Repression of both cathepsins K and S by siRNA knockdown in our screen resulted in elevation of ApoA-I levels in the HepG2 culture medium presumably by reducing degradation of ApoA-I.
Another gene target that warranted follow-up was FNTA. Repression of FNTA mRNA by siRNA SMARTpool and individual siRNAs resulted in a significant increase in secreted ApoA-I from HepG2 cells (Fig. 3C). Notably, the profile of apolipoprotein secretion was favorable because ApoA-I levels, but not ApoB levels, were increased (Fig. 3D). ApoB is the major protein constituent of LDL-c, and one expression of cardiovascular health and risk that is gaining importance in diagnostic medicine is the ApoA-I:ApoB ratio. In the INTERHEART study of cardiovascular risk factors in patients from 52 countries, the non-fasting ApoB:ApoA-I ratio was superior to any of the cholesterol ratios for estimation of the risk of acute myocardial infarction in all ethnic groups, in both sexes, and at all ages (13). Knockdown of FNTA by siRNA specifically raises ApoA-I levels, suggesting its potential as a target for reducing cardiovascular risk that warrants further follow-up by a variety of methods.
FNTA has been a well studied molecular target for anticancer therapeutics because FNTA-mediated isoprenylation of Ras proteins is necessary for their transforming activities (14). Several small molecules, such as lonafarnib and tipifarnib, have been tested in preclinical and clinical studies as cancer therapeutics. Another FNTA inhibitor, manumycin A, has been studied in a preclinical model of atherosclerosis. When the high fat-fed ApoE-null mouse that is highly prone to atherosclerosis was treated with manumycin A for 22 weeks, fatty streak lesion burden and the area of vascular smooth muscle-like cells in the aortic neointima were reduced (9). When manumycin A was administered in our HepG2 cell system, ApoA-I secretion levels increased in a dose-responsive manner (Fig. 4).
We reflected on our overall data set and the enzymatic activity of FNTA to generate a hypothesis to describe the mechanisms by which FNTA could be involved in ApoA-I secretion. Generally, maintaining intracellular lipid homeostasis requires that sterols and associated lipids move between cellular compartments by vesicular and non-vesicular pathways. FNTA post-translationally adds a farnesyl group to the –SH of the cysteine near the end of proteins, typically targeting the modified protein to membranes based on the hydrophobicity of the farnesyl group. Centromeric protein F (CENPF) is a farnesylated protein (15), and the siRNA directed toward CENPF caused a 1.98× increase in ApoA-I secretion in our study. We hypothesize that this initial farnesylation event is important for downstream events necessary for appropriate ApoA-I secretion (Fig. 6). This hypothesis is consistent with a study describing CENPF direct interaction with syntaxin 4, revealing a role for CENPF in vesicular transport. Indeed, depletion of cells of CENPF altered GLUT4-mediated glucose transfer (16).
FIGURE 6.
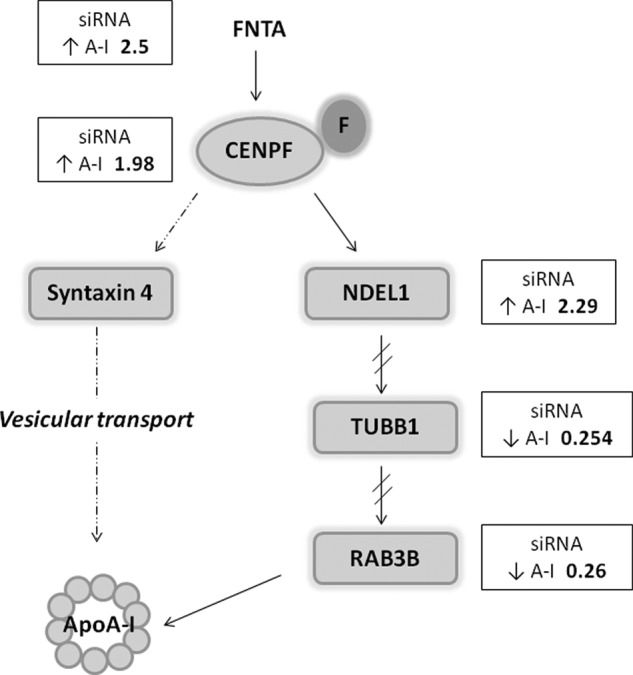
Mechanistic hypothesis by which FNTA regulates ApoA-I secretion. FNTA post-translationally adds a farnesyl group (F) to the –SH of the cysteine near the end of proteins, typically targeting the modified protein to membranes based on the hydrophobicity of the farnesyl group. CENPF is a farnesylated protein, and the siRNA directed toward CENPF caused a 1.98× increase in ApoA-I secretion in our study. We hypothesize that this initial farnesylation event is important for downstream events necessary for appropriate ApoA-I secretion. Through indirect (dotted arrows) and direct (solid arrows) interactions with downstream proteins known to be involved in vesicle transport and exocytosis, FNTA directs downstream events that lead to ApoA-I secretion.
A second alternative pathway that begins with CENPF is also plausible for explaining our observations. CENPF interacts directly with Ndel1, a cytoplasmic dynein-binding protein that regulates bidirectional vesicle transport (17, 18). In our study, siRNA directed toward Ndel1 increased ApoA-I secretion by 2.29-fold. These data suggest that the coordinated expression and/or subcellular localization of either CENPF or Ndel1 may be necessary for controlling ApoA-I secretion rates. Through functional interactions with other proteins, such as CDC42 and dynein, Ndel1 indirectly influences tubulin (TUBB1) and RAB3B. Both proteins are required for vesicular transport and exocytosis, and in our siRNA screen, knockdown of TUBB1 and RAB3B caused a 75 and 74% reduction in ApoA-I secretion. Interestingly, RAB3B is prenylated and could be a direct target of FNTA (19). In total, our data support a hypothesis whereby FNTA orchestrates protein activities that direct intracellular trafficking of ApoA-I, leading to overall modulation of secreted ApoA-I levels. These findings should be verified in the context of a polarized cell like that of a functional primary hepatocyte. Because these studies were conducted in HepG2 cells that are not polarized, the interactions between these proposed proteins may be real, but they may interact differently in a polarized cell setting.
To translate the cell-based findings into a preclinical model of human lipoprotein metabolism, we selected mice that express both human ApoA-I and CETP transgenes for studying in vivo effects of manumycin A. Following 1 day of treatment with manumycin A, circulating levels of human ApoA-I were increased in the double transgenic mice, and the elevated human ApoA-I levels persisted for the 14-day treatment course (Fig. 6). These data illustrate similar regulation of human ApoA-I in both cell culture and in vivo.
Interventions that increase HDL-c by reducing its catabolism in the circulation through inhibition of CETP activity are in late stage clinical development; however, an alternative approach such as enhancing de novo production of HDL-c in the liver and/or small intestine may provide even greater therapeutic benefit. The anticipated successful reduction of cardiovascular events attributed to elevated HDL-c via CETP inhibition is based on findings from the DEFINE (determining the efficacy and tolerability of CETP inhibition with anacetrapib) cardiovascular safety trial of anacetrapib (20). In the 1623-patient trial of 76-week duration, treatment with anacetrapib, as compared with placebo, increased HDL-c levels by 138.1%, decreased LDL-c levels by 39.8%, and decreased non-HDL-c levels by 31.7%. Importantly, only 2.0% of subjects receiving anacetrapib suffered prespecified, adjudicated cardiovascular safety end points as compared with 2.6% receiving placebo. Although the data are not conclusive in determining the likelihood of overall reduction in cardiovascular events in longer term trials, the data suggest potential outcome benefits. Similarly, infusion of recombinant ApoA-I or ApoA-I peptide mimetics is under development, and in some studies, regression of atherosclerotic plaque area has been observed by intravascular ultrasound measurement (4); however, no direct findings of outcome benefits are available (21).
With increasing insight into emerging qualitative factors that influence HDL functionality with respect to anti-inflammatory properties, capacity to promote reverse cholesterol transport, antithrombotic activity, whether any of the current HDL-elevating strategies will be beneficial remains to be determined in large, outcome-based clinical trials. The results of our study provide new functional genomics-based data for exploring new hypotheses and mechanisms by which ApoA-I levels may be regulated.
Supplementary Material
Acknowledgments
We thank Tonghuan (Angela) Hu, Jeffrey S. Arnold, Doug Perkins, and Patrick Forler for expert technical and scientific support.

This article contains supplemental Tables 1 and 2.
- CAD
- coronary artery disease
- LDL-c
- LDL cholesterol
- HDL-c
- HDL cholesterol
- Apo
- apolipoprotein
- h
- human
- FNTA
- farnesyltransferase
- CETP
- cholesterol ester transfer protein
- NTC
- non-targeting control
- ADRA1A
- adrenergic receptor α1A
- ACTB
- β-actin
- MC4R
- melanocortin 4 receptor
- PLTP
- phospholipid transfer protein
- CENPF
- centromeric protein F
- ANOVA
- analysis of variance.
REFERENCES
- 1. Gordon D. J., Probstfield J. L., Garrison R. J., Neaton J. D., Castelli W. P., Knoke J. D., Jacobs D. R., Jr., Bangdiwala S., Tyroler H. A. (1989) High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation 79, 8–15 [DOI] [PubMed] [Google Scholar]
- 2. Rubins H. B., Robins S. J., Collins D., Fye C. L., Anderson J. W., Elam M. B., Faas F. H., Linares E., Schaefer E. J., Schectman G., Wilt T. J., Wittes J. (1999) Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N. Engl. J. Med. 341, 410–418 [DOI] [PubMed] [Google Scholar]
- 3. Steiner G. (2001) Treating lipid abnormalities in patients with type 2 diabetes mellitus. Am. J. Cardiol. 88, 37N–40N [DOI] [PubMed] [Google Scholar]
- 4. Nissen S. E., Tsunoda T., Tuzcu E. M., Schoenhagen P., Cooper C. J., Yasin M., Eaton G. M., Lauer M. A., Sheldon W. S., Grines C. L., Halpern S., Crowe T., Blankenship J. C., Kerensky R. (2003) Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA 290, 2292–2300 [DOI] [PubMed] [Google Scholar]
- 5. Sittampalam G. S., Weidner J., Auld D., Glicksman M., Arkin M., Napper A., Inglese J. (2012) Assay Guidance Manual, Eli Lilly and Company and the National Center for Advancing Translational Sciences, Bethesda, MD: [PubMed] [Google Scholar]
- 6. Cartwright I. J., Higgins J. A. (1996) Intracellular degradation in the regulation of secretion of apolipoprotein B-100 by rabbit hepatocytes. Biochem. J. 314, 977–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wildsmith K. R., Han B., Bateman R. J. (2009) Method for the simultaneous quantitation of apolipoprotein E isoforms using tandem mass spectrometry. Anal. Biochem. 395, 116–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moskaug J. Ø., Borge G. I., Fagervoll A. M., Paur I., Carlsen H., Blomhoff R. (2008) Dietary polyphenols identified as intracellular protein kinase A inhibitors. Eur. J. Nutr. 47, 460–469 [DOI] [PubMed] [Google Scholar]
- 9. Sugita M., Sugita H., Kaneki M. (2007) Farnesyltransferase inhibitor, manumycin A, prevents atherosclerosis development and reduces oxidative stress in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 27, 1390–1395 [DOI] [PubMed] [Google Scholar]
- 10. Sniderman A. D., Williams K., Contois J. H., Monroe H. M., McQueen M. J., de Graaf J., Furberg C. D. (2011) A meta-analysis of low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B as markers of cardiovascular risk. Circ. Cardiovasc. Qual. Outcomes 4, 337–345 [DOI] [PubMed] [Google Scholar]
- 11. Teslovich T. M., Musunuru K., Smith A. V., Edmondson A. C., Stylianou I. M., Koseki M., Pirruccello J. P., Ripatti S., Chasman D. I., Willer C. J., Johansen C. T., Fouchier S. W., Isaacs A., Peloso G. M., Barbalic M., Ricketts S. L., Bis J. C., Aulchenko Y. S., Thorleifsson G., Feitosa M. F., Chambers J., Orho-Melander M., Melander O., Johnson T., Li X., Guo X., Li M., Shin Cho Y., Jin Go M., Jin Kim Y., Lee J. Y., Park T., Kim K., Sim X., Twee-Hee Ong R., Croteau-Chonka D. C., Lange L. A., Smith J. D., Song K., Hua Zhao J., Yuan X., Luan J., Lamina C., Ziegler A., Zhang W., Zee R. Y., Wright A. F., Witteman J. C., Wilson J. F., Willemsen G., Wichmann H. E., Whitfield J. B., Waterworth D. M., Wareham N. J., Waeber G., Vollenweider P., Voight B. F., Vitart V., Uitterlinden A. G., Uda M., Tuomilehto J., Thompson J. R., Tanaka T., Surakka I., Stringham H. M., Spector T. D., Soranzo N., Smit J. H., Sinisalo J., Silander K., Sijbrands E. J., Scuteri A., Scott J., Schlessinger D., Sanna S., Salomaa V., Saharinen J., Sabatti C., Ruokonen A., Rudan I., Rose L. M., Roberts R., Rieder M., Psaty B. M., Pramstaller P. P., Pichler I., Perola M., Penninx B. W., Pedersen N. L., Pattaro C., Parker A. N., Pare G., Oostra B. A., O'Donnell C. J., Nieminen M. S., Nickerson D. A., Montgomery G. W., Meitinger T., McPherson R., McCarthy M. I., McArdle W., Masson D., Martin N. G., Marroni F., Mangino M., Magnusson P. K., Lucas G., Luben R., Loos R. J., Lokki M. L., Lettre G., Langenberg C., Launer L. J., Lakatta E. G., Laaksonen R., Kyvik K. O., Kronenberg F., Konig I. R., Khaw K. T., Kaprio J., Kaplan L. M., Johansson A., Jarvelin M. R., Janssens A. C., Ingelsson E., Igl W., Kees Hovingh G., Hottenga J. J., Hofman A., Hicks A. A., Hengstenberg C., Heid I. M., Hayward C., Havulinna A. S., Hastie N. D., Harris T. B., Haritunians T., Hall A. S., Gyllensten U., Guiducci C., Groop L. C., Gonzalez E., Gieger C., Freimer N. B., Ferrucci L., Erdmann J., Elliott P., Ejebe K. G., Doring A., Dominiczak A. F., Demissie S., Deloukas P., de Geus E. J., de Faire U., Crawford G., Collins F. S., Chen Y. D., Caulfield M. J., Campbell H., Burtt N. P., Bonnycastle L. L., Boomsma D. I., Boekholdt S. M., Bergman R. N., Barroso I., Bandinelli S., Ballantyne C. M., Assimes T. L., Quertermous T., Altshuler D., Seielstad M., Wong T. Y., Tai E. S., Feranil A. B., Kuzawa C. W., Adair L. S., Taylor H. A., Jr., Borecki I. B., Gabriel S. B., Wilson J. G., Holm H., Thorsteinsdottir U., Gudnason V., Krauss R. M., Mohlke K. L., Ordovas J. M., Munroe P. B., Kooner J. S., Tall A. R., Hegele R. A., Kastelein J. J., Schadt E. E., Rotter J. I., Boerwinkle E., Strachan D. P., Mooser V., Stefansson K., Reilly M. P., Samani N. J., Schunkert H., Cupples L. A., Sandhu M. S., Ridker P. M., Rader D. J., van Duijn C. M., Peltonen L., Abecasis G. R., Boehnke M., Kathiresan S. (2010) Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lindstedt L., Lee M., Oörni K., Brömme D., Kovanen P. T. (2003) Cathepsins F and S block HDL3-induced cholesterol efflux from macrophage foam cells. Biochem. Biophys. Res. Commun. 312, 1019–1024 [DOI] [PubMed] [Google Scholar]
- 13. McQueen M. J., Hawken S., Wang X., Ounpuu S., Sniderman A., Probstfield J., Steyn K., Sanderson J. E., Hasani M., Volkova E., Kazmi K., Yusuf S. (2008) Lipids, lipoproteins, and apolipoproteins as risk markers of myocardial infarction in 52 countries (the INTERHEART study): a case-control study. Lancet 372, 224–233 [DOI] [PubMed] [Google Scholar]
- 14. Berndt N., Hamilton A. D., Sebti S. M. (2011) Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 11, 775–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ashar H. R., James L., Gray K., Carr D., Black S., Armstrong L., Bishop W. R., Kirschmeier P. (2000) Farnesyl transferase inhibitors block the farnesylation of CENP-E and CENP-F and alter the association of CENP-E with the microtubules. J. Biol. Chem. 275, 30451–30457 [DOI] [PubMed] [Google Scholar]
- 16. Pooley R. D., Moynihan K. L., Soukoulis V., Reddy S., Francis R., Lo C., Ma L. J., Bader D. M. (2008) Murine CENPF interacts with syntaxin 4 in the regulation of vesicular transport. J. Cell Sci. 121, 3413–3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vergnolle M. A., Taylor S. S. (2007) Cenp-F links kinetochores to Ndel1/Nde1/Lis1/dynein microtubule motor complexes. Curr. Biol. 17, 1173–1179 [DOI] [PubMed] [Google Scholar]
- 18. Segal M., Soifer I., Petzold H., Howard J., Elbaum M., Reiner O. (2012) Ndel1-derived peptides modulate bidirectional transport of injected beads in the squid giant axon. Biol. Open 1, 220–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johnston P. A., Archer B. T., 3rd, Robinson K., Mignery G. A., Jahn R., Südhof T. C. (1991) rab3A attachment to the synaptic vesicle membrane mediated by a conserved polyisoprenylated carboxy-terminal sequence. Neuron 7, 101–109 [DOI] [PubMed] [Google Scholar]
- 20. Cannon C. P., Shah S., Dansky H. M., Davidson M., Brinton E. A., Gotto A. M., Stepanavage M., Liu S. X., Gibbons P., Ashraf T. B., Zafarino J., Mitchel Y., Barter P. (2010) Safety of anacetrapib in patients with or at high risk for coronary heart disease. N. Engl. J. Med. 363, 2406–2415 [DOI] [PubMed] [Google Scholar]
- 21. Nicholls S. J. (2011) Apo A-I modulating therapies. Curr. Cardiol. Rep. 13, 537–543 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.