

Background: The two-component CovRS system is a GAS virulence gene regulator.
Results: A natural inactivating mutation has been found in CovS in a skin-invasive GAS strain, potentially affecting expression of key virulence genes.
Conclusion: The mutation in CovS showed substantial effects on regulation of virulence genes.
Significance: Expression of genes critical to GAS virulence can reveal reasons why specific strains of GAS are effective tissue specialists.
Keywords: Gene Expression, Gene Knock-out, Gene Regulation, Streptococcus pyogenes, Virulence Factors
Abstract
A skin-tropic invasive group A Streptococcus pyogenes (GAS) strain, AP53, contains a natural inactivating mutation in the covS gene (covSM) of the two-component responder (CovR)/sensor (CovS) gene regulatory system. The effects of this mutation on specific GAS virulence determinants have been assessed, with emphasis on expression of the extracellular protease, streptococcal pyrogenic exotoxin B (SpeB), capsular hyaluronic acid, and proteins that allow host plasmin assembly on the bacterial surface, viz. a high affinity plasminogen (Pg)/plasmin receptor, Pg-binding group A streptococcal M protein (PAM), and the human Pg activator streptokinase. To further illuminate mechanisms of the functioning of CovRS in the virulence of AP53, two AP53 isogenic strains were generated, one in which the natural covSM gene was mutated to WT-covS (AP53/covSWT) and a strain that contained an inactivated covR gene (AP53/ΔcovR). Two additional strains that do not contain PAM, viz. WT-NS931 and NS931/covSM, were also employed. SpeB was not measurably expressed in strains containing covRWT/covSM, whereas in strains with natural or engineered covRWT/covSWT, SpeB expression was highly up-regulated. Alternatively, capsule synthesis via the hasABC operon was enhanced in strain AP53/covSM, whereas streptokinase expression was only slightly affected by the covS inactivation. PAM expression was not substantially influenced by the covS mutation, suggesting that covRS had minimal effects on the mga regulon that controls PAM expression. These results demonstrate that a covS inactivation results in virulence gene alterations and also suggest that the CovR phosphorylation needed for gene up- or down-regulation can occur by alternative pathways to CovS kinase.
Introduction
The Gram-positive bacterium, Streptococcus pyogenes (or GAS),3 is the causative agent for a number of human infections. These common human pathogens colonize epithelial cells of the throat and the epidermal layer of skin, causing an array of mild to very severe diseases, ranging from simple pharyngitis to life-threatening necrotizing fasciitis and toxic shock syndrome. Serious sequela of GAS infections include post-streptococcal glomerulonephritis and rheumatic fever. More than 250 types of GAS have been identified based on serotyping of ubiquitously expressed cell wall-anchored M-proteins (1), which are major GAS virulence protein products of emm-type genes. The emm and emm-like genes are present in a major core virulence region under transcriptional control of a single-component upstream transcriptional regulator (2) encoded by the mga gene (3). The protein expression product of mga coordinately activates a number of core cell surface primary virulence genes. Included in this group are genes encoding M-protein and other homologous genes, e.g. fcR and enn, the products of which bind to the Fc regions of IgG and IgA (4), respectively, along with complement-inactivating C5a peptidase, encoded by the gene scpA (5) and the product of the fbp gene that allows adhesion of the bacteria to host fibronectin (6). The proteins produced by these genes assist the GAS in overcoming innate immune-based opsonization of the bacteria by macrophages and polymorphonuclear cells and/or anchor the bacteria to host cellular components (7), thus promoting their invasive properties.
In addition to these cell surface-associated virulence factors, a variety of GAS exoproducts are employed to promote virulence of the bacteria (8). One such example is streptokinase (SK), encoded by the bacterial sk gene. SK activates the critical host virulence factor human (h) plasma plasminogen (hPg), thus generating the fibrinolytic protease plasmin (hPm). This step assists in dissemination of the bacteria from local sterile fibrin-encased sites to deep tissue loci, the bloodstream, and the lymphatic system (9), and the invasiveness of GAS isolates has been correlated with direct binding of hPg/hPm to GAS (10). Subclasses of M-proteins have been shown to bind Pg/Pm directly (e.g. Pg-binding group A streptococcal M (PAM)-like protein-containing strains) (11) or indirectly via fibrinogen/fibrin-binding proteins, in each case allowing the bacteria to assemble a proteolytic surface (10). Although other streptococcal hPg/hPm receptors, e.g. the products of the sen and plr genes (12, 13), have been identified, their pathophysiological importance is unclear, especially in strains that produce functional PAM.
Two-component gene regulatory systems also exist in bacteria. In GAS, ∼13 such regulators are known (14), the most studied of which is the intracellular cluster of virulence (Cov) OmpR-like responder (covR) (15), together with the cell surface-bound EnvZ-type Cov sensor (covS) (16) system, two genes that are adjacent to each other on the GAS genome and are cotranscribed from a single promoter upstream of covR (17). Available evidence suggests that membrane-bound CovS functions as an autophosphorylase, kinase, and/or phosphatase (18), phosphorylating or dephosphorylating the cognate responder, CovR, to modulate the CovR-based repression of genes that are needed for GAS survival in response to host pressure, e.g. host temperature elevation (16). It has been reported that a relationship exists between CovRS and mga gene regulation (6), in this way greatly expanding the number of genes regulated by CovRS.
We find herein that skin-invasive AP53 and NS931 strains of GAS possess a similar arrangement of genes in the mga core regulon. However, the emm genes producing M-protein in these strains contain important differences. AP53 expresses the emm-like gene encoding PAM that specifically interacts with a binding locus within hPg/hPm (19), in this manner capturing host proteins to promote focal activation of hPg by bacterially secreted SK (20) to generate a cell-bound extracellular protease, hPm. We show that WT-NS931 contains a form of M-protein that does not specifically interact with hPg/hPm. During the course of our work, we discovered a natural inactivating gene mutation in the sensor component, covS, of covRS in WT-AP53 cells that was not present in NS931 cells, and we undertook an investigation of the relationship between this mutation and the nature of the virulence genes that are expressed by these GAS strains, especially those that assist in assembling hPm on the bacterial cell surface. The results of these studies are the subject of this report.
EXPERIMENTAL PROCEDURES
Bacterial Strains
All S. pyogenes strains were originally collected as primary isolates. The parental PAM+ isolate, AP53 (21), and the PAM− isolate, NS931 (10), have been described in previous studies. These strains were provided by Dr. M. J. Walker (Queensland, Australia) and Dr. M. Sanderson-Smith (Wollongong, Australia).
Isolation of Genomic DNA (gDNA)
Single colonies of the S. pyogenes strains were picked from streaks on horse blood agar and grown in THY (Todd-Hewitt broth supplemented with 1% (w/v) yeast extract) overnight at 37 °C. gDNA was isolated after treatment of the cells with lysozyme/proteinase K and cell lysis buffer (100 mm Tris, 5 mm EDTA, 0.2% SDS, 200 mm NaCl, pH 8.5) and extracted with phenol/chloroform/isoamyl alcohol (25:24:1, v/v/v). The gDNA was precipitated with isopropyl alcohol and washed with 70% ethanol.
Gene Sequencing and Identification
Traditional Sanger sequencing was accomplished on an ABI 3730xl 96-capillary sequencer using custom-designed primers. Genes were identified and assigned by BLAST searches.
Construction of Isogenic GAS Strains Containing Targeted Mutations of GAS Genes
To construct gene-deleted (Δ) mutants, targeting vectors for genes of AP53 and NS931 cells were constructed with the chloramphenicol acetyltransferase (cat) gene 5′-flanked by 300–400 bp of DNA upstream of the ATG for the gene of interest and 3′-flanked by 300–400 bp downstream of the TAA/TAG stop codon for the same gene. Restriction sites (typically 5′-NotI and a 3′-SalI) were cloned into the two ends of the entire DNA segment and were used for insertion into the same sites of the temperature-sensitive plasmid, pHY304 (from M. J. Walker), which also contained the erythromycin resistance (emr) gene. The resulting plasmid was then transformed into cells of interest by electroporation. Chromosomal integration via allelic replacement (22) was achieved by single crossover (SCO) at 30 °C for plasmid replication and then switched to 37 °C overnight for screening for Emr. For confirmation, SCO+ mutants were further screened by PCR with primers internal to the emr gene. The confirmed SCO+ cells were replicated at 30 °C and then switched to 37 °C overnight for double crossover (DCO). When DCO is successful, the emr and wild-type genes are lost. For such screening, the colonies were picked and replated on THY-agar and erythromycin/THY-agar simultaneously. The colonies that grew in THY, but not erythromycin/THY, were selected and evaluated by PCR. Successful DCO clones were also resistant to 2 μm chloramphenicol. Confirmation of the loss of the particular gene after DCO was made through use of null PCR results with gene-specific primers (Table 1).
In addition to the above gene-deleted mutants, GAS strains with targeted mutations in specific genes (e.g. NS931/covSM) were also generated. Instead of cat gene, the desired mutations of the gene of interest were first constructed by mutagenesis of the gene in the above targeting vectors. The SCO/DCO strategy was as described above.
Reverse Complementation of Genes
A DNA fragment comprising the entire gene of interest, along with 300–400 bp of 5′ and 3′ genomic flanking regions, was isolated by PCR. SacI and EcoRI restriction sites were incorporated by PCR into the 5′ and 3′ ends, respectively, of the fragment for ligation into the shuttle vector pDCem. The plasmid was then transformed into the GAS strain with the specific WT gene deleted and screened for emr. Positive colonies were tested for gene expression by RT-PCR with gene-specific primers (supplemental Table 1).
RNA Preparation
To prepare RNA, cells at the desired growth phase were centrifuged and resuspended in 200 μl of spheroplasting buffer (20 mm Tris-HCl, pH 6.8, 10 mm MgCl2, 26% raffinose), containing 100 μg/ml chloramphenicol and streptomycin. After adding 10 units of mutanolysin, the cells were pelleted and resuspended in RLT cell lysis buffer (Qiagen, Valencia, CA), and the isolation continued as described in the Qiagen RNeasy mini-kit. DNA contamination was further removed with two treatments of 4 units of DNase I (Qiagen).
Q-RT-PCR
Three independent extractions of total RNA from each of the strains were used. Real time reverse transcription-PCRs (RT-PCR) were performed essentially as described previously (23) with the primers of supplemental Table 1. The relative gene expression levels were analyzed by the 2−ΔΔCT method (24), in which CT represents the threshold cycle number of RT-PCR at which the amplified product was first detected. The statistical means of triplicate CT values were calculated for the target and reference genes (in this case, gapdh) from both WT and mutant strains. ΔCT was determined by CT of target gene − CT of reference gene, and then ΔΔCT was calculated by ΔCT of mutant − ΔCT of WT. The relative changes of a gene of interest (%) were calculated with the following equation: 100 × 2−ΔΔCT. We employed both gapdh (plr) and mutS as housekeeping genes. Although gapdh is normally used for this purpose, its translated product is a functional protein (Plr) in this system, and thus, we confirmed all relative expression data with a second loading gene mutS (25).
Western Blotting
Mid-log phase GAS cell suspensions (A600 nm ∼0.6) were centrifuged, washed with PBS, and resuspended in PBS to A600 nm ∼1.0. After treating with 100 units of mutanolysin, 10% SDS was added to lyse the cells. SDS-PAGE was performed, and the proteins were transferred to PVDF membranes. These membranes were then incubated with rabbit anti-streptococcal enolase (SEn) or rabbit anti-PLR (26), followed by incubation with alkaline phosphatase-conjugated goat anti-rabbit IgG. The protein was visualized using 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium detection.
Cloning and Expression of Proteins
Full-length sk was cloned from the gDNAs of WT-AP53 and WT-NS931 and expressed in Escherichia coli cells, as reported previously (27). Recombinant (r) hPg was expressed in Drosophila Schneider S2 cells as described (28).
Binding of hPg to GAS Cells by ELISA
GAS cells were grown to A600 nm ∼0.6 and collected by centrifugation. The cells were washed and resuspended in PBS to A600 nm ∼1.0. A volume of 50 μl of cell suspension containing 2 × 107 cfu of cells was used to coat individual wells of 96-well NUNC Maxisorb plates (NUNC Thermo, Rochester, NY). After blocking with 200 μl of 1% BSA, 20 μg/ml human nonimmune IgG, and washing with PBS, 100 μl of 20 μg/ml hPg in PBS was individually added. Mouse anti-hPg (ERL, South Bend, IN) in BSA blocking buffer was added, followed by horseradish peroxidase (HRP)-labeled goat anti-rabbit IgG. TMB substrate (R&D Systems, Minneapolis, MN) was used for detection at 450 nm after termination of the reaction with an equal volume of 2 m H2SO4. Negative controls were accomplished by this same procedure with PBS in place of cells.
Binding of hPg to GAS Cells Using Flow Cytometry
GAS cells were grown at 37 °C to an A600 nm ∼0.6. A 20-ml aliquot of the cells was centrifuged, washed, and resuspended to ∼5 × 107 cfu/ml in 5 ml of PBS, 1% BSA for blocking. Aliquots (200 μl) of cell suspensions were centrifuged, resuspended in 20 μg/ml hPg in PBS/1% BSA, and incubated for 1 h at room temperature. The cells were then washed with PBS, incubated with mouse anti-hPg (ERL) in PBS, 1% BSA for 30 min at room temperature, washed with PBS, and incubated with AlexaFluor488 goat anti-mouse IgG (Molecular Probes, Invitrogen) in PBS, 1% BSA for 30 min at room temperature. Finally, the cells were washed and resuspended in 200 μl of PBS, 1% paraformaldehyde for flow cytometric analysis (FCA).
FCA was conducted with a FACSAriaIII (BD Biosciences) using the 488-nm laser. Acquisition and analysis were performed by gating on side scatter (SSC-H) and fluorescence (FITC-A), setting the scales to logarithmic amplification. Cells in suspension were analyzed at a flow rate of 10 μl/min, and 10,000 events were recorded for analysis. Each histogram was analyzed using FCS Express Version 4 software (De Novo Software, Los Angeles, CA), which provided the Median, a statistical tool that was employed to calculate the events accepted by the gating formula that fall within the specified marker. This was used to calculate the % comparative binding of hPg, relative to WT-AP53 strain, arbitrarily set at 100%. The data were plotted using GraphPad PRISM 6 software.
Activation of hPg on Bacterial Cells
Cells of each strain were grown individually in THY medium to A600 nm ∼0.6 and collected by centrifugation (5,000 rpm, 10 min). The cells were washed with 10 mm Hepes, 150 mm NaCl, pH 7.4, followed by resuspension in the same buffer to A600 nm ∼1.0.
For hPg activation assays, 20 μl (∼1 × 107 cfu) of cells was added to individual wells of a 96-well Corning NBS nonbinding microwell plate, followed by 180 μl of 0.22 μm hPg, 0.28 mm S2251 (H-d-Val-Leu-Lys-p-nitroanilide; Chromogenix, Milan, Italy) in the same buffer. Finally, 5 nm r-SK was added. The A600 nm, which represents S2251 hydrolysis by the generated hPm, was continually measured at 30-s intervals using a plate reader.
SpeB Activity Assay
SpeB activity was measured as described (29) with overnight stationary phase cell-free culture supernatants (A600 nm ∼1.0–1.2). A volume of 50 μl of the filtered supernatants was incubated with 50 μl of the SpeB activation buffer (1 mm EDTA, 10 mm DTT, 0.1 m NaOAc, pH 5.0) for 30 min at 37 °C. Next, an equal volume of 0.6 mm N-benzoyl-proline-phenylalanine-arginine-p-nitroanilide hydrochloride (Sigma) in 0.1 m phosphate buffer, pH 7.4, was then added to the activated culture supernatant. After 1 h 30 min at 37 °C, the A405 nm was determined. All assays were performed in triplicate. Protein preparations were tested at least twice. Parallel assays were performed with addition of 10 μm of cysteine protease inhibitor E64 (Sigma) to confirm the specificity (30).
Hyaluronic Acid Capsule Assays
Overnight GAS cultures (A600 nm ∼1.0–1.2) in THY medium were inoculated into fresh THY and grown to the mid-log phase (A600 nm ∼0.6). The cells were then centrifuged and resuspended in H2O (0.5 ml). The capsule was liberated by shaking with CHCl3 (1 ml). The hyaluronic acid (HA) content in the aqueous phase was determined by the A640 nm after addition of an equal volume of Stains-All solution (20 mg of 1-ethyl-2-[3-(1-ethylnaphtho[1,2-d]thiazolin-2-ylidene)-2-methylpropenyl]naptho-[1,2-d]thiazolium bromide (Sigma)), 60 ml of HOAc, 100 ml of 50% formamide, as detailed previously (31). Standard curves were constructed using known concentrations of commercial HA (Sigma).
SK Assays in Washed Cell Culture Supernatants
Single colonies from plates of WT or mutant GAS strains were transferred to 2 ml of THY media and grown overnight at 37 °C. The cells were collected and washed two times with fresh THY media and resuspended in this same medium. Next, 1 ml of an overnight culture was inoculated into 35 ml of pre-warmed THY media, and the cultures were grown at 37 °C to A600 nm ∼0.6 in the presence of 28 μm aSpeB inhibitor, E64 (Sigma). The cultures were centrifuged at 10,000 rpm for 20 min to remove cells, and 10 ml of supernatants were collected. This supernatant was concentrated 20× using 10,000 nominal molecular weight limit membrane centrifugation filters. Supernatants were washed four times with 0. 1 m phosphate buffer, pH 7.2, to remove media components. To measure SK activity in these GAS culture supernatants, hPg activation assays were performed in 10 mm Hepes, pH 7.4, at 25 °C, using 0.25 mm S2251 substrate (Diapharma, West Chester, OH). The assay contained 150 mm NaCl, 0.2 μm Glu1-hPg, 0.2 μm PAM, and 0.25 mm S2251. The reaction was accelerated by adding 10 μl of cell supernatants. The A405 nm was measured at 1-min intervals for 2 h. Control reactions were performed in the absence of hPg to ensure that the proteolytic activity of supernatants was due to hPm.
Mouse Survival Studies
C57Bl/6 male mice containing the hPg transgene (32), at 6–10 weeks of age, along with WT controls, were employed for survival studies. On the day prior to injection with bacteria, depilatory cream was used to remove hair from the right rear flank of the mouse. Mice were anesthetized with isoflurane and injected subcutaneously in the right flank with 4–6 × 108 GAS cells/mouse. Mice were observed twice daily for survival up to 10 days.
RESULTS
The mga Regulatory Regions of GAS Strains AP53 and NS931
Sequencing began with completing the full DNA sequence of the AP53-pam gene using information from its partial sequence (33) and employing a strategy described earlier for generally identifying gene loci (34). The published partial DNA sequence of pam (GenBankTM accession number Z22219.1) was used to design internal primers, and HindIII restriction sites were used for the self-circularization of genomic DNA with T4 DNA ligase. Forward and reverse primers internal to known sequences within pam allowed amplification of fragments from these internal primers through upstream 5′ and downstream 3′ unknown sequences. From sequence data obtained with amplicons from these primers, the ATG and TAA start and stop codons for pam were located, thus completing its gene sequence with codons for 12 amino acids, including the start site at the N terminus, and 31 amino acids at the C terminus, along with flanking sequences at each end.
This expanded sequence information of pam, along with the complete genomic sequence data of other S. pyogenes strains in the GenBankTM database, allowed primers to be designed to identify genes flanking pam. When these amplifications were successful, the clones were sequenced, and this information was used in BLAST searches to assign genes. The new sequences obtained allowed continuation of this approach to ultimately obtain the entire sequence and gene identification of the virulence region of GAS strain AP53.
The 13.4-kb DNA sequence of overlapping genomic clones revealed eight potential structural genes downstream of the stand-alone transcriptional regulator, mga (35), that sequentially translate to proteins with homology to IgG-binding proteins (FcR) (4), hPg-binding proteins (PAM), IgA-binding proteins (Enn) (4), a serine protease that inactivates C5a (ScpA) (5), fibronectin-binding proteins (Fbp) (36), and laminin-binding proteins (Lbp) (30), all of which play potential roles in GAS virulence. From 5′ to 3′, these genes are mga-fcR-pam-enn-scpA-fbp-lbp (Fig. 1A). The identities of these genes have been identified by homologies with previous genomic sequence results in GenBankTM, by Shine-Dalgarno sequences proximal to a translation initiation codon (supplemental Table 2), by translation stop regions, and by −35 and −10 transcription initiation sites in the 5′-proximal promoters of these putative genes. However, the AP53-fcR gene cannot be fully translated in this GAS strain because a single base mutation in the position corresponding to amino acid 85 of fcR placed a translation stop signal (TAA) at that location, in an otherwise complete gene. Similar PCR amplifications of the fcR gene in numerous other GAS strains did not show this stop codon.
FIGURE 1.

Gene arrangement and expression within the mga regulon region of GAS strains AP53 and NS931. A, total of 13,383 bp from the WT-AP53 genome was sequenced from a genomic clone (black bar; 1–13,383). The red arrows show the genes identified, their directions from 5′ to 3′, and their sequence positions (beginning at the ATG initiation codon to the end of the translation stop codon), arbitrarily starting at position 1 of the assembled genomic clone. The numbering above each arrow shows the range of bp from its translation initiation codon to the translation stop codon. orf is the open reading frame of the most 5′ gene with unknown function; mga is the trans-acting positive regulatory gene for this core group of virulence proteins; fcR, which in the AP53 strain and GAS strain has a stop codon after amino acid 84 from the translation initiation site in an otherwise complete fcR gene, is the antiphagocytic IgG receptor gene; pam refers to the direct Pg-binding gene; downstream of pam is the enn gene, encoding an IgA-binding protein; which is sequentially followed by genes encoding C5a peptidase (scpA), fibronectin-binding protein (fbp), and laminin-binding protein (lbp). Subclones generated by PCR from homologous sequences found in other GAS strains were used to obtain overlapping gene and flanking sequences in order that the genes in the entire genomic clone were identified and positioned. B, PCR amplicons of individual genes in gDNA of WT-AP53 (black) and WT-NS931 (red) gDNA using the PCR primers (supplemental Table 1) for WT-AP53 gene sequences in A. The pam gene is not observed in NS931 cells. C, Q-RT-PCR analysis of the relative transcript levels of the mga core regulon genes in total mRNA isolated from AP53/ΔMga cells (black bars), AP53/ΔPAM (white bars), AP53/ΔEnn (gray bars), and AP53/ΔScpA (striped bars), based on each gene of WT-AP53 arbitrarily set at 100% (not shown in the figure). The WT-AP53 data were first normalized to the gapdh transcript level or that of a second housekeeping gene, mutS, in the individual total mRNA. No significant differences were found whether gapdh or mutS was used for this purpose. D, Q-RT-PCR analysis of the relative transcript levels of the mga regulon genes in total mRNA isolated from WT-NS931 cells (white bars) and NS931/ΔMga cells (striped bars), based on each gene of WT-NS931 arbitrarily set at 100%, as normalized to the gapdh transcript level in the individual total mRNA. The same primers for each gene were used in each WT and isogenic strains. Specific primers for the emm gene were used in this case. E, RT-PCR amplicons for mga, fcR, pam, enn, scpA, fbp, lbp, and plr (gapdh) examined in the isogenic strains, WT-AP53, AP53/ΔMga, and AP53/Mga.R. F, effect of inactivation of the mga gene on critical virulence genes outside of the mga core regulon in WT-AP53 and WT-NS931 cells. The % expression of the specific transcripts in total mRNA is relative to each gene set at 100% in WT-AP53 cells. Primers for individual genes between AP53 and NS931 strains were exact matches to the respective gene sequences. Data are shown for mRNA of sk, the hasA component of the hasABC regulon, and the cysteine protease speB.
The gene components of the proximal mga regulon of GAS strain NS931 have been compared with those of AP53. Although we did not completely sequenced all of these genes in NS931 gDNA (full sequences have been accomplished for mga and emm and partial sequences for the remainder of the NS931 genes), we show, by using the PCR primers of supplemental Table 1, that gDNA amplicons for mga-fcR-emm-enn-scpA-fbp-lbp were sequentially arranged in this same order in NS931 cells and display approximately the same molecular sizes as the AP53 genes (Fig. 1B). Importantly, a different M-protein gene (emm) in strain NS931 replaces pam of strain AP53 in this same locus and does not contain the hPg-binding site that is present in PAMAP53, as our translated protein sequence data shown in Sequence 1.
 |
The a1(italics)/a2(boldface) locus, which contains the hPg-binding site in PAMAP53 (amino acids sufficient and critical for hPg binding are underlined) (19, 37–39), is not present in EMMNS931, thus rendering this M-protein incapable of directly binding to hPg.
Transcription of Genes within the Core mga Regulon
RT-PCR analysis of the isolated mRNA amplicons from this isogenic AP53 strain with gene-specific primers (supplemental Table 1) demonstrated that fcR, pam/emm, enn, spcA, and fbp were severely down-regulated in mRNA from log phase (A600 nm ∼0.6) cultures of AP53/Δmga (Fig. 1C) and NS931/Δmga (Fig. 1D), as compared with their respective WT strains. The mRNA level of lbp was not significantly altered by the mga deletion in either strain (Fig. 1, C and D). In addition, deletions of genes for pam, enn, and scpA did not influence genes of any other Mga core regulon genes (Fig. 1C). For these experiments, we arbitrarily set the relative expression of each tested gene of WT-AP53 and WT-NS931 at 100%, because our interest here was to only compare the effect of a mga gene inactivation on the expression of individual genes in each strain. Loading controls were accomplished with gapdh and mutS, with the results unaffected by the nature of the housekeeping gene used.
The data from Fig. 1, C and D, show that the boundaries of the mga core regulon in both AP53 and NS931 strains are 5′-mga-through-fbp-3′. The down-regulation of genes for fcR, pam, enn, spcA, and fbp seen in strain AP53/Δmga was reversed in the mutant bacterial strain that was reverse-complemented with the WT-mga gene yielding AP53/mga.R (Fig. 1E). This recomplemented cell line was shown by Q-RT-PCR to produce 118% of mga and 115% of pam, compared with these transcripts in WT-AP53, taken as 100%. Thus, both WT-AP53 and WT-NS931 GAS strains contain the large vir (mga) core regulon (40), with identical components except for the nature of the M-protein.
Genes outside of this core mga regulon are controlled directly or indirectly by Mga. Specifically, we examined the influence of Mga on three critical virulence genes of interest to this work, viz. the hPg activator, streptokinase (sk), hasA, the first gene of the hyaluronate synthetase operon that provides the bacterial capsule, and speB, a gene that encodes a bacterial cysteine protease that can degrade SK, as well as other bacterial surface virulence-resistance genes, e.g. PAM. Minimal effects (≤2-fold) of an mga gene inactivation were observed on transcript levels of sk, hasA, and speB, when compared with their parent AP53 and NS931 cells (Fig. 1F). In Fig. 1F, the relative expression of each gene in WT-AP53 cells was set at 100%, because we wished to obtain a general idea of gene-specific expression levels between strains, a valid approach for this gene set because the primer sequences used for amplification of these genes were exact matches between strains AP53 and NS931.
Binding of hPg to GAS Cells
Using FCA (Fig. 2, A and B), as well as a sandwich ELISA with in-house antibodies against hPg (Fig. 2C), we show that mid-log phase cultures (A600 nm ∼0.6) of WT-AP53 bind hPg. Under these same conditions, WT-NS931, AP53/Δmga, and AP53/Δpam show a reduction of ∼80–90% in hPg binding, undoubtedly because of the absence, inactivation, and/or down-regulation of PAM expression in these cell lines. When AP53/Mga.R cells were examined in this regard, binding of hPg was very similar to WT-AP53 (Fig. 2, A–C). This suggests that other hPg receptors and/or hPg-binding proteins, e.g. SEn and Plr, which are expressed similarly in each of these cell lines (Fig. 2D), are relatively less important than PAM as cellular hPg receptors, at least when PAM is present as the M-protein in the strains. A similar observation using another approach has been reported in another GAS strain, NS88.2, that contains a PAM-related protein (22).
FIGURE 2.

Binding and activation of hPg to GAS cells. A, binding of hPg to the indicated strains of GAS was examined by FCA. Cells of each GAS line (1 × 107 cfu) were incubated with hPg (20 μg/ml). Mouse anti-human Pg was added, followed by AlexaFluor488 goat anti-mouse IgG. The cells were then resuspended in PBS, 1% paraformaldehyde for FCA. FCA histograms for each of the cell lines are shown at 488 nm, with gating on side scatter (SSC-H) and fluorescence (FITC-A), using logarithmic amplification. The cell suspensions were analyzed at a flow rate of 10 μl/min with 10,000 events used for analysis. IT represents the antibody isotype control. B, % binding of hPg calculated from A. The hPg binding to WT-AP53 was taken as 100%; the binding by other strains was calculated using the Median statistical value provided from analysis of each FCA histogram by FCS Express version 4 software. C, hPg binding measured by ELISA. Cells (∼2 × 107 cfu) from log phase growth (A600 nm ∼0.6) of the indicated strains of GAS were added to individual wells of 96-well microtiter plates. Next, hPg was added, followed sequentially by rabbit anti-human Pg and HRP-conjugated goat anti-rabbit IgG, with washes between additions. After addition of the HRP substrate TMB, for 20 min, the reaction was terminated with 2 m H2SO4, and the A405 nm was determined and plotted directly. D, SEn and Plr expression detected by Western blot analysis of whole cell extracts of each cell line. The A280 nm was adjusted to 0.3, and 10 μl was applied to each lane. The antibodies employed were anti-SEn (top) and anti-Plr (bottom). Lane 1, molecular weight markers; lane 2, WT-AP53; lane 3, AP53/ΔPAM; lane 4, AP53/ΔMga; lane 5, AP53/Mga.R; lane 6, WT-NS931. E, cells of the GAS strains indicated were grown to A600 nm ∼0.6 in 10 mm Hepes, 150 mm NaCl, pH 7.0. Aliquots of cells (1 × 107 cfu) were added to individual wells of 96-well low protein binding microtiter plates. hPg was then added followed by a solution containing 5 nm r-SK2b/0.25 mm S-2251. The A405 nm was continuously monitored using a plate reader. The data were collected at room temperature. The red line, represented as −Cells, is a control activation in the absence of cells and is very similar to the activation rate seen with WT-NS931 cells. There was no difference in the curves when E64 was placed in the cell growth medium and the assay.
Activation of hPg by r-SK2b on S. pyogenes Cells
We examined whether the binding of hPg to PAM on AP53 cells stimulates its activation by exogenous r-SK2b, cloned from AP53 cells. The data (Fig. 2E) show that the activation of hPg by r-SK2b, which is the form of SK that optimally acts on cell-bound hPg, is highly stimulated by AP53 cells. With AP53/ΔPAM, AP53/ΔMga, or WT-NS931 cells, none of which produce PAM, very little hPg is activated under these conditions by r-SK2b (Fig. 2E) and is similar to the control activation without cells (−Cells). The low activation capacities of these cell lines demonstrate that other hPg receptors, e.g. SEn and Plr on AP53 cells (Fig. 2D), do not stimulate hPg activation by SK2b to nearly the same extent as PAM. The isogenic strain, AP53/Mga.R, which elevates the highly diminished PAM levels of AP53/ΔMga cells to WT-AP53 levels, also restores the activation rates of hPg by SK2b to those of WT-AP53 cells (Fig. 2E). Thus, SK2b-producing invasive GAS strains require PAM or a PAM-like analog for optimal activation of hPg.
CovRS Regulatory System in AP53 Cells
The two-component CovRS gene regulatory system, the genes of which are consecutively present on all GAS genomes studied, including WT-AP53 and WT-NS931 cells (Fig. 3A), was examined. The covRS operon, controlled by a single promoter upstream of covR (41), has been recently linked to positive regulation of the mga gene, via rivRX (23). If this is the case, the expression of pam mRNA should also be indirectly regulated by covRS. To examine this point, we first sought to evaluate the integrity of the covRS system in WT-AP53 and WT-NS931 cells that originated from GAS isolates. Full genomic sequences of the entire covRS bicistronic locus from these AP53 and NS931 strains yielded >99% identical WT-covR sequences, compared with each other and to covR from numerous GAS strains in the GenBankTM database, and also contained an intact WT-covS gene in WT-NS931 DNA. However, covS from AP53 DNA showed a deletion of Thr1404 (numbered from ATG of covS), resulting in the following translated CovS protein sequence differences shown in Sequence 2.
FIGURE 3.

Effect of the CovS mutation on CovRS gene regulation. A, PCR using RT-PCR primers (supplemental Table 1) showing gDNA amplicons of some typical genes in WT-AP53 and WT-NS931 strains that are typically regulated by CovRS. B, SDS-PAGE of late-log phase (A600 nm ∼1.0) GAS culture supernatants in the absence of E64. Lane 1, WT-AP53 (AP53/CovSM); lane 2, AP53/ΔMga (AP53/CovSM/ΔMga); lane 3, AP53/ΔPAM (AP53/CovSM/ΔPAM); lane 4, WT-NS931 (NS931/CovSWT); lane 5, a blot of the WT-NS931 culture supernatant of lane 4 with commercial anti-SpeB; lane 6, a gel of the culture supernatant of WT-NS931 cells, grown in the presence of 28 μm of the SpeB inhibitor E64. Gels 5 and 6 were run at different times from each other and from the gels of lanes 1–4. The molecular mass of activated mature SpeB (aSpeB) is ∼28,000 and that of SpeB zymogen (pSpeB) is ∼39,000. C, aSpeB enzymatic activity, using the substrate benzoyl-proline-phenylalanine-arginine-p-nitroanilide-HCl of late-LP (A600 nm ∼1.0) GAS culture supernatants of the indicated cell lines in the absence (−) of E64 and in the presence (+) of 28 μm E64. D–F, restoration of CovRS system in AP53 cells. D, Q-RT-PCR measurements displaying relative levels of speB transcripts in late-log phase (A600 nm ∼1.0) GAS cell total mRNA, relative to WT-AP53 (AP53/CovSM), arbitrarily set at 100%. E, SDS-PAGE of the late-log phase (A600 nm ∼1.0) cell culture supernatants of the following: lane 1, molecular weight markers (M); lane 2, AP53/CovSM (WT-AP53); lane 3, AP53/CovSWT; lane 4, AP53/CovSWT*, except that the cells represented in lane 3 were grown in the presence of the SpeB inhibitor E64; lane 5, NS931/CovSWT (WT-NS931); lane 6, NS931/CovSM. F, aSpeB enzymatic activity at late-log phase (A600 nm ∼1.0) growth of whole cell extracts of the indicated cell lines toward the aSpeB substrate, benzoyl-proline-phenylalanine-arginine-p-nitroanilide-HCl, in the absence (−) and presence (+) of the aSpeB inhibitor, E64.
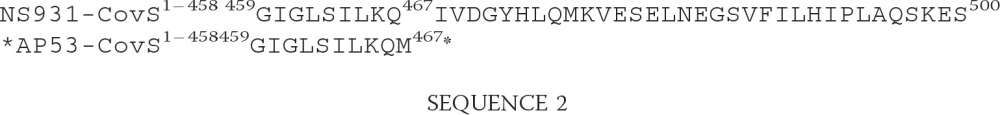 |
Effect of the Truncation Mutant of CovS (CovSM) on Function of CovRS
To determine whether this particular mutation in CovS affected its ability to regulate CovR, we examined several of the genes that are known to be regulated by the CovRS system, e.g. speB, hasA, and sk. Many of these essential genes exist in both AP53 and NS931, as seen by the presence of predicted gDNA amplicons generated with specific primers for these genes (Fig. 3A).
The gene encoding the cysteine protease, speB, is present in all GAS strains investigated and is known to be highly up-regulated by WT-CovRS. CovR represses transcription of speB (42), whereas CovRS enhances transcription of this gene (43). The zymogen form of SpeB (pSpeB), of Mr ∼40,000, is produced in the late-LP of GAS cell growth and is proteolytically cleaved to the active form of SpeB (aSpeB), of Mr ∼28,000 (30, 44). The protein gels of concentrated culture supernatants of late-LP GAS cells (A600 nm ∼1.0) (Fig. 3B) do not show evidence for SpeB in either AP53/CovSM (WT-AP53) or its WT isogenic mutants, AP53/ΔMga, AP53/CovSM/ΔMga (AP53/ΔMga), and AP53/CovSM/ΔMga (AP53/ΔPAM) (lanes 1–3), likely due to repression by CovR that is not controlled by the CovS mutant found in WT-AP53 cells. However, WT-NS931 cells (NS931/CovSWT), containing WT-CovS, produce comparatively large amounts of protein of the appropriate size of aSpeB (Fig. 3B, lane 4), which reacts with anti-SpeB (lane 5). When WT-NS931 cells are grown in the presence of the aSpeB inhibitor E64, only pSpeB is produced (Fig. 3B, lane 6). These results are confirmed in enzymatic assays of these late-LP culture supernatants (Fig. 3C), wherein no aSpeB activity toward the substrate, benzoyl-proline-phenylalanine-arginine-p-nitroanilide-HCl, is seen in culture supernatants from AP53 cells and its indicated isogenic variants. However, high aSpeB activity is observed in late LP supernatants from NS931 cells in the absence of the aSpeB inhibitor E64 (−) but not in the presence (+) of this inhibitor.
The effect of the natural CovS mutation found in WT-AP53 cells on SpeB expression has been examined (Fig. 3, D–F). Q-RT-PCR of total cell mRNA, using specific speB primers (supplemental Table 1), shows that total mRNA of AP53 cells with the natural truncation mutant corrected to that of CovSWT (AP53/CovSWT) produces ∼5 times more speB transcript than that contained in total mRNA from AP53/CovSM cells. Similarly, total mRNA from NS931/CovSWT cells expresses ∼8 times higher levels of the mRNA for speB than the total mRNA from NS931/CovSM cells. The SDS-PAGE protein gel data (Fig. 3E) are in agreement with this conclusion. Here, late-LP culture supernatants of the mutated cell line, AP53/CovSWT (Fig. 3E, lane 3), produce much more aSpeB than culture supernatants from WT-AP53 cells (lane 2), whereas the converse is true in WT-NS931 (lane 5) and NS931/CovSM (lane 6) cells. Growth of AP53/CovSWT cells in the presence of 28 μm E64 (Fig. 3E, lane 4), as with growth of WT-NS931 cells under these same conditions (Fig. 3B, lane 6), produce only pSpeB. aSpeB activity assays (Fig. 3F) fully confirm these findings in that only late-LP AP53/CovSWT and NS931/CovSWT cell supernatants produce aSpeB, when grown in the absence (−) of E64, but not in its presence (+).
Whereas WT-CovRS stimulates speB expression, this system represses the hasABC regulon, thus potentially reducing capsule production (45). We show that under nonstress conditions CovSWT is required to repress the repression of hasA by CovRWT, a conclusion that is not in agreement with previous work (16). Specifically, the data (Fig. 4, A and B) demonstrate that the amount of hasA present in AP53/CovSM (WT-AP53) cells is ∼10 times higher in hasA mRNA levels (Fig. 4A) and ∼12-fold higher in hyaluronic acid (HA) content, when compared with its isogenic mutant cell line, AP53/CovSWT. Similarly, the amount of hasA is ∼2-fold higher in NS931/CovSM cells (Fig. 4A) and is ∼5-fold higher in HA (Fig. 4B) content than in WT-NS931/CovSWT (WT-NS931) cells (Fig. 4A). These data show that whereas WT-CovRS indeed represses hasA and HA, the dominant role is provided by CovR, as is seen by the dramatically increased expression of hasA and HA in AP53/CovSM/ΔCovR cells (Fig. 4, A and B).
FIGURE 4.

Regulation of virulence genes by CovRS in GAS. A, effect of the inactivation of CovS (CovSM) and CovR (ΔCovR) on the transcription levels of hasA, sk, and sen genes. The levels of each specific transcript in the total mRNA of the indicated cell lines, relative to expression of each of the genes in AP53/CovSM (WT-AP53) cells arbitrarily set at 100%, are shown. Both gapdh and mutS were used as loading controls, with the results of A not differing with either housekeeping gene. B, amount of HA-containing capsule in AP53/CovSM (WT-AP53), AP53/CovSWT, AP53/CovSM/ΔCovR, NS931/CovSWT (WT-NS931), and NS931/CovSM. C, activation of hPg by mid-log phase culture supernatants (A600 nm ∼0.6) of the cell lines indicated on the graph.
In a similar vein, it has been shown previously that sk transcription is repressed by CovR (46), but the specific influence of CovS on CovR repression of sk2b regulation is more uncertain. Accordingly, we investigated the functional effect of CovS on SK2b production in AP53 cells through examination of the activation of hPg by dialyzed whole cell culture supernatants of mid-log phase (A600 nm ∼0.6) GAS cells. E64 (28 μm) was maintained in the inoculated cultures and in assays to inhibit the known degradation of SK by aSpeB (47). The data obtained show that AP53 cells mutated to display the WT-CovRS system, viz. AP53/CovSWT cells, possess slightly higher mRNA levels of sk (Fig. 4A), and corresponding higher activation rates (∼1.5 times) of hPg are seen (Fig. 4C), as compared with isogenic cells with intact CovR and inactive CovS (e.g. WT-AP53 cells). Similar small differences were observed in NS931/CovSWT (WT-NS931) and NS931/CovSM cells (data not shown). Deletion of CovR from WT-AP53 (AP53/CovSM/ΔCovR) cells shows an ∼4 times increase in mRNA levels of sk (Fig. 4A) and a corresponding larger amount of SK2b protein in the supernatant (Fig. 4C). Because the overall differences in the AP53 strain made by CovS inactivation are small (∼1.5–2-fold), the data suggest that CovS plays a minor role with regard to regulation of the sk gene under normal cell growth conditions, and the repressor role of CovR on sk transcription and corresponding protein expression is dominant, albeit not large (3.5–4.5 times).
Interaction of the covRS Operon With mga Regulon
It has been proposed that the CovRS system indirectly regulates expression of the mga gene, through rivRX (23). If that is the case, indirect effects should be observed with expression of pam, and we have assessed the functional consequences of changes in PAM expression through binding of hPg to the various mutated cell lines and through the corresponding activation of hPg by SK2b in the presence of these same GAS strains. WT-AP53 cells have been used as the reference for comparisons of the binding of hPg to other GAS cells. Relative mRNA levels of another hPg receptor, sen, are not affected by covS or covR gene inactivations (Fig. 4A). The data for hPg binding to various isogenic cell lines obtained by FCA (Fig. 5, A and B) and specific ELISA (Fig. 5C) show that AP53/CovSM (WT-AP53) cells and AP53/CovSWT cells bind hPg very similarly, with only ∼10% increased binding to AP53/CovSWT cells. This small increase in binding is apparently not productive, because it is seen (Fig. 5D) that activation by exogenous SK2b in the presence of AP53/CovSWT cells is even lower than this same activation in the presence of AP53/CovSM cells. These assays were performed both in the absence of E64 and in the presence of 28 μm E64 to inhibit the possible degradation of SK by aSpeB and the release of PAM from the cell surface, another known activity of aSpeB. Thus, under normal laboratory growth conditions, CovS does not play a regulatory role on CovR with regard to mga regulation. In fact, Q-RT-PCR results (data not shown) for levels of the mga mRNA in total mRNA isolated from AP53/CovSM cells and AP53/CovSWT cells show no significant differences.
FIGURE 5.

Effect of the CovSM on functional expression of PAM. A, binding of hPg to AP53/CovSM (WT-AP53) cells and AP53/CovSWT cells as assessed with FCA. Cells (1 × 107 cfu) were incubated with hPg (20 μg/ml). Mouse anti-human Pg was added, followed by AlexaFluor488 goat anti-mouse IgG. The cells were then resuspended in PBS, 1% paraformaldehyde for FCA. FCA histograms for each of the cell lines are shown at 488 nm, with gating on side scatter (SSC-H) and fluorescence (FITC-A), using logarithmic amplification. The cell suspensions were analyzed at a flow rate of 1 ml/s with 10,000 events used for analysis. IT represents the antibody isotype control. B, % binding of hPg to AP53/CovSWT cells, relative to hPg binding to AP53/CovSM set at 100%, was calculated using the median statistical value provided from analysis of each FCA histogram by FCS Express version 4 software. C, cells (∼2 × 107 cfu) of the indicated strains of GAS were added to individual wells of 96-well microtiter plates. Next, hPg was added, followed sequentially by rabbit anti-human Pg and then HRP-conjugated goat anti-rabbit IgG. After addition of the HRP substrate TMB for 20 min, the reaction was terminated with 2 m H2SO4, and the A450 nm was determined. D, cells (1 × 107 cfu) were added to individual wells of 96-well low protein binding microtiter plates. hPg was then added followed by a solution containing 5 nm r-SK2b/0.25 mm S-2251. The A405 nm was continuously monitored on a plate reader. The data were collected at room temperature. The lines represented as −Cells and −SK are control activations in the absence of cells and SK, respectively. All cells were presented at mid-log phase growth (A600 nm ∼0.6). Assays were performed both in the absence (−E64) and presence (+E64, 28 μm) of the aSpeB inhibitor, E64, in each cell line during cell growth and during the hPg activation assay.
We considered whether the CovS-inactivating mutation in WT-AP53 cells was a result of genetic drift of this bacterium or whether it resulted in the AP53 isolate in the patient from an in vivo phase shift in CovRS of the cells under stress. This latter phenomenon occurs in GAS and is employed by the bacteria to rapidly up- or down-regulate genes that assist its survival in the host at different stages of infection. It has been reported that in an M1T1 GAS strain, M1, the HA capsule, and/or a DNase I (sda1) expressed by a bacteriophage acquired by the GAS during evolution are necessary for this hyperinvasive genetic phase switching to occur (48, 49). To determine whether genotypic switching in covRS could occur in the AP53 GAS strain, which has M-like protein and capsule, but not Sda1, we infected mice, subcutaneously, with AP53/CovSWT cells and reisolated the cells from the skin lesion that developed 3 days after infection. We then examined aSpeB production in the GAS colonies obtained (Fig. 6, A–C). aSpeB production would be down-regulated if CovR and/or CovS was inactivated during infection. The results obtained from screening 1,500 colonies/mouse from the active infection in each of three mice demonstrated that aSpeB was produced in all colonies, as seen by the lytic zones surrounding each single colony (Fig. 6C). That this assay would detect such a mutation in CovRWT or CovSWT is also demonstrated in Fig. 6. Here, noninjected AP53/CovSM cells do not show lysis zones around the clones (Fig. 6A), but noninjected AP53/CovSWT isogenic cells do show these lytic zones (Fig. 6B). In addition to these experiments, we accomplished total nucleotide sequencing of the entire covR-covS genomic region, amplified by RT-PCR, from five randomly chosen clones from each of three of the plates of passaged AP53/CovSWT cells of Fig. 6C. The results demonstrated that only WT-covRS was present. Thus, CovR and CovS retained their WT status during infection and continued to up-regulate speB in AP53/CovSWT cells during the active infection.
FIGURE 6.

Genetic switching of CovS in AP53 cells. AP53/CovSWT cells (7 × 109 cfu) were injected subcutaneously into C57Bl/6 mice. Skin lesions were harvested after 3 days and homogenized. Dilutions were plated on THY/milk medium, and production of aSpeB from individual colonies was observed by the lytic zones surrounding the cells. A total of 1,500 colonies/mouse was screened for each of three mice. The unpassaged cells were prepared similarly to the injected cells. A, unpassaged AP53/CovSM (WT-AP53) cells. No aSpeB was observed. B, unpassaged AP53/CovSWT cells. aSpeB was produced in all clones. C, passaged AP53/CovSWT cells. aSpeB was produced in all clones, suggesting that CovR/CovS remained as WT proteins and stimulated aSpeB production.
Finally, the effects of these variant cell lines on survival of mice containing the hPg transgene (hPg(Tg)) have been investigated. The data obtained are shown in Fig. 7. After subcutaneous administration of ∼5 × 108 AP53/CovSM (WT-AP53) cells, 14/15 C57Bl/6(hPg(Tg)) mice expired within 5 days, and C57Bl/6 mice that did not contain hPg(Tg) survived for the full 10 days of observation (data not shown), in agreement with earlier observations (32). Deletion of the scpA or enn genes from AP53/CovSM cells did not protect mice against lethality in this model; 8/8 and 6/7 of the AP53/CovSM/ΔScpA and AP53/CovSM/ΔEnn, respectively, died at the same rate as AP53/CovSM mice (Fig. 7A). A total of 5/9 and 4/7 AP53/CovSM/ΔPAM and AP53/CovSM/ΔMga treated mice survived the entire 10 days (Fig. 7A), demonstrating the protection against lethality by deletion of these genes. Alteration of CovSM in AP53/CovSM cells to AP53/CovSWT offered nearly complete protection from lethality in C57Bl/6(hPg(Tg)) mice (Fig. 7A). This result further supports the point that CovSWT is not being mutated in the animal during the infection. These data also suggest that hPg and PAM are important for GAS lethality in this model, which also requires an intact and functional CovRS system. A similar trend is observed using a strain of PAM− bacteria, NS931, with C57Bl/6(hPg(Tg)) mice (Fig. 7B). In this case, NS931/CovSM-injected mice die at a much faster rate than mice injected with the NS931/CovSWT strain. Although hPm is not produced by the same route with this latter bacterial strain, it is nonetheless produced in plasma rather than the bacterial surface, because the SK1 generated by these bacteria do not require PAM for surface activation of hPg and will generate hPm in the solution state.
FIGURE 7.

Lethality of AP53 GAS strains. GAS cells (∼4–6 × 109 cfu for each GAS strain) of the indicated GAS cell lines were injected in C57Bl/6-hPg(Tg) mice (n = 7–10 mice for each GAS strain). Survival was monitored up to 10 days. A, bacterial strains isogenic with AP53/CovSM (WT-AP53) cells. B, NS931/CovSWT (WT-NS931) and NS931/CovSM cells. The CovS mutant generated in this strain is the same as found in WT-AP53 cells.
DISCUSSION
A 13.4-kb genomic fragment of S. pyogenes pattern D strain AP53, bound by the mga and lbp genes, contains sequentially mga-fcR-pam-enn-scpA-fbp-lbp, within which the M protein pathogenicity region of the genome is found. It has been reported that SOF− strains of S. pyogenes rarely contain this full complement of M-protein-related genes in this DNA island and typically lack fcR and enn (50, 51). AP53, which is SOF−, violates this paradigm, as does NS931, which also contains this same gene pattern in the mga-dependent virulence region. Upon comparing 5′-promoter sequences of each of the genes with the consensus Mga-binding sequence (52), we find homologies with promoter sequences of mga, fcR, pam, enn, and scpA but not with 5′-proximal sequences of fbp and lbp. Although lbp is not regulated by Mga, as would be predicted, we show that fbp transcription is strongly down-regulated by an inactivation of the mga gene in both AP53 and NS931 strains of GAS. Thus, it is possible that fbp is a member of a polycistronic gene, regulated by a promoter of an upstream gene, most likely scpA, or is more indirectly influenced by mga. Thus, with the possible exception of bicistronic scpA-fbp transcription, all of the genes in this region are most likely monocistronic, because they independently possess proximal promoter homologies to the Mga-binding consensus polynucleotide. As has been found in other S. pyogenes strains, disruption of the mga gene directly and indirectly affects transcription of many other genes outside of this M-protein virulence island (53).
This study expands the previous fingerprint and restriction mapping analyses of this virulence region of a number of isolates with pam-like genes (54–56) to an identification of its gene structure and regulation. Our interest in the AP53 bacterial strain was governed by the direct dependence of its virulence on the integrity of the human host fibrinolytic system. Cell surface acquisition of hPg is important to the virulence of this class of bacteria in humans, and in the case of GAS, hPg binding is a critical virulence determinant (10, 56). Other virulence mechanisms surely exist, and the importance of the host Pg pathway may be a property of those strains that can assemble host hPg on the surface of the bacteria via direct binding to PAM or indirect binding to the host fibrinogen that itself interacts with fibrinogen-binding M-proteins. In fact, another strain used in this report, NS931, closely resembles the gene arrangement within the mga regulon. This strain is also an invasive isolate from skin. It does not contain PAM, and thus functions somewhat differently. Thus, although PAM is an essential virulence factor for AP53, other strains do not require PAM and likely function at different end points of virulence as mechanisms for survival of GAS, as the host adapts.
During sequence analysis of AP53, we discovered a truncation mutation in CovS (CovSM), the sensor constituent of the two-component CovRS system, that allows GAS gene expression to respond to a variety of environmental challenges by the host, such as [Mg2+], osmolality, temperature, etc. (16). It is likely that differences in gene regulation by the CovRS system (41), which both up- and down-regulates a large number of genes in the bacterial genome (57), is responsible for some of the different properties of these two GAS strains. We focused on some key virulence genes of importance to our work, viz. speB, hasA, sk, and pam. It is possible that this latter gene is influenced indirectly via effects of CovRS on the mga gene.
Using the important virulence factor, speB, the gene that encodes the cysteine proteinase precursor, pSpeB, as one model of gene up-regulation wherein WT-CovS derepresses CovR on gene expression (43), we find that CovR attenuates speB expression in GAS strains possessing the truncated CovS found in WT-AP53 and in the NS931 strain in which that same mutation was engineered in NS931 (NS931/CovSM). However, in WT-CovRS systems, e.g. WT-NS931, and an engineered GAS strain, AP53/CovSWT, speB expression is highly up-regulated. Because CovS does not function in the absence of CovR, but CovR can function in the absence or presence of CovS (43), the natural CovS mutation found in WT-AP53 must be considered a mechanism used by GAS to activate the repressor activity of CovR toward the speB gene. To be effective, this mode of regulation of gene expression by genetic switching also would be required to occur rapidly at different stages of an active infection, as has been found (58). Because aSpeB assists in the initial stages of infection in terms of matrix degradation, but inhibits subsequent bacterial dissemination by digesting virulence factors, e.g. PAM and SK, a rapid switch exists to enhance and attenuate expression of such virulence proteins. The CovRS system that chemically responds to environmental changes, via CovS, and can directly regulate gene expression through phosphorylation/dephosphorylation of CovR, is such as system (59).
The opposite activity for CovS on CovR is found with regard to the hasA gene, which is part of the hasABC operon that provides the capsular material, HA, to certain GAS strains (60). We confirm that hasA expression is repressed in the WT-CovRS strains, WT-NS931 and AP53/CovSWT, and is de-repressed in strains with the particular CovS mutant that we find naturally in WT-AP53 or that we have engineered in strain NS931/CovSM. This mutation leads to higher levels of HA in these latter two strains, along with an increase in capsular material. Similar findings have been made with respect to SK expression.
The assembly of hPg on bacterial surfaces facilitates its activation to the protease, hPm, by bacterially secreted SK, and it protects hPm from inhibition by the host plasma inhibitor, α2-antiplasmin. This mechanism of cell surface hPm assembly thereby allows the bacteria assembly to possess a broad spectrum protease, which, in-turn, can degrade fibrin, as well as extracellular matrices, both directly and through activation of matrix metalloproteinases (61), thereby disrupting barriers that potentially inhibit bacterial dissemination. By expression of the protein product of the pam gene, hPg is able to bind directly to the surface of the microorganism and, after activation, to generate this highly focalized proteolytic activity. This feature is an important characteristic of skin-tropic GAS strains, wherein PAM and PAM-related proteins are present (56). In several other GAS lines, e.g. the globally disseminated M1T1 and the closely related SF370 (62) strains, M-proteins do not bind hPg directly but have the ability to bind fibrinogen (63), which then serves as a template for hPg binding and activation. Interestingly, the nature of the SK produced also coordinates with the Pg binding properties of the M-proteins. AP53 and analogous strains containing functional PAM, produces SK2b, which maximally activates Pg bound to PAM via the kringle 2 region of hPg. Strains with fibrinogen-binding M-proteins, e.g. SF370, can assemble hPg via the Pg-kringle 1/kringle 4 domains and produce SK2a. GAS strains that contain M-proteins that cannot directly or indirectly assemble hPg (e.g. NS931), produce SK1, which maximally activates hPg in solution. Thus, GAS is a highly coordinated organism with respect to host-specific infection, and many strains rely in various ways on an intact fibrinolytic system, and strong trends are present that hPg assembly on GAS is necessary, but not sufficient, for GAS invasiveness. This general trend is also reflected in the lethality induced by intradermal injection of GAS lines, in a murine model of bacterial dissemination. The GAS lines that do not contain PAM, e.g. NS931, or are engineered not to produce PAM, e.g. AP53/ΔPAM and AP53/ΔMga, are less invasive than lines that express PAM, e.g. WT-AP53. Interestingly, when the inactivating mutation of CovS found in WT-AP53 was restored to CovSWT, the lethality of WT-AP53 was reversed. This could be related to the fact that hPg activation was also attenuated in this strain, although hPg binding was unaffected, and the fact that aSpeB expression is up-regulated and not down-regulated by genetic switching in this strain. In the early stages of development, aSpeB assists in colonization, and this enzyme should therefore inhibit dissemination because of its inactivation by proteolysis of PAM and SK.
Although the major hPg receptor-binding site on S. pyogenes is PAM (11), other hPg receptors are also present in bacterial strains, e.g. PLR (12) and SEn (13). However, these receptors do not appear to play major roles in hPg binding or activation by SK in strains that also contain PAM or PAM-like proteins. This is shown through the nonproductive, with regard to SK2b stimulation, and low binding capacity of hPg to strains wherein PAM is absent or down-regulated, e.g. WT-NS931, AP53/ΔMga, AP53/ΔPAM, despite the fact that all these strains contain SEn and PLR.
Invasive GAS infections and related sequela account for morbidity and mortality of nearly 1 million humans/year (64), and thus are serious worldwide health risks. Counteracting these potential infections with vaccines is a goal that has not been effectively achieved as yet. Thus, it is critical to identify and understand the regulatory properties of genes that have evolved to render these bacteria so effective and specific for the human host.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant HL013423 (to F. J. C.).

This article contains supplemental Tables 1 and 2.
- GAS
- group A streptococcus
- SOF
- serum opacity factor
- cat
- chloramphenicol acetyltransferase
- h
- human
- Pg
- plasminogen
- Pm
- plasmin
- covRS
- two-component cluster of virulence (Cov) sensor (S) and gene regulator (R)
- emm
- a gene family encoding M-proteins
- sen
- streptococcal enolase gene
- gapdh
- glyceraldehyde-3-phosphate dehydrogenase gene
- plr
- gene encoding plasmin(ogen) receptor
- mga
- multiple gene regulator
- fcR
- gene coding IgG Fc receptor
- pam
- hPg-binding M-like protein gene
- enn
- gene encoding IgA Fc receptor
- scpA
- C5a peptidase gene
- fbp
- fibronectin-binding protein gene
- lbp
- laminin-binding protein gene
- mutS
- dam-directed mutator S gene
- hasABC
- hyaluronic acid synthetase operator
- speB
- streptococcal erythrogenic toxin B gene
- SK
- streptokinase
- TMB
- tetramethylbenzidine
- emr
- erythromycin resistance gene
- SCO
- single crossover
- DCO
- double crossover
- Q-RT
- quantitative RT
- FCA
- flow cytometric analysis
- r
- recombinant.
REFERENCES
- 1. Smeesters P. R., McMillan D. J., Sriprakash K. S. (2010) The streptococcal M protein. A highly versatile molecule. Trends Microbiol. 18, 275–282 [DOI] [PubMed] [Google Scholar]
- 2. Perez-Casal J., Caparon M. G., Scott J. R. (1991) Mry, a trans-acting positive regulator of the M protein gene of Streptococcus pyogenes with similarity to the receptor proteins of two-component regulatory systems. J. Bacteriol. 173, 2617–2624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hondorp E. R., McIver K. S. (2007) The Mga virulence regulon: infection where the grass is greener. Mol. Microbiol. 66, 1056–1065 [DOI] [PubMed] [Google Scholar]
- 4. Stenberg L., O'Toole P., Lindahl G. (1992) Many group A streptococcal strains express two different immunoglobulin-binding proteins, encoded by closely linked genes. Characterization of the proteins expressed by four strains of different M-type. Mol. Microbiol. 6, 1185–1194 [DOI] [PubMed] [Google Scholar]
- 5. Simpson W. J., LaPenta D., Chen C., Cleary P. P. (1990) Coregulation of type 12 M protein and streptococcal C5a peptidase genes in group A streptococci: evidence for a virulence regulon controlled by the virR locus. J. Bacteriol. 172, 696–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roberts S. A., Churchward G. G., Scott J. R. (2007) Unraveling the regulatory network in Streptococcus pyogenes. The global response regulator CovR represses rivR directly. J. Bacteriol. 189, 1459–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bisno A. L., Brito M. O., Collins C. M. (2003) Molecular basis of group A streptococcal virulence. Lancet Infect. Dis. 3, 191–200 [DOI] [PubMed] [Google Scholar]
- 8. Kalia A., Bessen D. E. (2004) Natural selection and evolution of streptococcal virulence genes involved in tissue-specific adaptations. J. Bacteriol. 186, 110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lähteenmäki K., Kuusela P., Korhonen T. K. (2001) Bacterial plasminogen activators and receptors. FEMS Microbiol. Rev. 25, 531–552 [DOI] [PubMed] [Google Scholar]
- 10. McKay F. C., McArthur J. D., Sanderson-Smith M. L., Gardam S., Currie B. J., Sriprakash K. S., Fagan P. K., Towers R. J., Batzloff M. R., Chhatwal G. S., Ranson M., Walker M. J. (2004) Plasminogen binding by group A streptococcal isolates from a region of hyperendemicity for streptococcal skin infection and a high incidence of invasive infection. Infect. Immun. 72, 364–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Berge A., Sjöbring U. (1993) PAM, a novel plasminogen-binding protein from Streptococcus pyogenes. J. Biol. Chem. 268, 25417–25424 [PubMed] [Google Scholar]
- 12. Winram S. B., Lottenberg R. (1996) The plasmin-binding protein Plr of group A streptococci is identified as glyceraldehyde-3-phosphate dehydrogenase. Microbiology 142, 2311–2320 [DOI] [PubMed] [Google Scholar]
- 13. Pancholi V., Fischetti V. A. (1998) α-Enolase, a novel strong plasmin(ogen) binding protein on the surface of pathogenic streptococci. J. Biol. Chem. 273, 14503–14515 [DOI] [PubMed] [Google Scholar]
- 14. Churchward G. (2007) The two faces of Janus. Virulence gene regulation by CovR/S in group A streptococci. Mol. Microbiol. 64, 34–41 [DOI] [PubMed] [Google Scholar]
- 15. Kondo H., Nakagawa A., Nishihira J., Nishimura Y., Mizuno T., Tanaka I. (1997) Escherichia coli positive regulator OmpR has a large loop structure at the putative RNA polymerase interaction site. Nat. Struct. Biol. 4, 28–31 [DOI] [PubMed] [Google Scholar]
- 16. Dalton T. L., Scott J. R. (2004) CovS inactivates CovR and is required for growth under conditions of general stress in Streptococcus pyogenes. J. Bacteriol. 186, 3928–3937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gryllos I., Levin J. C., Wessels M. R. (2003) The CsrR/CsrS two-component system of group A Streptococcus responds to environmental Mg2+. Proc. Natl. Acad. Sci. U.S.A. 100, 4227–4232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alves R., Savageau M. A. (2003) Comparative analysis of prototype two-component systems with either bifunctional or monofunctional sensors. Differences in molecular structure and physiological function. Mol. Microbiol. 48, 25–51 [DOI] [PubMed] [Google Scholar]
- 19. Wistedt A. C., Kotarsky H., Marti D., Ringdahl U., Castellino F. J., Schaller J., Sjöbring U. (1998) Kringle 2 mediates high affinity binding of plasminogen to an internal sequence in streptococcal surface protein PAM. J. Biol. Chem. 273, 24420–24424 [DOI] [PubMed] [Google Scholar]
- 20. Ben Nasr A., Wistedt A., Ringdahl U., Sjöbring U. (1994) Streptokinase activates plasminogen bound to human group C and G streptococci through M-like proteins. Eur. J. Biochem. 222, 267–276 [DOI] [PubMed] [Google Scholar]
- 21. Bessen D. E., Sotir C. M., Readdy T. L., Hollingshead S. K. (1996) Genetic correlates of throat and skin isolates of group A streptococci. J. Infect. Dis. 173, 896–900 [DOI] [PubMed] [Google Scholar]
- 22. Sanderson-Smith M. L., Dinkla K., Cole J. N., Cork A. J., Maamary P. G., McArthur J. D., Chhatwal G. S., Walker M. J. (2008) M protein-mediated plasminogen binding is essential for the virulence of an invasive Streptococcus pyogenes isolate. FASEB J. 22, 2715–2722 [DOI] [PubMed] [Google Scholar]
- 23. Roberts S. A., Scott J. R. (2007) RivR and the small RNA RivX: the missing links between the CovR regulatory cascade and the Mga regulon. Mol. Microbiol. 66, 1506–1522 [DOI] [PubMed] [Google Scholar]
- 24. Schmittgen T. D., Livak K. J. (2008) Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 25. Enright M. C., Spratt B. G., Kalia A., Cross J. H., Bessen D. E. (2001) Multilocus sequence typing of Streptococcus pyogenes and the relationships between emm type and clone. Infect. Immun. 69, 2416–2427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cole J. N., McArthur J. D., McKay F. C., Sanderson-Smith M. L., Cork A. J., Ranson M., Rohde M., Itzek A., Sun H., Ginsburg D., Kotb M., Nizet V., Chhatwal G. S., Walker M. J. (2006) Trigger for group A streptococcal M1T1 invasive disease. FASEB J. 20, 1745–1747 [DOI] [PubMed] [Google Scholar]
- 27. Zhang Y., Liang Z., Hsueh H. T., Ploplis V. A., Castellino F. J. (2012) Characterization of streptokinases from group A streptococci reveals a strong functional relationship that supports the coinheritance of plasminogen-binding M protein and cluster 2b streptokinase. J. Biol. Chem. 287, 42093–42103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nilsen S. L., Castellino F. J. (1999) Expression of human plasminogen in Drosophila Schneider S2 cells. Protein Expr. Purif. 16, 136–143 [DOI] [PubMed] [Google Scholar]
- 29. Hytönen J., Haataja S., Gerlach D., Podbielski A., Finne J. (2001) The SpeB virulence factor of Streptococcus pyogenes, a multifunctional secreted and cell surface molecule with strepadhesin, laminin-binding and cysteine protease activity. Mol. Microbiol. 39, 512–519 [DOI] [PubMed] [Google Scholar]
- 30. Zimmerlein B., Park H. S., Li S., Podbielski A., Cleary P. P. (2005) The M protein is dispensable for maturation of streptococcal cysteine protease SpeB. Infect. Immun. 73, 859–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schrager H. M., Rheinwald J. G., Wessels M. R. (1996) Hyaluronic acid capsule and the role of streptococcal entry into keratinocytes in invasive skin infection. J. Clin. Invest. 98, 1954–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun H., Ringdahl U., Homeister J. W., Fay W. P., Engleberg N. C., Yang A. Y., Rozek L. S., Wang X., Sjöbring U., Ginsburg D. (2004) Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science 305, 1283–1286 [DOI] [PubMed] [Google Scholar]
- 33. Ringdahl U., Svensson M., Wistedt A. C., Renné T., Kellner R., Müller-Esterl W., Sjöbring U. (1998) Molecular co-operation between protein PAM and streptokinase for plasmin acquisition by Streptococcus pyogenes. J. Biol. Chem. 273, 6424–6430 [DOI] [PubMed] [Google Scholar]
- 34. Liang Z., Breman A. M., Grimes B. R., Rosen E. D. (2008) Identifying and genotyping transgene integration loci. Transgenic Res. 17, 979–983 [DOI] [PubMed] [Google Scholar]
- 35. Kreikemeyer B., McIver K. S., Podbielski A. (2003) Virulence factor regulation and regulatory networks in Streptococcus pyogenes and their impact on pathogen-host interactions. Trends Microbiol. 11, 224–232 [DOI] [PubMed] [Google Scholar]
- 36. Ferretti J. J., McShan W. M., Ajdic D., Savic D. J., Savic G., Lyon K., Primeaux C., Sezate S., Suvorov A. N., Kenton S., Lai H. S., Lin S. P., Qian Y., Jia H. G., Najar F. Z., Ren Q., Zhu H., Song L., White J., Yuan X., Clifton S. W., Roe B. A., McLaughlin R. (2001) Complete genome sequence of an M1 strain of Streptococcus pyogenes. Proc. Natl. Acad. Sci. U.S.A. 98, 4658–4663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cnudde S. E., Prorok M., Castellino F. J., Geiger J. H. (2006) X-ray crystallographic structure of the angiogenesis inhibitor, angiostatin, bound to a peptide from the group A streptococcal surface protein PAM. Biochemistry 45, 11052–11060 [DOI] [PubMed] [Google Scholar]
- 38. Fu Q., Figuera-Losada M., Ploplis V. A., Cnudde S., Geiger J. H., Prorok M., Castellino F. J. (2008) The lack of binding of VEK-30, an internal peptide from the group A streptococcal M-like protein, PAM, to murine plasminogen is due to two amino acid replacements in the plasminogen kringle-2 domain. J. Biol. Chem. 283, 1580–1587 [DOI] [PubMed] [Google Scholar]
- 39. Schenone M. M., Warder S. E., Martin J. A., Prorok M., Castellino F. J. (2000) An internal histidine residue from the bacterial surface protein, PAM, mediates its binding to the kringle-2 domain of human plasminogen. J. Pept. Res. 56, 438–445 [DOI] [PubMed] [Google Scholar]
- 40. Podbielski A. (1993) Three different types of organization of the vir regulon in group A streptococci. Mol. Gen. Genet. 237, 287–300 [DOI] [PubMed] [Google Scholar]
- 41. Levin J. C., Wessels M. R. (1998) Identification of csrR/csrS, a genetic locus that regulates hyaluronic acid capsule synthesis in group A Streptococcus. Mol. Microbiol. 30, 209–219 [DOI] [PubMed] [Google Scholar]
- 42. Heath A., DiRita V. J., Barg N. L., Engleberg N. C. (1999) A two-component regulatory system, CsrR-CsrS, represses expression of three Streptococcus pyogenes virulence factors, hyaluronic acid capsule, streptolysin S, and pyrogenic exotoxin B. Infect. Immun. 67, 5298–5305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Treviño J., Perez N., Ramirez-Peña E., Liu Z., Shelburne S. A., 3rd, Musser J. M., Sumby P. (2009) CovS simultaneously activates and inhibits the CovR-mediated repression of distinct subsets of group A Streptococcus virulence factor-encoding genes. Infect. Immun. 77, 3141–3149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hauser A. R., Stevens D. L., Kaplan E. L., Schlievert P. M. (1991) Molecular analysis of pyrogenic exotoxins from Streptococcus pyogenes isolates associated with toxic shock-like syndrome. J. Clin. Microbiol. 29, 1562–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Federle M. J., Scott J. R. (2002) Identification of binding sites for the group A streptococcal global regulator CovR. Mol. Microbiol. 43, 1161–1172 [DOI] [PubMed] [Google Scholar]
- 46. Churchward G., Bates C., Gusa A. A., Stringer V., Scott J. R. (2009) Regulation of streptokinase expression by CovR/S in Streptococcus pyogenes. CovR acts through a single high affinity binding site. Microbiology 155, 566–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rezcallah M. S., Boyle M. D., Sledjeski D. D. (2004) Mouse skin passage of Streptococcus pyogenes results in increased streptokinase expression and activity. Microbiology 150, 365–371 [DOI] [PubMed] [Google Scholar]
- 48. Cole J. N., Pence M. A., von Köckritz-Blickwede M., Hollands A., Gallo R. L., Walker M. J., Nizet V. (2010) M protein and hyaluronic acid capsule are essential for in vivo selection of covRS mutations characteristic of invasive serotype M1T1 group A Streptococcus. MBio. 1, e00191–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Walker M. J., Hollands A., Sanderson-Smith M. L., Cole J. N., Kirk J. K., Henningham A., McArthur J. D., Dinkla K., Aziz R. K., Kansal R. G., Simpson A. J., Buchanan J. T., Chhatwal G. S., Kotb M., Nizet V. (2007) DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat. Med. 13, 981–985 [DOI] [PubMed] [Google Scholar]
- 50. Haanes E. J., Heath D. G., Cleary P. P. (1992) Architecture of the vir regulons of group A streptococci parallels opacity factor phenotype and M protein class. J. Bacteriol. 174, 4967–4976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Podbielski A., Flosdorff A., Weber-Heynemann J. (1995) The group A streptococcal virR49 gene controls expression of four structural vir regulon genes. Infect. Immun. 63, 9–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McIver K. S., Heath A. S., Green B. D., Scott J. R. (1995) Specific binding of the activator Mga to promoter sequences of the emm and scpA genes in the group A Streptococcus. J. Bacteriol. 177, 6619–6624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ribardo D. A., McIver K. S. (2006) Defining the Mga regulon. Comparative transcriptome analysis reveals both direct and indirect regulation by Mga in the group A Streptococcus. Mol. Microbiol. 62, 491–508 [DOI] [PubMed] [Google Scholar]
- 54. Gardiner D., Hartas J., Currie B., Mathews J. D., Kemp D. J., Sriprakash K. S. (1995) Vir typing: a long-PCR typing method for group A streptococci. PCR Methods Appl. 4, 288–293 [DOI] [PubMed] [Google Scholar]
- 55. Sanderson-Smith M. L., Walker M. J., Ranson M. (2006) The maintenance of high affinity plasminogen binding by group A streptococcal plasminogen-binding M-like protein is mediated by arginine and histidine residues within the a1 and a2 repeat domains. J. Biol. Chem. 281, 25965–25971 [DOI] [PubMed] [Google Scholar]
- 56. Svensson M. D., Sjöbring U., Bessen D. E. (1999) Selective distribution of a high affinity plasminogen-binding site among group A streptococci associated with impetigo. Infect. Immun. 67, 3915–3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Federle M. J., McIver K. S., Scott J. R. (1999) A response regulator that represses transcription of several virulence operons in the group A Streptococcus. J. Bacteriol. 181, 3649–3657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Engleberg N. C., Heath A., Miller A., Rivera C., DiRita V. J. (2001) Spontaneous mutations in the CsrRS two-component regulatory system of Streptococcus pyogenes result in enhanced virulence in a murine model of skin and soft tissue infection. J. Infect. Dis. 183, 1043–1054 [DOI] [PubMed] [Google Scholar]
- 59. Graham M. R., Smoot L. M., Migliaccio C. A., Virtaneva K., Sturdevant D. E., Porcella S. F., Federle M. J., Adams G. J., Scott J. R., Musser J. M. (2002) Virulence control in group A Streptococcus by a two-component gene regulatory system. Global expression profiling and in vivo infection modeling. Proc. Natl. Acad. Sci. U.S.A. 99, 13855–13860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bernish B., van de Rijn I. (1999) Characterization of a two-component system in Streptococcus pyogenes which is involved in regulation of hyaluronic acid production. J. Biol. Chem. 274, 4786–4793 [DOI] [PubMed] [Google Scholar]
- 61. Werb Z. (1997) ECM and cell surface proteolysis. Regulating cellular ecology. Cell 91, 439–442 [DOI] [PubMed] [Google Scholar]
- 62. Cleary P. P., Kaplan E. L., Handley J. P., Wlazlo A., Kim M. H., Hauser A. R., Schlievert P. M. (1992) Clonal basis for resurgence of serious Streptococcus pyogenes disease in the 1980s. Lancet 339, 518–521 [DOI] [PubMed] [Google Scholar]
- 63. Schmidt K. H., Wadström T. (1990) A secreted receptor related to M1 protein of Streptococcus pyogenes binds to fibrinogen, IgG, and albumin. Zentralbl. Bakteriol. 273, 216–228 [DOI] [PubMed] [Google Scholar]
- 64. Carapetis J. R., Steer A. C., Mulholland E. K., Weber M. (2005) The global burden of group A streptococcal diseases. Lancet Infect. Dis. 5, 685–694 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.