

Background: Hydralazine and organic nitrates have clinical benefits for heart failure, but the underlying mechanism is controversial.
Results: Hydralazine reduced sarcoplasmic reticulum Ca2+ leak and improved Ca2+ cycling and contractility; nitroglycerin enhanced contractile efficiency; both were impaired by nitroso-redox imbalance.
Conclusion: These agents exert complementary effects on nitroso-redox imbalance.
Significance: New mechanistic insights for redox-targeted treatments of heart failure.
Keywords: Heart Failure, Nitric-oxide Synthase, Oxidative Stress, Redox Regulation, Sarcoplasmic Reticulum (SR), Cardiomyocytes, Hydralazine, Nitroso-Redox Imbalance, SR Calcium Leak, Organic Nitrates
Abstract
Although the combined use of hydralazine and isosorbide dinitrate confers important clinical benefits in patients with heart failure, the underlying mechanism of action is still controversial. We used two models of nitroso-redox imbalance, neuronal NO synthase-deficient (NOS1−/−) mice and spontaneously hypertensive heart failure rats, to test the hypothesis that hydralazine (HYD) alone or in combination with nitroglycerin (NTG) or isosorbide dinitrate restores Ca2+ cycling and contractile performance and controls superoxide production in isolated cardiomyocytes. The response to increased pacing frequency was depressed in NOS1−/− compared with wild type myocytes. Both sarcomere length shortening and intracellular Ca2+ transient (Δ[Ca2+]i) responses in NOS1−/− cardiomyocytes were augmented by HYD in a dose-dependent manner. NTG alone did not affect myocyte shortening but reduced Δ[Ca2+]i across the range of pacing frequencies and increased myofilament Ca2+ sensitivity thereby enhancing contractile efficiency. Similar results were seen in failing myocytes from the heart failure rat model. HYD alone or in combination with NTG reduced sarcoplasmic reticulum (SR) leak, improved SR Ca2+ reuptake, and restored SR Ca2+ content. HYD and NTG at low concentrations (1 μm), scavenged superoxide in isolated cardiomyocytes, whereas in cardiac homogenates, NTG inhibited xanthine oxidoreductase activity and scavenged NADPH oxidase-dependent superoxide more efficiently than HYD. Together, these results revealed that by reducing SR Ca2+ leak, HYD improves Ca2+ cycling and contractility impaired by nitroso-redox imbalance, and NTG enhanced contractile efficiency, restoring cardiac excitation-contraction coupling.
Introduction
Heart failure (HF)3 is a common and increasingly prevalent cause of morbidity and mortality. Prior to the African-American heart failure trial, the only pharmacologic strategy that improved survival in HF was neurohormonal blockade therapy. This trial revealed increased survival among African-American patients with advanced HF treated with the combination of isosorbide dinitrate (ISDN) and hydralazine (HYD) (1–3). However, the precise mechanism(s) by which this regimen reduced mortality remains unclear.
HF is characterized by impaired excitation-contraction (E-C) coupling. For instance, increased contractile force upon increasing frequency of stimulation (force-frequency relationship, FFR) is compromised in failing hearts, giving rise to a blunted or negative FFR (4–6). Defects in calcium (Ca2+) handling may be responsible for this phenomenon (7, 8). It is increasingly appreciated that altered signaling in both Ca2+ reuptake mechanisms and Ca2+ leak through the ryanodine receptor (RyR2) contribute to impairment of the Ca2+ cycle in the failing heart. In this regard, Ca2+ leak is believed to be mediated by post-translational modifications of the RyR2, associated with nitroso-redox (NO/redox) imbalance (9, 10), a linked abnormality in the failing myocardium (11, 12). As a consequence of the sarcoplasmic reticulum (SR) Ca2+ leak, an altered E-C coupling occurs by depleting the SR Ca2+ stores resulting in possible impaired contractile function of the heart.
Increased xanthine oxidoreductase (XOR)-, and possibly NADPH oxidase (NOX)-mediated reactive oxygen species (ROS) production also occurs in failing hearts (13–15). Importantly, disruption in signaling pathways due to oxidative stress can be intensified by NO deficiency, a situation of NO/redox disequilibrium (16, 17). One possible explanation of the clinical benefits of HYD-ISDN is restoration of the balance between formation of reactive oxygen and reactive nitrogen species (2).
We used two established models of cardiac NO/redox imbalance to test the hypothesis that HYD and organic nitrates restore NO bioavailability and reactivity, ameliorate NO/redox disequilibrium, and improves myocardial contractility as measured by FFR. Accordingly, we examined the effects of each drug, alone and in combination, on sarcomere length (SL) shortening, Ca2+ cycling, and SR Ca2+ leak in neuronal NO synthase-deficient (NOS1−/−) mice and spontaneously hypertensive heart failure (SHHF) rats. ROS production by XOR and NOX were determined in vitro in NOS1−/− hearts.
EXPERIMENTAL PROCEDURES
Animal Models and Myocyte Isolation
All protocols and experimental procedures were approved by the Institutional Animal Care and Use Committee of The University of Miami following the Guide for the Care and Use of Laboratory Animals (NIH Publication 85–234, revised 1996).
Cardiomyocytes (CMs) were isolated from C57BL/6J mice (WT, 3–5 months old, n = 25; Jackson Laboratories, Bar Harbor, ME) and transgenic mice with homozygous deletions for NOS1 (B6;129S4-Nos1<tm1Plh>J, 3–5 months old, n = 41) or male SHHF rats (22–24 months old, n = 4; Charles River Laboratories Inc., Wilmington, MA) and their normotensive controls, Wistar Kyoto rats (WKY, n = 5) hearts as described in detail (18). Briefly, hearts were harvested and retrograde perfused through the aorta in a modified Langendorf system with an isolation solution containing collagenase type 2 (Worthington Biochemical Corp.) and protease type XIV (Sigma).
Protocols
Cells were loaded with Fura-2. The SL and intracellular Ca2+ concentration ([Ca2+]i) were measured simultaneously in CMs stimulated at 0.5, 1, 2, 3, and 4 Hz. Experiments were repeated after a 10-min incubation with HYD (0.1, 1, and 10 μm; Sigma), nitroglycerin (NTG, 0.1, 1 and 10 μm; American Regent Laboratories, Inc., Shirley, NY), 10 μm ISDN (Alexis Biochemicals, Enzo Life Sciences, Inc., PA), or HYD plus either NTG or ISDN. Experiments in SHHF rat CMs were carried out similarly, with the exception that the concentration of drugs was as follows: 10 μm HYD, 10 μm NTG, and their combination. All experiments were conducted at 37 °C.
Contractility and Ca2+ Measuring
SL shortening (%ΔSL) was recorded with an IonOptix iCCD camera and calculated as: (resting SL − peak SL) × 100/resting SL. Ca2+i was measured using a dual excitation (340/380 nm) spectrofluorometer (IonOptix LLC, Milton, MA). The “in vivo” calibration was performed using solutions containing 10 μm ionomycin (Sigma) as described by Grynkiewicz et al. (19) and the [Ca2+]i was calculated using the following equation,
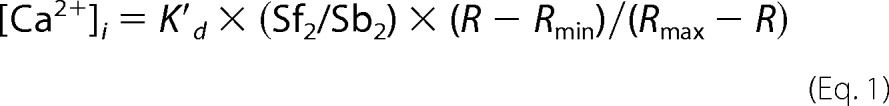 |
where Kd′ (apparent dissociation constant for Fura-2) in adult myocytes was 224 nm. Rmin and Rmax as well as the scaling factors (Sf2 and Sb2) were extracted from the calibration curves. Δ[Ca+2]i amplitude was considered as: peak [Ca+2]i − resting [Ca+2]i.
SR Ca2+ Leak and SR Ca2+ Load Measurement
SR Ca2+ leakage was assessed with tetracaine as described by Shannon et al. (20). Briefly, after pacing was stopped, a fast switch to a 0 Na+, 0 Ca2+ Tyrode solution (Na+ was replaced by an equimolar amount of Li+) was performed. After 60 s, similar to Bassani et al. (21), a rapid switching to 0 Na+, 0 Ca2+ solution containing 20 mm caffeine to assess SR Ca2+ content was applied. Following recovery of the cell, the same pacing protocol was assessed. After stop pacing, a switch to 0 Na+, 0 Ca2+ Tyrode solution containing 1 mmol/liter of tetracaine (Sigma) was performed. The observed drop in the Fura-2 ratio compared with the nontetracaine treated condition was considered the Ca2+ leak for a particular CM. After assessing the Ca2+ leak, tetracaine was washed out by superfusing fresh 0 Na+, 0 Ca2+ Tyrode solution and then a new caffeine challenge was applied to estimate the SR Ca2+ load. SR Ca2+ contents were calculated considering that SR represents 3.5% and cytosol 65% of the CM volumes. The following equation from Shannon et al. (20) was used: [Ca2+]SR = [Ca2+]caff + (βmax-SR × [Ca2+]caff)/([Ca2+]caff + Kd-SR). [Ca2+]SR is the SR Ca2+ content, [Ca2+]caff is the SR Ca2+ released by caffeine, βmax-SR and Kd-SR are the usual Michaelis parameters for SR Ca2+ binding. SR leak-SR load pairs were grouped by comparable SR Ca2+ loads and expressed as a leak-load relationship.
Detection of Superoxide by DHE Staining
Fresh isolated mouse CMs were incubated at room temperature for 30 min with dihydroethidium (DHE, 3 μm; Molecular Probes). After washing with PBS, cells were fixed with 2% paraformaldehyde for 5 min at 4 °C. Finally, the samples were mounted in Prolong Gold anti-fade (Invitrogen). The images were obtained using a Zeiss LSM-710 confocal microscope. Nuclear fluorescence captured at 562 nm was quantified using the Image-Pro plus software (MediaCybernetics, Silver Spring, MD) and normalized by cytosolic fluorescence.
NOX-dependent Superoxide Production
NOX-dependent superoxide production was measured in heart homogenates from NOS1−/− mice using lucigenin (5 μm)-enhanced chemiluminescence (β-NADPH 300 μm; room temperature) on a microplate luminometer (Veritas, Turner Biosystems, Sunnyvale, CA). Chemiluminescence readings were expressed as integrated light units. Experiments were performed in the presence of increasing concentrations of HYD and NTG (0.01, 0.1, and 1 mm).
XOR Activity
XOR activity was investigated by measuring uric acid (22) and superoxide production. Heart homogenates from NOS1−/− mice were passed through a Sephadex G-25 column (GE Healthcare) and XOR-dependent superoxide production was measured using Amplex Red (Molecular Probes, Eugene, OR). The effluent was assessed at 295 nm for uric acid. Both assay plates were incubated at 37 °C for 30 min in the presence or absence of allopurinol, and HYD and NTG at increasing concentrations.
Assessment of Ca2+ Myofilament Responsiveness
Myofilament responsiveness to Ca2+ was assessed using the steady-state relationship between SL and [Ca2+]i in intact single CMs tetanized by high-frequency (10 Hz) stimulation after exposure to thapsigargin (0.2 μm for 15 min), as described previously (23). With this approach, the Ca2+i was reversibly clamped during the tetanic contracture for 20 s and then rapidly returned to resting levels upon cessation of electrical stimulation via the Na+/Ca2+ exchanger. The steady-state levels of Ca2+ achieved during tetanus exposure were regulated by subjecting CMs to Tyrode solutions containing increasing concentrations of Ca2+ (0.1, 1.0, 5.0, 10, and 20 mm CaCl2).
Western Blot Immunoanalysis
Hearts from 12 NOS1−/− mice were perfused with Krebs solution (30 min; n = 6), or Krebs plus 10 μm HYD (30 min; n = 4), or 10 μm ISDN (30 min; n = 2). Hearts were homogenized in cold RIPA buffer containing the protease inhibitor mixture and phosphatase inhibitors. Samples were electrophoresed using a NuPAGE 10% Bis-Tris gel (Invitrogen) and transferred to PVDF membranes (Bio-Rad Laboratories). Immunoblot analysis was performed with goat polyclonal antibody for SERCA2 (Santa Cruz Biotechnology, Inc.), mouse monoclonal antibody for phospholamban (PLB; Pierce, Thermo Scientific), rabbit polyclonal for phospho-PLB(Ser16) (Pierce, Thermo Scientific), PLB phospho Thr-17 (Badrilla, Leeds, UK), NCX1 (RDI, Flanders, NJ), and GAPDH (as loading control; Santa Cruz). Phosphorylated PLB were expressed as a ratio of Ser(P)-16/PLB or Thr(P)-17/PLB and SERCA2, NCX1, or total PLB expression compared with GAPDH.
Statistical Analysis
Data are reported as mean ± S.E. Statistical significance was determined by unpaired Student's t test, one-way ANOVA, or two-way ANOVA followed by Student's-Newman-Keuls or Bonferonni's post hoc tests, as appropriate, using the GraphPad Prism version 4.02 (GraphPad Prism Software Corporation, San Diego, CA). The null hypothesis was rejected at p < 0.05.
RESULTS
Hydralazine and Organic Nitrates Improved Contractility in NOS1−/− CMs
To determine the effect of the drugs on CM contractility, we measured SL shortening in cells from WT and NOS1−/− mice paced at 0.5, 1, 2, 3, and 4 Hz (Fig. 1, A and B). At baseline, SL shortening was similar in NOS1−/− and WT CMs (0.5 Hz; Table 1). However, as previously described (12), the increased SL shortening due to augmented pacing frequency was blunted in NOS1−/− CMs compared with WT (4Hz, Table 1, and Fig. 1, A and B, p < 0.0001 versus WT, two-way ANOVA).
FIGURE 1.

Contractile performance in NOS1−/− cardiomyocytes. A, representative traces of SL in WT (black) and NOS1−/− (red) CMs under increasing rates of pacing from 0.5 to 4 Hz. B, CM contractility expressed as % SL shortening versus resting SL, in NOS1−/− (■; n = 50–54 cells) compared with WT (□; n = 34–41 cells). C, SL shortening at 4 Hz expressed as the percentage of shortening versus resting SL, in WT control or NOS1−/− CMs in the absence (bl) or presence of HYD (0.1, 1 or 10 μm), NTG (0.1, 1, or 10 μm), ISDN (10 μm), and a combination (HYD + NTG, 0.1, 0.5 or 10 μm each and HYD + ISDN; 10 μm each). **, p < 0.01; ***, p < 0.001 versus WT control, two-way ANOVA.
TABLE 1.
Baseline myocyte performance
| WT |
NOS1−/− |
|||
|---|---|---|---|---|
| 0.5 Hz | 4 Hz | 0.5 Hz | 4 Hz | |
| Cellsa | 40–41 | 32–34 | 50–54 | 40–50 |
| SL shortening (%)b | 4.75 ± 0.30 | 9.30 ± 0.50 | 4.27 ± 0.30 | 5.90 ± 0.40c |
| Diastolic [Ca2+]i (nm) | 234 ± 16 | 298 ± 26 | 284 ± 19d | 337 ± 19 |
| Δ[Ca2+]i transient (nm) | 160 ± 13 | 315 ± 40 | 254 ± 19e | 278 ± 21 |
| Δ[Ca2+]i decay (τ s) | 0.198 ± 0.009 | 0.091 ± 0.004 | 0.231 ± 0.013d | 0.101 ± 0.006 |
| SR Ca2+ load (μm) | 42.0 ± 7.4 | 70.6 ± 7.5 | 37.9 ± 7.2 | 52.4 ± 4.7d |
a Five to 10 cells per heart were studied.
b SL shortening is calculated as: (resting SL − peak SL)/resting SL × 100.
c p < 0.001 versus WT (unpaired Student's t test).
d p < 0.05 versus WT (unpaired Student's t test).
e p < 0.01 versus WT (unpaired Student's t test).
HYD augmented the frequency-dependent SL shortening in NOS1−/− CMs in a concentration-dependent manner (Figs. 1C and 2E, supplemental Table S1). In contrast, NTG alone did not affect the SL shortening in NOS1−/− (Fig. 1C, supplemental Table S1). To test whether the drug combination would have a synergistic effect, NOS1−/− myocytes were incubated with HYD plus NTG. Although this treatment restored the FFR in a dose-dependent manner, the response was not greater than hydralazine alone (Figs. 1C and 2E, Supplemental Table 1). In WT CMs, neither HYD nor NTG, alone or in combination, affected frequency-dependent SL shortening (data not shown).
FIGURE 2.

Intracellular Ca2+ transient (Δ[Ca2+]i) in NOS1−/− cardiomyocytes. A, Δ[Ca2+]i (nm) in NOS1−/− (■; n = 40–50 cells) compared with WT (□; n = 32–40 cells). B, Δ[Ca2+]i in NOS1−/− (■) and WT (□) CMs, expressed as % increase versus 0.5 Hz. C, Δ[Ca2+]i at 4 Hz in WT control or NOS1−/− CMs in the absence (bl) or presence of HYD (0.1, 1 or 10 μm), NTG (0.1, 1 and 10 μm), ISDN (10 μm), and a combination (HYD + NTG; 0.1, 0.5, and 10 μm each; and HYD + ISDN; 10 μm each). D, ΔCa2+ frequency response in WT CMs (□) under baseline conditions or NOS1−/− baseline (■) or treated with 10 μm HYD (red circle) or 10 μm HYD + NTG (blue circle). E, FFR in CMs under the same conditions as mentioned above. The combination induces a significant increase in FFR but does not change ΔCa2+ significantly compared with NOS1−/− CMs. However, the contractile reserve is restored. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 versus NOS1−/− control; †, p < 0.05 versus HYD alone; two-way ANOVA.
We next assessed whether ISDN, the pharmacologically employed nitrate, had similar effects as NTG. Indeed, the combination of HYD and ISDN also induced an increased frequency-dependent SL shortening in NOS1−/− compared with untreated NOS1−/− CMs (Fig. 1C and supplemental Table S1; p = 0.004) at an equimolar concentration of 10 μm. Similar to NTG, 10 μm ISDN did not alter contractility in NOS1−/− (Fig. 1C and supplemental Table S1).
Divergent Effects of Hydralazine and Nitroglycerin on Ca2+ Transient in NOS1−/− CMs
Resting diastolic [Ca2+]i values were slightly elevated in NOS1−/− compared with WT CMs, over the range of studied frequencies (Table 1; p < 0.01) as previously described (12, 24). When the [Ca2+]i transient (Δ[Ca2+]i) was studied, [Ca2+]i-FFR was clearly depressed in NOS1−/− CMs compared with WT, as previously described (Fig. 2, A and B, p < 0.0001 versus WT; two-way ANOVA) (18, 24, 25). HYD induced a Δ[Ca2+]i increase in a concentration-dependent manner (Fig. 2C) as well as significantly increased the [Ca2+]i-FFR (Fig. 2D, supplemental Table S2, p < 0.05). Thus, the increase in contractility in response to HYD was mirrored by a concomitant increase in Δ[Ca2+]i (Fig. 2E).
Although NTG did not affect contractility, it induced a concentration-dependent decrease in the Δ[Ca2+]i amplitude compared with control NOS1−/− myocytes most clearly seen at 4 Hz. This decrease was reproduced using 10 μm ISDN (Fig. 2C and supplemental Table S2). The combination of HYD and NTG (Fig. 2D) or ISDN (supplemental Table S2) on NOS1−/− CMs restored the increasing [Ca2+]i-FFR (frequency p = 0.0213 or p = 0.0014, respectively; two-way ANOVA) by reducing the Δ[Ca2+]i amplitude toward the levels in WT CMs (p = 0.042 or p = 0.0005 versus HYD alone, respectively). Thus, for any given improvement in cardiac contraction due to HYD, organic nitrates offset the increase in [Ca2+]i, thereby enhancing contractile efficiency (Fig. 2, D and E). Neither HYD nor NTG, alone or in combination (10 μm), altered the [Ca2+]i-FFR in WT CMs (data not shown).
We also examined the peak [Ca2+]i decay time constant (τ), which was significantly higher in NOS1−/− than WT CMs (p < 0.0001, Table 1, Fig. 3, A and B). All treatments accelerated τ (particularly at 0.5 Hz, Fig. 3B and supplemental Table S3) in a dose-dependent manner (p < 0.0001 HYD alone or in combination with NTG versus NOS1−/− baseline; p = 0.001 NTG alone versus NOS1−/− baseline, two-way ANOVA). ISDN was equally effective in reducing τ, either alone or combined with 10 μm HYD (p < 0.001; supplemental Table S3). Reuptake of Ca2+ assessed by decay of caffeine-induced Ca2+ transients in a Na+ and Ca2+-free solution, which avoids any Ca2+ flux throughout the sarcolemma, was slower in NOS1−/− (K = 0.1888/s; T50 = 3.65 ± 0.12 s, fitted by an one-phase exponential equation) compared with WT cells (K = 0.2724/s; T50 = 2.53 ± 0.08 s; p = 0.0005). Treatment with either 10 μm HYD or NTG increased the SR Ca2+ reuptake rate in NOS1−/− (K = 0.240/s, T50 = 2.88 ± 0.17 s, p = 0.0003 and K = 0.2392/s, T50 = 2.88 ± 0.11 s, p < 0.0001, respectively). The combination exerted an additive effect (K = 0.2919/s, T50 = 2.36 ± 0.06 s, p < 0.0001) suggesting that the improvement in the regular field-stimulated Δ[Ca2+]i decay was mediated by enhanced SERCA2 activity. This speculation was confirmed by assessing phosphorylation of PLB in cardiac homogenates from NOS1−/− hearts treated with either 10 μm HYD or ISDN. HYD increased phosphothreonine 17 (Thr(P)-17) for pentameric PLB (p = 0.0393); but not on total PLB (p = 0.0597) or monomeric PLB (Fig. 3, C and D). ISDN increased total Thr(P)-17 (p = 0.011; Fig. 3, E and F). Neither treatment significantly increased phosphoserine 16 (Ser(P)-16; Fig. 3, D–F).
FIGURE 3.

Hydralazine and organic nitrates enhance Ca2+ influx to the SR in NOS1−/− cardiomyocytes. A, Ca2+ decay time constant (tau, τ) versus frequency in NOS1−/− (■) compared with WT (□) CMs. B, Tau values at 0.5 Hz in WT control or NOS1−/− CMs treated with or without HYD, NTG, or a combination (HYD + NTG) at the indicated concentrations (μm) (n = 5–21 cells) (*, p < 0.05; **, p < 0.01; and ***, p < 0.001 versus NOS1−/− control; two-way ANOVA). C, PLB phosphorylation at threonine 17 (Thr(P)-17) in baseline or 10 μm HYD-treated NOS1−/− hearts (total and pentameric Thr(P)-17 PLB). D, representative Western blots for all studied proteins in the absence or presence of 10 μm HYD. E, PLB Thr(P)-17 in baseline or 10 μm ISDN-treated NOS1−/− hearts (total and monomeric Thr(P)-17 PLB). Phosphorylated PLB at Ser-16 (Ser(P)-16, bottom) was also measured. F, representative Western blots. No significant changes were observed in Ser(P)-16, SERCA2, or NCX1 with either treatment. †, p < 0.05 versus NOS1−/− control; Student's t test.
Hydralazine Alone and in Combination with Nitroglycerin, Lowers SR Ca2+ Leakage Toward Normal
We next assessed SR Ca2+ leak and the SR Ca2+ leak-load relationship, using the technique of Shannon et al. (20) using tetracaine, a RyR2 blocker (Fig. 4A). SR Ca2+ leak was augmented in NOS1−/− (R2 = 0.5225 for WT and R2 = 0.8827 for NOS1−/−, Fig. 4B; p = 0.0002). The decreased SR Ca2+ content and increased diastolic Ca2+ exhibited by NOS1−/− compared with WT CMs are consistent with the elevated Ca2+ leak. HYD reduced the leak in a concentration-dependent manner in NOS1−/− myocytes (Fig. 4C; p = 0.0012). Neither 1 nor 10 μm HYD significantly affected the SR Ca2+ leak in WT CMs (data not shown).
FIGURE 4.

Assessment of SR diastolic Ca2+ leakage in the presence of hydralazine, organic nitrates, or their combination. A, protocol to assess the SR Ca2+ leak using tetracaine to block RyR2 and caffeine to estimate the SR Ca2+ content. B, SR Ca2+ load-leak relationship in WT (□) and NOS1−/− (■) CMs. C, SR Ca2+ load-leak relationship in NOS1−/− CMs in the absence (red ■) or presence of HYD (0.1 μm, red ▾, 1 μm red ▴ or 10 μm, red ●). D, in the presence of NTG (0.1 μm, green ▾; 1 μm, green ▴; or 10 μm, green ●). E, in the presence HYD plus NTG (0.1 μm, blue ▾; 0.5 μm, blue ▴; or 10 μm, blue ●). NOS1−/− CMs exhibited an increased SR Ca2+ leak and HYD alone or a combination reduced it in a dose-dependent manner. F, dose-response relationship; effect of increasing concentrations of HYD (red ▴), NTG (green ●), and a combination of HYD + NTG (blue ▾) on SR Ca2+ leak in NOS1−/− CMs (**, p < 0.01 versus nontreated NOS1−/− CMs; one-way ANOVA). G, SR Ca2+ load-leak curves in NOS1−/− (■) CMs in the absence or presence of 10 μm ISDN alone (dark green ●) or in combination with 10 μm HYD (dark blue ●). Load-leak relationship of WT was included as normal control (□). H, SR Ca2+ content at 4 Hz. HYD alone and in combination with NTG restored the SR Ca2+ content in NOS1−/− in a concentration-dependent manner toward normal levels as exhibited by WT CMs. NTG alone had no significant effects. *, p < 0.05 and **, p < 0.01 versus NOS1−/− control; two-way ANOVA.
NTG did not significantly affect the SR Ca2+ leak in NOS1−/− cells (Fig. 4D). In contrast, the combination of NTG plus HYD reduced the leak in a concentration-dependent manner as shown by the leak-load relationship (Fig. 4E; p = 0.0001). Fig. 4F shows the drug concentration-SR Ca2+ leak response relationship. The IC50 for HYD was 0.0705 ± 0.0473 μm (at matched SR load of 50 μm). The combination exerted a comparable effect to HYD alone (IC50 = 0.160 ± 0.128 μm).
The combination of 10 μm HYD and 10 μm ISDN reduced the SR Ca2+ leak in NOS1−/− equal to HYD plus NTG (Fig. 4G; p = 0.0186). ISDN alone exhibited differential effects at the low SR Ca2+ load (reduced leak) compared with the higher load (no effect) in NOS1−/− CMs (Fig. 4G). We also assessed the effects of 10 μm HYD plus 10 μm NTG on the SR Ca2+ leak in WT CMs but found no significant difference compared with control, whereas 10 μm NTG significantly reduced the degree of the SR Ca2+ leak in WT (data not shown).
Cardiomyocyte SR Ca2+ Content
Because SR Ca2+ storage is the key determinant of FFR, we estimated the Ca2+ content by rapidly infusing caffeine after pacing the myocytes. Although increasing the pacing augmented the SR Ca2+ content in both NOS1−/− and WT CMs (p < 0.05, Table 1), the increase was smaller in NOS1−/− (38.2%) than WT (66.6%), which was consistent with previous studies (25). In the presence of HYD, NOS1−/− CMs also exhibited a rise in SR Ca2+ content in response to increasing frequency (p < 0.05, data not shown). SR Ca2+ content evaluated at 4 Hz was increased by HYD in a concentration-dependent manner, reaching a load similar to WT CMs (p < 0.01) (Fig. 4H). We also determined that, similar to HYD alone, the combination of equal concentrations of NTG and HYD increased the SR Ca2+ load in a concentration-dependent manner in NOS1−/− CMs (Fig. 4H). However, 10 μm HYD alone or combined with 10 μm NTG abolished the frequency-induced rise in SR Ca2+ content in WT CMs (p = 0.001 and p = 0.005, respectively; data not shown). NTG alone did not affect the SR Ca2+ load in NOS1−/− (Fig. 4H) or WT myocytes (data not shown).
Detection of Superoxide by DHE Staining
To determine the molecular underpinnings for improved E-C coupling in NOS1−/− myocytes in response to these drugs, we used the superoxide-sensitive dye DHE to assess the scavenging capacity of HYD and NTG alone, or in combination at 0.1, 1, or 10 μm (Fig. 5, A and B). As DHE is oxidized by superoxide and then translocated to the nucleus (Fig. 5B), the ratio between fluorescence in the nucleus and cytoplasm indexes superoxide production. Although the lowest drug concentrations did not affect superoxide detection, increasing concentrations of HYD as well as NTG scavenged superoxide (p < 0.05; Fig. 5A). In combination they had an additive effect (p < 0.01; Fig. 5A). Thus, at concentrations that reduced the SR Ca2+ leak, HYD alone or in combination with NTG exhibited an antioxidant effect.
FIGURE 5.

Oxidative stress is attenuated by hydralazine and nitroglycerin. A, superoxide-mediated oxidation of DHE. The ratio of fluorescence in the nucleus/cytoplasm preincubated 30 min with HYD (red ▴), NTG (green ●) and a combination of HYD + NTG (blue ▾) is shown. B, representative pictures of mice ventricular CMs stained with 3 μm DHE (superoxide-sensitive dye) in the absence (control) or presence (0.1, 1, or 10 μm) of the drugs (Bar, 10 μm). Yellow arrows indicate nuclei highly stained and green arrows show nuclei with little or no dye. C, NOX-dependent superoxide production in NOS1−/− cardiac homogenates. Effects of HYD and NTG on the NOX enzymatic system containing 0.3 mm NADPH as substrate. D, XOR activity in WT and NOS1−/− mouse hearts in terms of superoxide production (left) or uric acid generation (right). E, effect of allopurinol (♦), HYD, and NTG on XOR-dependent superoxide production in NOS1−/− cardiac homogenates. F, effects of allopurinol, HYD, and NTG on uric acid production in the XOR enzymatic system containing 0.2 mm xanthine as substrate. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 versus baseline, one-way ANOVA; †, p < 0.05 versus nontreated NOS1−/−, Student's t test.
NOX-dependent Superoxide Production
We also studied activation of specific oxidases, XOR and NOX, in mouse heart homogenates in the presence of either HYD or NTG. NOX activity of WT (n = 3) and NOS1−/− (n = 4) hearts was similar (2.29 ± 0.15 versus 2.19 ± 0.13 integrated light units/μg of protein, respectively). HYD (0.1–1 mm, IC50 0.42 ± 0.23 mm) decreased NOX activity (Fig. 5C, p < 0.01 for both concentrations versus control) in NOS1−/− cardiac homogenates. NTG (>0.01 mm, IC50 0.013 ± 0.06 mm) also inhibited NOX-dependent superoxide production (Fig. 5C; p < 0.05 for 0.01 mm, p < 0.01 for 0.1 and 1 mm versus control).
XOR Activity
NOS1−/− hearts (n = 3) exhibited up-regulated XOR activity compared with WT (n = 3) using the Amplex Red detection of XOR-mediated ROS production assay (Fig. 5D; 2.1 ± 0.18 versus 1.63 ± 0.09 milliunits/μg of protein, respectively; p < 0.05), confirming previous findings in this mouse (25–27). Amplex Red XOR activity was inhibited by allopurinol (>0.01 mm, IC50 0.19 ± 0.06 mm, Fig. 5D; n = 3) and HYD (IC50 of 0.7 ± 0.3 mm, Fig. 5E; n = 3) in NOS1−/− cardiac homogenates. Interestingly, NTG (IC50 1.0 ± 0.4 mm, Fig. 5E, n = 3) also inhibited XOR. Because the Amplex Red assay measures superoxide levels, reduced activity could also reflect increased O2˙̄ scavenging. To address this possibility, we measured uric acid conversion as a direct measurement of enzymatic activity. With this approach, NOS1−/− hearts (n = 3) again exhibited increased XOR activity compared with WT (n = 3) (Fig. 5D; 9.8 ± 0.46 versus 8.5 ± 0.21 milliunits/mg tissue, respectively, p < 0.05). Allopurinol (>0.1 mm) inhibited XOR enzyme activity in NOS1−/− mice (Fig. 5F, n = 3). Interestingly, HYD did not, but NTG did display XOR-inhibitory activity in NOS1−/− (Fig. 5F, n = 3).
Organic Nitrates Increased Myofilament Responsiveness to Ca2+ in NOS1−/− Myocytes
Because our FFR data suggested that organic nitrates improve efficiency in myofilament responsiveness to Ca2+i, we assessed the mechanism by which NTG offsets the HYD-induced Δ[Ca2+]i increase toward normal, and directly tested the myofilament responsiveness in NOS1−/− as well as WT CMs in the presence of 10 μm NTG. Responsiveness of myofilaments to Ca2+ (both sensitivity and maximal response) was lower in NOS1−/− compared with WT CMs (EC50,NOS1−/− = 0.627 ± 0.0185 and EC50,WT = 0.398 ± 0.018 μm, respectively; p = 0.0009, Fig. 6, A–D). In agreement with previous studies using different NO donors (23, 28), NTG reduced myofilament responsiveness to Ca2+i in WT (not shown). Surprisingly, but consistent with our results, NTG increased responsiveness to Ca2+ in NOS1−/− (Fig. 6, B and D), as shown by a leftward shift (EC50,NOS1−/−,NTG = 0.427 ± 0.0186 μm, p = 0.0016; Fig. 6C). Therefore, NTG restored the impaired myofilament sensitivity in NOS1−/− toward normal levels observed in WT CMs. Analysis of [Ca2+]i-SL shortening loops confirm that sensitivity to Ca2+ is deficient in NOS1−/− CMs but treatment with 10 μm ISDN improves it toward WT (Fig. 6E).
FIGURE 6.

Myofilament sensitivity to Ca2+ in NOS1−/− cardiomyocytes. A, representative traces of SL shortening (top) in response to similar increases of Ca2+i (bottom) obtained by high frequency-induced tetanus of the CMs in the presence of thapsigargin. Response of NOS1−/− was compared with WT cells in the absence (left) or presence of 10 μm NTG (right). B, curves of myofilament sensitivity to [Ca2+]i in NOS1−/− baseline (■) or 10 μm NTG-treated (green ■) CMs compared with WT (□) as normalized to the maximal response in each group (sigmoidal dose-response − variable slope − fitting). C, EC50 values of the curves. D, myofilament sensitivity curves expressed as absolute values; the fitting provides not only higher EC50 values but also a smaller maximal response in the NOS1−/− control. E, SL shortening − [Ca2+]i loops of WT (□; n = 27 cells) or NOS1−/− CMs in the absence (■; n = 30 cells) or presence of 10 μm ISDN (dark green ▾; n = 10 cells). The slopes that fit the relaxation phases of the loops correlate with the sensitivity of myofilaments to Ca2+. Sensitivity of NOS1−/− (—) is lower than WT (- - -); however, treatment with ISDN (—) increased the fitted slope toward the values observed in WT CMs. *, p < 0.05 versus NOS1−/−, Student's t test.
Frequency-stimulated Contractility and Ca2+ Transients Are Improved by Hydralazine and Nitroglycerin in CMs from Heart Failure Rats
To study the effects of HYD and NTG in a model of heart failure (dilated cardiomyopathy), we used SHHF rats. Usually, the SL shortening amplitude in rat CMs is flat or negative in response to increasing the pacing rate. We recently showed that SL shortening drops more abruptly in SHHF than WKY control CMs (13). Here, we also observed a significant decrease in contractility in SHHF compared with WKY CMs (p = 0.0002, Fig. 7, A and B). Treatment with 10 μm HYD significantly improved SL shortening in SHHF myocytes (p = 0.0414). Consistent with observations in NOS1−/−, 10 μm NTG did not affect the SL shortening in SHHF myocytes (p = 0.2553). The combination of HYD and NTG showed a strong trend toward increased contractility in SHHF myocytes (p = 0.0839), which also exhibited an FFR pattern similar to HYD alone (Fig. 7A). Furthermore, there was a significant difference at 4 Hz compared with the SHHF control (p = 0.0359, Fig. 7B). Normalization of SL shortening as a percentage of 0.5 Hz pacing showed that HYD alone as well as in combination with NTG significantly improved FFR in failing CMs (p = 0.035 and p = 0.0013, respectively, data not shown).
FIGURE 7.

SL shortening and Ca2+ transient amplitude-frequency response in SHHF cardiomyocytes. A, FFR in SHHF rat CMs in the absence (■) or presence of 10 μm HYD (red ●), 10 μm NTG (green ●), or a combination (HYD + NTG, 10 μm each; blue ●) compared with WKY control CMs (□). B, bar graph showing contractility data at 4 Hz. C, Ca2+-FFR in SHHF rat CMs treated with 10 μm HYD or 10 μm NTG or their combination (HYD + NTG, 10 μm each). D, bar graph showing Δ[Ca2+]i data at 4 Hz. In SHHF CMs, similar effects to those observed in NOS1−/− were obtained with these treatments (n = 5–15 cells). *, p < 0.05; ***, p < 0.001 versus SHHF control; two-way ANOVA. †, p < 0.05; ††, p < 0.01 versus SHHF control, Student's t test.
The ΔCa2+ increased in WKY CMs in response to pacing (p = 0.0019, Fig. 7C), compared with the flat pattern in SHHF CMs. In agreement with the results obtained in NOS1−/− mice, in failing CMs Δ[Ca2+]i amplitude was augmented by 10 μm HYD (p = 0.0124) in comparison to SHHF controls (Fig. 7C). In this setting, the combination of HYD with 10 μm NTG also induced a nonsignificant rise in Δ[Ca2+]i primarily at the highest frequency (p = 0.1756, Fig. 7D). On the other hand, consistent with the effect observed in contractility in this model, Δ[Ca2+]i was not affected by NTG (Fig. 7, C and D). Therefore treatment with HYD and NTG in a genetic model of heart failure enhances E-C coupling in a similar fashion (yet species appropriate) as observed in NOS1−/− cells, a model of NO/redox imbalance.
DISCUSSION
Our major new finding is that HYD and NTG corrected the increased Ca2+ leak from the SR, thus normalizing contractility-frequency responses by modulating Ca2+ cycling in models of NO/redox imbalance. Although HYD improved contractile force in parallel with increasing Ca2+ transients and total restoration of leak, NTG offset Ca2+ cycling without impairing contractility, thereby improving contractile efficiency. Direct measure of myofilament sensitivity confirmed that NTG restores depressed myofilament sensitivity to Ca2+ in NOS1−/− myocytes. Importantly, NTG inhibited two major sources of cellular ROS production, NOX and XOR, and also exhibited ROS scavenging capacity. HYD scavenged ROS but did not inhibit XOR. Although NOX activity appeared to be inhibited by HYD, this effect could be an epiphenomenon due to the superoxide scavenging properties of HYD, because NOX activity was only assessed by NADPH-induced superoxide production. Despite the lack of NOS1-derived NO and the fact that NOS3 is uncoupled under oxidative stress (29), peroxynitrite (ONOO−) was measurable in NOS1−/− homogenates and was reduced by both HYD and NTG, in a dose-dependent manner (data not shown).
Model of NO-Redox Imbalance
NOS1−/− mice are widely used to investigate the involvement of NO in E-C coupling because CMs exhibit a particularly impaired performance due to deficient NO production and consequent NO/redox imbalance (12, 25). Sears et al. (30) showed that NOS1−/− CMs have larger ΔCa2+, which is consistent with larger ICa and augmented SL shortening compared with control animals, at low frequencies. However, we (12, 18) and others (31) have shown that the contractile reserve is impaired in NOS1−/− as the increase in ΔCa2+ and contractility in response to pacing are depressed. Consistently, we have found that the reduced SR Ca2+ load was associated with augmented SR Ca2+ leak. In contrast, Sears et al. (30) found that the SR Ca2+ load was higher in NOS1−/−, when measured at 1 Hz. In our hands, SR Ca2+ loads at low frequencies were not different between strains, but during pacing with rates more physiologic for murine myocytes, were depressed versus WT (25). Similarly, Wang and colleagues (32) demonstrated that the SR Ca2+ leak is reduced in NOS1−/− myocytes, suggesting that NOS1-derived NO induces RyR2 to leak. However, and in contrast to our experiments, this group studied the SR Ca2+ leak at room temperature (22 °C), whereas, we assessed the leak at 37 °C. This simple detail may be responsible for the differences. We recently demonstrated that SHHF rats, a representative heart failure model that exhibits oxidative stress as a hallmark (13), share certain specific features with NOS1−/− mice. Both models exhibit increased superoxide production associated with elevated XOR activity (25) and blunted NOS1 activity, evidenced by hyponitrosylation of RyR2 (12). Moreover, increased SR Ca2+ leak, reduced SR Ca2+ load, and depressed Ca2+ and force-frequency responses are characteristics of these models (13, 18). Unlike SHHF rats, NOS1−/− mice are not considered a representative model for heart failure. Rather, they develop age-related cardiac hypertrophy (a pre-heart failure stage) and exhibit accelerated mortality (33, 34). Additionally, after myocardial infarction, NOS1−/− also undergo exaggerated remodeling and higher mortality than WT controls (27). Consequently, NOS1−/− mice are an excellent model of NO/redox imbalance that share features with a model of heart failure, the SHHF rat.
Therapeutic Use of Hydralazine and Organic Nitrates
Recently, Cole et al. (2) reviewed the use of HYD-ISDN as a treatment for heart failure (1, 3) highlighting the fact that the mechanistic basis for this regimen to reduce all-cause mortality remains to be established. We show for the first time that this regimen restores disrupted NO/redox equilibrium and in so doing restores E-C coupling derangements.
The effects of HYD and NTG resemble, in part, those of XOR inhibitors. Allopurinol improves myocyte contraction without changing Δ[Ca2+]i enhancing contractile efficiency in both NOS1−/− mice and heart failure models (25, 35, 36). In large animal models, allopurinol restores mechano-energetic uncoupling (15). Interestingly, XOR inhibitors did not have clinical benefits in a broad heart failure population, although it may improve outcomes in patients with high uric acid levels (37) and may benefit patients with hypertension and ischemic heart disease (38–40).
Mechanism of Action and Pharmacology of Hydralazine
HYD is a peripheral vasodilator used for decades to treat essential hypertension and heart failure. The mechanism of its effect on the vasculature remains unknown. It is proposed to inhibit the endoplasmic reticulum Ca2+ release in vascular smooth muscle (41, 42), although the molecular mechanism by which HYD regulates the IP3 receptor (a member of the superfamily of homotetrameric ligand-gated intracellular Ca2+ channels, which includes RyR2) has not been elucidated. Because HYD increases contractility and Ca2+ transients in myocardial fibers (43), it may positively affect the unregulated RyR2 activity in NOS1−/−.
We showed that HYD enhanced depressed NOS1−/− CM contractility and [Ca2+]i responses resulting from NO/redox imbalance. Importantly, HYD restored the increased SR Ca2+ leakage both alone or in combination with NTG. This feature suggests a regulatory action of HYD on RyR2 gating, possibly depressing Ca2+ release in diastole and enhancing gating in systole.
Accordingly, a more efficient handling of Ca2+ would underlie the recovery of the SR Ca2+ content observed in CMs treated with HYD or HYD plus NTG. Biochemically, HYD + NTG exhibited an additive superoxide scavenging capacity at concentrations as low as 1 μm, as shown by DHE staining. This effect may be correlated with the additive acceleration in SR Ca2+ reuptake in NOS1−/− induced by combining HYD with NTG. Peroxynitrite links superoxide production to Ca2+ reuptake. Although ONOO− generation depends on superoxide availability, PLB phosphorylation is decreased due to ONOO−-dependent activation of phosphatases, impairing Ca2+ decay and relaxation in NOS1−/− (31, 44, 45). Furthermore, HYD and NTG scavenged ONOO− in NOS1−/− heart homogenates (data not shown). Together these findings suggest a role for scavenging of ONOO− on the recovery of the Ca2+ reuptake rate, a key process in E-C coupling in mouse CMs (44, 46). Considerable evidence supports the hypothesis that oxidative stress induces cardiac injury by oxidizing cellular constituents, including proteins critical for E-C coupling, and largely by diminishing NO bioactivity (9), thus playing a role in heart failure pathophysiology (47). Therefore, the effect of HYD, which reduced ONOO− in NOS1−/− hearts (data not shown), and scavenged XOR-mediated superoxide production may impact the redox regulation of RyR2 gating and the pharmacology of the response. Importantly, our results agree with Leiro et al. (48), who suggested antioxidant properties for HYD.
Mechanism of Action and Pharmacology of Organic Nitrates
Janero et al. (49) showed that NTG increases cardiac nitrosylation in vivo, and this effect was potentiated by xanthine oxidase inhibition with allopurinol, highlighting the interactions between NO and XOR-derived superoxide. Importantly, decreased NOS1 in cardiac SR contributes to depressed contractile reserve in response to pacing. It has been hypothesized that NTG may restore ion channel S-nitrosylation in NOS1−/− myocytes (24). We showed that NTG and ISDN restored the normal amplitude of calcium transients toward WT levels in NOS1−/− cells treated with HYD. This effect would be consistent with a cGMP-dependent regulation of L-type Ca2+ channels and/or restoration of RyR2 S-nitrosylation (33, 50) and occurs without affecting cardiac contractility, which correlates with the observed improvement in myofilament sensitivity induced by NTG in NOS1−/− CMs. This improvement may also be attributed to the restoration of the S-nitrosylation/normal redox state of redox-sensitive sites on sarcomeric proteins. Thus, this responsiveness is evident across the broad range of pacing frequencies tested, confirming that organic nitrates enhance contractile efficiency. It is reasonable to speculate that the lack of NOS1 and the resulting NO/redox imbalance reduces responsiveness of myofilaments to Ca2+ (as confirmed by our results). This reduction was restored by NO donors. These findings suggest that a feasible mechanism for the positive clinical outcomes of organic nitrates (when combined with HYD) may be by regulating intracellular Ca2+ handling and restoring myofilament sensitivity impaired by NO/redox imbalance, thereby modulating E-C coupling toward a more efficient performance.
Inhibition of NOX and XOR by NO has been previously demonstrated (26, 51–53), supporting our results. In contrast to the low concentrations required to scavenge superoxide production in isolated myocytes, the effective concentrations of NTG to inhibit the substrate-induced activity of these enzymes in cardiac homogenates exceeded achievable blood levels of this drug (10 ng/ml, 44 nm) (54).
Despite the successful use of nitrates such as NTG or ISDN in the treatment of a variety of cardiovascular diseases, nitrate tolerance during long-term use in heart failure has been described. Co-treatment with HYD has been demonstrated to prevent this effect (55) and several authors have tried to explain the mechanism of this phenomenon (56–58).
The effects of organic nitrates on SR Ca2+ leak appear to be uncertain and depend strongly on several variants such as the concentration or nature of the NO donor. As shown in the supplemental material, NTG exerted differential effects along the range of concentrations from 0.1 to 100 μm. On the other hand, ISDN reduced the SR leak in NOS1−/− at very low SR Ca2+ contents but did not affect it at increasing Ca2+ loads. We speculate that this differential response may correspond to the delicate equilibrium of the nitrosylation/denitrosylation cycle involved in the regulation of RyR2 activity (50).
Combination of HYD and NTG
The effect of NTG in NOS1−/− hearts may appear puzzling. At lower concentrations (0.1 and 1 μm), NTG did not significantly affect SL shortening, Ca2+ transient, SR Ca2+ leak or Ca2+ decline. It reduced the Ca2+ frequency response with a concomitant acceleration of Ca2+ decline at 10 μm. However, despite 10 μm NTG improving Ca2+ reuptake, there was a trend toward increasing SR Ca2+ leak (or at least effecting no change on SR Ca2+ leak; Fig. 3E), which would counteract the improvement in Ca2+ influx by the SR, thereby abolishing the SR Ca2+ content recovery. This balancing may explain why there was no change in SR Ca2+ load, although this was not reflected on the Ca2+ amplitude, which was reduced. Because Ca2+ influx (ICa) was enhanced in NOS1−/− (30), the reduced RyR2 gating in the presence of 10 μm NTG (or ISDN) may be caused by the down-regulation of the enhanced ICa in NOS1−/− by exogenous NO (as previously shown) (24). Thus, whereas hydralazine alone reduced RyR2 leak, when combined with NTG (or ISDN), down-regulation of the Ca2+-induced Ca2+ release (via inhibition of ICa) would oppose the effect of hydralazine, thereby leading to a reduced Ca2+ transient amplitude. Together with the reduced leak and increased reuptake under these conditions, the contractile reserve was recovered in NOS1−/−. Thus, the evidence of improved EC coupling in this model using the HYD-NTG treatment would be restoration of Ca2+ levels toward those exhibited by WT CMs and recovery of the increasing Ca2+ frequency response and myofilament responsiveness to Ca2+, where NTG played a key role.
Both HYD and NTG scavenged superoxide but were more potent in combination. However, we must consider that the chemical reactions of each molecule with superoxide yield different products. Although HYD can effectively scavenge the radical, the combination of NO and superoxide yields ONOO−, a reactive nitrogen species even more harmful than superoxide. In this sense, we speculate that ONOO− may target RyR2 in a similar manner as superoxide. Therefore, the SR Ca2+ leak was still elevated in the presence of NTG. Fig. 4F showed that despite 10 μm NTG inducing a trend toward an increased leak in NOS1−/−, 100 μm NTG restored it to baseline. This observation suggests that higher concentrations of NTG not only scavenge superoxide but also the surplus would counteract the deficiency of NO in NOS1−/− and is perhaps the mechanism of the combination of NTG and HYD.
Effects in SHHF Rats
Although this study is focused on NOS1−/− myocytes as a model of NO/redox imbalance, we also tested the effect of these drugs alone or in combination on failing CMs from SHHF rats. The results obtained in SHHF CMs correlate with those in NOS1−/− mice. HYD alone or in combination with NTG improved the Ca2+-frequency response and FFR. NTG alone induced a trend to increase contractility at 4 Hz with no changes in ΔCa2+, suggesting an improvement in myofilament sensitivity. These results allow us to speculate that our approach may be extrapolated to heart failure in humans.
Conclusion
Our data show novel effects of HYD and NTG. In combination they quench superoxide in isolated cells, concomitantly with physiologic functional effects, including improved myocardial contractility and Ca2+ cycling. At higher in vitro concentrations, this combination scavenges superoxide and peroxynitrite production from two major ROS generating enzymatic systems, NOX and XOR. This improvement in myocyte performance, normalization of SR Ca2+ leak, regulation of RyR2 gating, and myofilament sensitivity by NTG may be associated in part with normalizing NO/redox equilibrium. Thus, these drugs in combination exert direct myocardial effects that provide a mechanistic basis for the favorable functional and structural responses in the treatment of congestive heart failure.
Supplementary Material
Acknowledgments
We thank Drs. Ivonne Schulman and Wayne Balkan for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants HL-65455, 5R01AG025017, and R01 HL094849 (to J. M. H.).

This article contains supplemental Tables S1–S3.
- HF
- heart failure
- CM
- cardiomyocyte
- E-C
- excitation-contraction
- FFR
- force-frequency response
- HYD
- hydralazine
- ISDN
- isosorbide dinitrate
- NTG
- nitroglycerin
- NO/redox
- nitroso-redox
- NOS1−/−
- neuronal nitric-oxide synthase deficient mice
- NOX
- NADPH oxidase
- ONOO−
- peroxynitrite
- PLB
- phospholamban
- ROS
- reactive oxygen species
- RyR2
- cardiac ryanodine receptor
- SERCA2
- SR Ca2+-ATPase 2
- SHHF
- spontaneously hypertensive heart failure rats
- SL
- sarcomere length
- SR
- sarcoplasmic reticulum
- WKY
- Wistar Kyoto
- XOR
- xanthine oxidoreductase
- DHE
- dihydroethidium
- ANOVA
- analysis of variance.
REFERENCES
- 1. Taylor A. L., Ziesche S., Yancy C., Carson P., D'Agostino R., Jr., Ferdinand K., Taylor M., Adams K., Sabolinski M., Worcel M., Cohn J. N. (2004) Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N. Engl. J. Med. 351, 2049–2057 [DOI] [PubMed] [Google Scholar]
- 2. Cole R. T., Kalogeropoulos A. P., Georgiopoulou V. V., Gheorghiade M., Quyyumi A., Yancy C., Butler J. (2011) Hydralazine and isosorbide dinitrate in heart failure. Historical perspective, mechanisms, and future directions. Circulation 123, 2414–2422 [DOI] [PubMed] [Google Scholar]
- 3. Taylor A. L., Sabolinski M. L., Tam S. W., Ziesche S., Ghali J. K., Archambault W. T., Worcel M., Cohn J. N. (2012) Effect of fixed-dose combined isosorbide dinitrate/hydralazine in elderly patients in the African-American heart failure trial. J. Card. Fail. 18, 600–606 [DOI] [PubMed] [Google Scholar]
- 4. Antoons G., Vangheluwe P., Volders P. G., Bito V., Holemans P., Ceci M., Wuytack F., Caroni P., Mubagwa K., Sipido K. R. (2006) Increased phospholamban phosphorylation limits the force-frequency response in the MLP−/− mouse with heart failure. J. Mol. Cell Cardiol. 40, 350–360 [DOI] [PubMed] [Google Scholar]
- 5. Lamberts R. R., Hamdani N., Soekhoe T. W., Boontje N. M., Zaremba R., Walker L. A., de Tombe P. P., van der Velden J., Stienen G. J. (2007) Frequency-dependent myofilament Ca2+ desensitization in failing rat myocardium. J. Physiol. 582, 695–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rossman E. I., Petre R. E., Chaudhary K. W., Piacentino V., 3rd, Janssen P. M., Gaughan J. P., Houser S. R., Margulies K. B. (2004) Abnormal frequency-dependent responses represent the pathophysiologic signature of contractile failure in human myocardium. J. Mol. Cell Cardiol. 36, 33–42 [DOI] [PubMed] [Google Scholar]
- 7. O'Rourke B., Kass D. A., Tomaselli G. F., Kääb S., Tunin R., Marbán E. (1999) Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure. I: experimental studies. Circ. Res. 84, 562–570 [DOI] [PubMed] [Google Scholar]
- 8. Yano M., Ikeda Y., Matsuzaki M. (2005) Altered intracellular Ca2+ handling in heart failure. J. Clin. Invest. 115, 556–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hare J. M. (2004) Nitroso-redox balance in the cardiovascular system. N. Engl. J. Med. 351, 2112–2114 [DOI] [PubMed] [Google Scholar]
- 10. Hare J. M., Stamler J. S. (2005) NO/redox disequilibrium in the failing heart and cardiovascular system. J. Clin. Invest. 115, 509–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Terentyev D., Györke I., Belevych A. E., Terentyeva R., Sridhar A., Nishijima Y., de Blanco E. C., Khanna S., Sen C. K., Cardounel A. J., Carnes C. A., Györke S. (2008) Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ. Res. 103, 1466–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gonzalez D. R., Beigi F., Treuer A. V., Hare J. M. (2007) Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc. Natl. Acad. Sci. U.S.A. 104, 20612–20617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gonzalez D. R., Treuer A. V., Castellanos J., Dulce R. A., Hare J. M. (2010) Impaired S-nitrosylation of the ryanodine receptor caused by xanthine oxidase activity contributes to calcium leak in heart failure. J. Biol. Chem. 285, 28938–28945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cappola T. P., Kass D. A., Nelson G. S., Berger R. D., Rosas G. O., Kobeissi Z. A., Marbán E., Hare J. M. (2001) Allopurinol improves myocardial efficiency in patients with idiopathic dilated cardiomyopathy. Circulation 104, 2407–2411 [DOI] [PubMed] [Google Scholar]
- 15. Saavedra W. F., Paolocci N., St John M. E., Skaf M. W., Stewart G. C., Xie J. S., Harrison R. W., Zeichner J., Mudrick D., Marbán E., Kass D. A., Hare J. M. (2002) Imbalance between xanthine oxidase and nitric-oxide synthase signaling pathways underlies mechanoenergetic uncoupling in the failing heart. Circ. Res. 90, 297–304 [DOI] [PubMed] [Google Scholar]
- 16. Saraiva R. M., Minhas K. M., Zheng M., Pitz E., Treuer A., Gonzalez D., Schuleri K. H., Vandegaer K. M., Barouch L. A., Hare J. M. (2007) Reduced neuronal nitric oxide synthase expression contributes to cardiac oxidative stress and nitroso-redox imbalance in ob/ob mice. Nitric. Oxide. 16, 331–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tziomalos K., Hare J. M. (2009) Role of xanthine oxidoreductase in cardiac nitroso-redox imbalance. Front. Biosci. 14, 237–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khan S. A., Skaf M. W., Harrison R. W., Lee K., Minhas K. M., Kumar A., Fradley M., Shoukas A. A., Berkowitz D. E., Hare J. M. (2003) Nitric oxide regulation of myocardial contractility and calcium cycling. Independent impact of neuronal and endothelial nitric oxide synthases. Circ. Res. 92, 1322–1329 [DOI] [PubMed] [Google Scholar]
- 19. Grynkiewicz G., Poenie M., Tsien R. Y. (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450 [PubMed] [Google Scholar]
- 20. Shannon T. R., Ginsburg K. S., Bers D. M. (2002) Quantitative assessment of the SR Ca2+ leak-load relationship. Circ. Res. 91, 594–600 [DOI] [PubMed] [Google Scholar]
- 21. Bassani R. A., Bassani J. W., Bers D. M. (1992) Mitochondrial and sarcolemmal Ca2+ transport reduce [Ca2+]i during caffeine contractures in rabbit cardiac myocytes. J. Physiol. 453, 591–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Robak J., Gryglewski R. J. (1988) Flavonoids are scavengers of superoxide anions. Biochem. Pharmacol. 37, 837–841 [DOI] [PubMed] [Google Scholar]
- 23. Vila-Petroff M. G., Younes A., Egan J., Lakatta E. G., Sollott S. J. (1999) Activation of distinct cAMP-dependent and cGMP-dependent pathways by nitric oxide in cardiac myocytes. Circ. Res. 84, 1020–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Burger D. E., Lu X., Lei M., Xiang F. L., Hammoud L., Jiang M., Wang H., Jones D. L., Sims S. M., Feng Q. (2009) Neuronal nitric-oxide synthase protects against myocardial infarction-induced ventricular arrhythmia and mortality in mice. Circulation 120, 1345–1354 [DOI] [PubMed] [Google Scholar]
- 25. Khan S. A., Lee K., Minhas K. M., Gonzalez D. R., Raju S. V., Tejani A. D., Li D., Berkowitz D. E., Hare J. M. (2004) Neuronal nitric-oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling. Proc. Natl. Acad. Sci. U.S.A. 101, 15944–15948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kinugawa S., Huang H., Wang Z., Kaminski P. M., Wolin M. S., Hintze T. H. (2005) A defect of neuronal nitric oxide synthase increases xanthine oxidase-derived superoxide anion and attenuates the control of myocardial oxygen consumption by nitric oxide derived from endothelial nitric oxide synthase. Circ. Res. 96, 355–362 [DOI] [PubMed] [Google Scholar]
- 27. Saraiva R. M., Minhas K. M., Raju S. V., Barouch L. A., Pitz E., Schuleri K. H., Vandegaer K., Li D., Hare J. M. (2005) Deficiency of neuronal nitric oxide synthase increases mortality and cardiac remodeling after myocardial infarction. Role of nitroso-redox equilibrium. Circulation 112, 3415–3422 [DOI] [PubMed] [Google Scholar]
- 28. Layland J., Li J. M., Shah A. M. (2002) Role of cyclic GMP-dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J. Physiol. 540, 457–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen C. A., Wang T. Y., Varadharaj S., Reyes L. A., Hemann C., Talukder M. A., Chen Y. R., Druhan L. J., Zweier J. L. (2010) S-Glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 468, 1115–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sears C. E., Bryant S. M., Ashley E. A., Lygate C. A., Rakovic S., Wallis H. L., Neubauer S., Terrar D. A., Casadei B. (2003) Cardiac neuronal nitric-oxide synthase isoform regulates myocardial contraction and calcium handling. Circ. Res. 92, e52–e59 [DOI] [PubMed] [Google Scholar]
- 31. Wang H., Kohr M. J., Traynham C. J., Wheeler D. G., Janssen P. M., Ziolo M. T. (2008) Neuronal nitric-oxide synthase signaling within cardiac myocytes targets phospholamban. Am. J. Physiol. Cell Physiol. 294, C1566–C1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang H., Viatchenko-Karpinski S., Sun J., Györke I., Benkusky N. A., Kohr M. J., Valdivia H. H., Murphy E., Györke S., Ziolo M. T. (2010) Regulation of myocyte contraction via neuronal nitric oxide synthase. Role of ryanodine receptor S-nitrosylation. J. Physiol. 588, 2905–2917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barouch L. A., Harrison R. W., Skaf M. W., Rosas G. O., Cappola T. P., Kobeissi Z. A., Hobai I. A., Lemmon C. A., Burnett A. L., O'Rourke B., Rodriguez E. R., Huang P. L., Lima J. A., Berkowitz D. E., Hare J. M. (2002) Nitric oxide regulates the heart by spatial confinement of nitric-oxide synthase isoforms. Nature 416, 337–339 [DOI] [PubMed] [Google Scholar]
- 34. Barouch L. A., Cappola T. P., Harrison R. W., Crone J. K., Rodriguez E. R., Burnett A. L., Hare J. M. (2003) Combined loss of neuronal and endothelial nitric-oxide synthase causes premature mortality and age-related hypertrophic cardiac remodeling in mice. J. Mol. Cell Cardiol. 35, 637–644 [DOI] [PubMed] [Google Scholar]
- 35. Amado L. C., Saliaris A. P., Raju S. V., Lehrke S., St John M., Xie J., Stewart G., Fitton T., Minhas K. M., Brawn J., Hare J. M. (2005) Xanthine oxidase inhibition ameliorates cardiovascular dysfunction in dogs with pacing-induced heart failure. J. Mol. Cell Cardiol. 39, 531–536 [DOI] [PubMed] [Google Scholar]
- 36. Ekelund U. E., Harrison R. W., Shokek O., Thakkar R. N., Tunin R. S., Senzaki H., Kass D. A., Marbán E., Hare J. M. (1999) Intravenous allopurinol decreases myocardial oxygen consumption and increases mechanical efficiency in dogs with pacing-induced heart failure. Circ. Res. 85, 437–445 [DOI] [PubMed] [Google Scholar]
- 37. Hare J. M., Mangal B., Brown J., Fisher C., Jr., Freudenberger R., Colucci W. S., Mann D. L., Liu P., Givertz M. M., Schwarz R. P. (2008) Impact of oxypurinol in patients with symptomatic heart failure. Results of the OPT-CHF study. J. Am. Coll. Cardiol. 51, 2301–2309 [DOI] [PubMed] [Google Scholar]
- 38. Bergamini C., Cicoira M., Rossi A., Vassanelli C. (2009) Oxidative stress and hyperuricaemia. Pathophysiology, clinical relevance, and therapeutic implications in chronic heart failure. Eur. J. Heart Fail. 11, 444–452 [DOI] [PubMed] [Google Scholar]
- 39. Noman A., Ang D. S., Ogston S., Lang C. C., Struthers A. D. (2010) Effect of high-dose allopurinol on exercise in patients with chronic stable angina. A randomised, placebo controlled crossover trial. Lancet 375, 2161–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zoccali C., Maio R., Mallamaci F., Sesti G., Perticone F. (2006) Uric acid and endothelial dysfunction in essential hypertension. J. Am. Soc. Nephrol. 17, 1466–1471 [DOI] [PubMed] [Google Scholar]
- 41. Gurney A. M., Allam M. (1995) Inhibition of calcium release from the sarcoplasmic reticulum of rabbit aorta by hydralazine. Br. J. Pharmacol. 114, 238–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ellershaw D. C., Gurney A. M. (2001) Mechanisms of hydralazine induced vasodilation in rabbit aorta and pulmonary artery. Br. J. Pharmacol. 134, 621–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miao L., Perreault C. L., Travers K. E., Morgan J. P. (1997) Mechanisms of positive inotropic action of flosequinan, hydralazine, and milrinone on mammalian myocardium. Eur. J. Pharmacol. 321, 201–208 [DOI] [PubMed] [Google Scholar]
- 44. Kohr M. J., Wang H., Wheeler D. G., Velayutham M., Zweier J. L., Ziolo M. T. (2008) Targeting of phospholamban by peroxynitrite decreases β-adrenergic stimulation in cardiomyocytes. Cardiovasc. Res. 77, 353–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Y. H., Zhang M. H., Sears C. E., Emanuel K., Redwood C., El-Armouche A., Kranias E. G., Casadei B. (2008) Reduced phospholamban phosphorylation is associated with impaired relaxation in left ventricular myocytes from neuronal NO synthase-deficient mice. Circ. Res. 102, 242–249 [DOI] [PubMed] [Google Scholar]
- 46. Katori T., Donzelli S., Tocchetti C. G., Miranda K. M., Cormaci G., Thomas D. D., Ketner E. A., Lee M. J., Mancardi D., Wink D. A., Kass D. A., Paolocci N. (2006) Peroxynitrite and myocardial contractility. In vivo versus in vitro effects. Free Radic. Biol. Med. 41, 1606–1618 [DOI] [PubMed] [Google Scholar]
- 47. Giordano F. J. (2005) Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Invest. 115, 500–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Leiro J. M., Alvarez E., Arranz J. A., Cano E., Orallo F. (2004) Antioxidant activity and inhibitory effects of hydralazine on inducible NOS/COX-2 gene and protein expression in rat peritoneal macrophages. Int. Immunopharmacol. 4, 163–177 [DOI] [PubMed] [Google Scholar]
- 49. Janero D. R., Bryan N. S., Saijo F., Dhawan V., Schwalb D. J., Warren M. C., Feelisch M. (2004) Differential nitros(yl)ation of blood and tissue constituents during glyceryl trinitrate biotransformation in vivo. Proc. Natl. Acad. Sci. U.S.A. 101, 16958–16963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Beigi F., Gonzalez D. R., Minhas K. M., Sun Q. A., Foster M. W., Khan S. A., Treuer A. V., Dulce R. A., Harrison R. W., Saraiva R. M., Premer C., Schulman I. H., Stamler J. S., Hare J. M. (2012) Dynamic denitrosylation via S-nitrosoglutathione reductase regulates cardiovascular function. Proc. Natl. Acad. Sci. U.S.A. 109, 4314–4319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Harrison C. B., Drummond G. R., Sobey C. G., Selemidis S. (2010) Evidence that nitric oxide inhibits vascular inflammation and superoxide production via a p47phox-dependent mechanism in mice. Clin. Exp. Pharmacol. Physiol. 37, 429–434 [DOI] [PubMed] [Google Scholar]
- 52. Shinyashiki M., Pan C. J., Lopez B. E., Fukuto J. M. (2004) Inhibition of the yeast metal reductase heme protein fre1 by nitric oxide (NO). A model for inhibition of NADPH oxidase by NO. Free Radic. Biol. Med. 37, 713–723 [DOI] [PubMed] [Google Scholar]
- 53. Cote C. G., Yu F. S., Zulueta J. J., Vosatka R. J., Hassoun P. M. (1996) Regulation of intracellular xanthine oxidase by endothelial-derived nitric oxide. Am. J. Physiol. 271, L869–L874 [DOI] [PubMed] [Google Scholar]
- 54. Thadani U., Whitsett T. (1988) Relationship of pharmacokinetic and pharmacodynamic properties of the organic nitrates. Clin. Pharmacokinet. 15, 32–43 [DOI] [PubMed] [Google Scholar]
- 55. Gogia H., Mehra A., Parikh S., Raman M., Ajit-Uppal J., Johnson J. V., Elkayam U. (1995) Prevention of tolerance to hemodynamic effects of nitrates with concomitant use of hydralazine in patients with chronic heart failure. J. Am. Coll. Cardiol. 26, 1575–1580 [DOI] [PubMed] [Google Scholar]
- 56. Daiber A., Mülsch A., Hink U., Mollnau H., Warnholtz A., Oelze M., Münzel T. (2005) The oxidative stress concept of nitrate tolerance and the antioxidant properties of hydralazine. Am. J. Cardiol. 96, 25i–36i [DOI] [PubMed] [Google Scholar]
- 57. Münzel T., Daiber A., Mülsch A. (2005) Explaining the phenomenon of nitrate tolerance. Circ. Res. 97, 618–628 [DOI] [PubMed] [Google Scholar]
- 58. Bauer J. A., Fung H. L. (1991) Concurrent hydralazine administration prevents nitroglycerin-induced hemodynamic tolerance in experimental heart failure. Circulation 84, 35–39 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.