

Background: The distal histidine in myoglobin is thought to act as a gate regulating oxygen uptake.
Results: Neither the open nor closed conformation hinders oxygen uptake; a hydrophobic site is more apparent in the open conformation.
Conclusion: The driving force for ligand uptake is the hydrophobic effect.
Significance: This amplifies our understanding of the mechanisms through which proteins regulate ligand uptake.
Keywords: Computer Modeling, Histidine, Myoglobin, Oxygen Binding, Protein Conformation, E7 Channel, Free Energy, Histidine Gate
Abstract
Since the elucidation of the myoglobin (Mb) structure, a histidine residue on the E helix (His-E7) has been proposed to act as a gate with an open or closed conformation controlling access to the active site. Although it is believed that at low pH, the His-E7 gate is in its open conformation, the full relationship between the His-E7 protonation state, its conformation, and ligand migration in Mb is hotly debated. We used molecular dynamics simulations to first address the effect of His-E7 protonation on its conformation. We observed the expected shift from the closed to the open conformation upon protonation, but more importantly, noted a significant difference between the conformations of the two neutral histidine tautomers. We further computed free energy profiles for oxygen migration in each of the possible His-E7 states as well as in two instructive Mb mutants: Ala-E7 and Trp-E7. Our results show that even in the closed conformation, the His-E7 gate does not create a large barrier to oxygen migration and permits oxygen entry with only a small rotation of the imidazole side chain and movement of the E helix. We identify, instead, a hydrophobic site in the E7 channel that can accommodate an apolar diatomic ligand and enhances ligand uptake particularly in the open His-E7 conformation. This rate enhancement is diminished in the closed conformation. Taken together, our results provide a new conceptual framework for the histidine gate hypothesis.
Introduction
In the crystal structures of hemoglobin (Hb)7 and myoglobin (Mb) (1, 2), the histidine perched at the seventh position along the E helix resembles a gate guarding the entrance to the active site on the distal side of the heme group. Based on this observation, Perutz et al. (3) proposed that a swinging motion of this single amino acid side chain could control the migration rate for ligand entry into either Mb or Hb (Fig. 1), an idea that thus became known as the histidine gate hypothesis. More than 50 years of research have culminated in Mb becoming one of the most studied proteins in the field of structural biology (4). The histidine gate hypothesis and the role played by alternative ligand migration paths have been examined in detail by both experimental and theoretical methodologies but are still surrounded by controversy (5). It would be impractical to describe all the available data, and instead, we will attempt to summarize the key observations and discrepancies. Mb crystal structures have resolved the His-E7 side chain in the closed and open conformations (6). At neutral pH, the Mb crystal structures show the closed structure, which was originally observed by Perutz and Kendrew (1–3). Lowering the pH of Mb crystals resulted in His-E7 adopting an open conformation, which is attributed to His-E7 protonation and increased solvent exposure of imidazolium cation. Rabenstein et al. (7) analyzed the effect of pH on the His-E7 rotameric state theoretically and theirs results also indicated that when His-E7 is protonated the fraction of the open conformation increases.
FIGURE 1.

A schematic representation of the His-E7 gate hypothesis, in which molecular oxygen can enter and exit the distal site due to the movement of the histidine at position 7 of helix E (His-E7). The heme group (yellow) and His-E7 in the open and closed conformations are shown. The protonated state was chosen arbitrarily for schematic proposes.
The rates of ligand migration into and out of Mb are characterized experimentally by the overall ligand association (k′X) and dissociation (kX) rate constants, where X refers to the ligand being studied. The dissociation rate constant is determined by the speed of the iron-ligand bond breakage and the competition between the ligand migration to the solvent and internal rebinding to the iron atom. The limiting step is generally the iron-ligand bond breaking, with rates on the order of 0.01 to 100 s−1, whereas rates of ligand migration out of the active site on the order of 106 to 107 s−1, making the observed value of kX a relatively poor indicator of ligand trajectories. The rate of ligand association is determined by the speed of ligand entry into the distal site and the extent of iron-ligand bond formation versus ligand escape and is often a more direct indicator of ligand pathways. Thus, for protein variants or changes in pH, observed changes in k′X can be often be used to estimate changes in the initial barrier to the ligand entry (8–10). If the fraction of geminate recombination, Fgeminate, is measured after photodissociation of the iron-ligand bond, the true bimolecular rate for ligand entry, k′entry, can be computed as k′entry = k′X/Fgeminate because Fgeminate provides the fraction of ligand molecules that bind to the iron atom after they enter the active site (8).
Site-directed mutagenesis studies in Mb have shown that both k′entry and k′X increase up to 10-fold when His-E7 is replaced with smaller apolar residues, such as Ala or Gly, whereas both rate constants decrease ∼3-fold when His-E7 is replaced with the larger Trp (11), highlighting the importance of the E7 pathway in ligand migration. These results also show that the Trp-E7 and Ala-E7 Mb mutants are two useful limiting cases for evaluating the E7 pathway (8, 12). Association rate constants for ligand binding to Mb also suggest a pH dependence in the opening of the His-E7 gate because both k′O2 and k′CO increase 2-fold upon lowering the pH from 7.0 to 4.6 (13, 14). This increase in the k′X is more dramatic upon further lowering the pH to 2.0, but under these conditions, the increase in rate is due to protonation of the proximal histidine and formation of a four-coordinate deoxyheme group (15). In addition to the mutant and pH kinetic work, numerous other studies have also lent support to the relevance of the His-E7 gate opening to ligand entry in Mb (16–19).
Arguments against the His-E7 gate being the primary pathway for ligand entry come from studies showing the existence of multiple hydrophobic cavities (the Xe binding sites), which are thought to play a role in ligand migration, especially because internal pathways connecting the cavities have been identified (20–28). In addition, all computational studies have even identified many possible ligand exit pathways involving the Xe cavities (29–31), as well as ligand exit through the E7 gate (31). However, mutagenesis studies of Mb residues lining the Xe cavities only resulted in minor impacts on either k′O2 or k′entry (8, 33), suggesting that the role of the Xe cavities in ligand uptake is likely secondary to that of the E7 pathway. Regardless of this controversy, there is a general consensus that the His-E7 gate is one of the principal pathways for ligand entry in myoglobin (11, 29, 30, 34).
Although correlations between pH, His-E7 conformation, and ligand migration rates have been identified, there is still a need for a thermodynamic characterization of ligand migration through the His-E7 gate and its relationship to the conformation of the imidazole side chain. To fill this gap, we performed molecular dynamics simulations to understand the relationships between the following: 1) the His-E7 protonation state and the side chain conformation (open or closed), 2) the open and closed conformations and the free energy barriers to oxygen movement, and 3) Ala-E7 and Trp-E7 substitutions and rates of ligand entry. To our knowledge, this study is the first to explicitly attempt to characterize the relationship between distal histidine tautomeric states, side chain orientations, amino acid replacement, and the free energy barriers for oxygen migration through the E7 channel.
MATERIALS AND METHODS
System Preparation
Classical molecular dynamics (MD) simulations were performed to sample the configurational space of the systems of study. Coordinates from the Protein Data Bank were used to create initial structural models (Table 1). Each conformation found in the crystal structures of the wild-type Mb (open and closed) was simulated in the three tautomeric states: the neutral δ-tautomer (HID), the neutral ϵ-tautomer (HIE), and the positively charged protonated state (HIP), giving us six different initial structures for the wild-type myoglobin.
TABLE 1.
Summary of structures used to generate starting conformations for MD simulations
| Mb variant | Protein Data Bank code |
|---|---|
| wt His-E7 (open conformation) | 1SPE (6)a |
| wt His-E7 (closed conformation) | 1VXF (6) |
| Ala-E7 | 102 m (16) |
| Trp-E7 | 3OGB (12) |
a References are in parentheses.
Aside from His-E7, all histidine protonation states were assigned based on the immediate environment in the crystal structures and cross-checked against the protonation prediction server H++ (35). Throughout the simulations, all of the histidines maintained the same immediate environments, validating the choice of protonation states.
All systems were built using the Amber ff99SB force field (36) and solvated in a box of TIP3P water molecules, where the minimum distance between the protein and the extreme of the box was 12 Å. Crystallographic waters were left as found in the original crystal structures. The heme parameters were determined using restrained electrostatic potential charges (37) and HF/6–31G(d) wave functions according to the Amber standard protocol, as described previously (38–45).
Molecular Dynamics Simulations
MD simulations were performed using the pmemd module within the AMBER11 simulation package. Periodic boundary conditions and Ewald sums were employed for the long range electrostatic interactions (46). With the SHAKE algorithm (47), we used a 2-fs time step for the propagation of coordinates in time in the isothermal-isobaric ensemble, using the Berendsen thermostat and barostat (48). The equilibration process was performed by slowly heating the system from 0 to 300 K over the course of 500 ps. We performed a total of 150 ns of MD simulations (isothermal-isobaric ensemble) for the HID and HIP system and 200 ns for the HIE system. To provide some evidence about the convergence the simulations, we used the last snapshot of each trajectory to perform (after an additional equilibration period) 50 ns of MD with switched protonation states: HIE to HID, HIE to HIP, HIP to HIE, and HID to HIE.
Multiple Steered Molecular Dynamics
In the multiple steered molecular dynamics method, the original potential is modified by adding an harmonic restraint called Vadd(t),
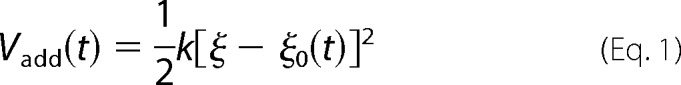 |
where k is the spring constant, ξ is an arbitrary reaction coordinate, and ξ0(t) is the time-dependent center of the spring, which is modified based on an arbitrary velocity (v) as follows in Equation 2.
Using this approach and integrating the force applied in each step, it is possible to calculate the irreversible work (W) through the reaction coordinate chosen. From multiple irreversible works obtained starting from different initial microconfigurations it is possible to employ Jarzynski's equation (49) to estimate the free profile of the process. Jarzynski's equation relates the irreversible works with free energy change as follows,
 |
where kB is the Boltzmann constant, T is the temperature, and W is the calculated irreversible work for each of the independent non-equilibrium process. A total of 40 independent perturbations were performed. This approach was used in a variety of studies such as ligand entry, escape, dynamic control of tunnel opening, and various mutagenesis studies, yielding results that are in excellent agreement with the experimentally determined rates (42–45, 50).
Because the reaction coordinate involves the formation of the O2-iron bond, a process not allowed in the standard MD force field, we introduced a Morse potential to describe this coordination (51),
where Do corresponds to the well depth (10 kcal/mol), r0 is the equilibrium bond length (1.8 Å), and the parameter β was 4 Å−1. Error analysis of the free energy profiles was done considering the bias inherent to the Jarzynski free energy estimator for a small number of independent simulations (n = 40) as proposed by Gore et al. (52).
RESULTS
Relationship between His-E7 Tautomeric and Conformational States
We performed extensive MD simulations to determine whether the His-E7 protonation state controls its conformation. To identify the protonation states of histidine, we use the following nomenclature: HIE, the neutral tautomer with the proton on the ϵ nitrogen; HID, the neutral tautomer with the proton on the δ nitrogen; and HIP, the histidine in its positively charged protonated state. As discussed below, the fixed protonation simulations afford an interesting analysis of the nonequivalence of the two neutral histidine tautomers. Given the two possible conformations of the imidazole side chain (open or closed), we performed a total of 150-ns MD simulations for each tautomer (HIE, HIP, HID) starting from the two possible conformations to remove any bias due to the initial structure. Moreover, we perturbed the protonation state after each 50-ns block of simulation time (HIE to HID, HIE to HIP, HIP to HIE, and HID to HIE) to ensure causality between the change in protonation state and the observed conformation of the His-E7 side chain. We classified the conformation as open or closed according to the His-E7 side chain dihedral angle (C-Cα-Cβ-Cγ) (∼160° open; ∼60° closed) according to the observed values in the neutral and low pH Mb crystal structures as well as in previous MD studies (26). The results show that the protonated (HIP) and the neutral ϵ-tautomer (HIE) sample exclusively either the open or closed conformation, respectively (data not shown). The third form (HID), however, was found primarily in the open conformation, although not as exclusively as in the HIP form. In the HIP and HID forms, the open conformation is stabilized by a strong hydrogen bond interaction between the HNδ of His-E7 and the backbone carbonyl group of nearby Asp-60 (Fig. 2), as previously observed (7, 53). Extending the gate analogy, this interaction serves as a “latch” for the His-E7 gate, maintaining it in the open conformation. Recall that the δ proton is present in both HIP and HID but not in the HIE state. These results suggest that His-E7 does not have to necessarily be in its charged (protonated) state to sample the open conformation. Thus, even at neutral pH, Mb should significantly populate the open state even if it cannot be “seen” in crystal structures.
FIGURE 2.

Schematic representation of His-E7 neutral δ-tautomer (HID) and protonated (HIP) after MD simulations. In both cases, His-E7 is in the open conformation forming a strong hydrogen bond with the carbonyl of Asp-60.
Structure and Dynamics of the Ala-E7 and Trp-E7 Mb Mutants
To have a better understanding of the steric constraints in the E7 channel, we also simulated the two experimentally well characterized Ala-E7 and Trp-E7 Mb mutants, which provide the upper and lower bounds for the rates of ligand entry (Table 2). Simulation of the Ala-E7 Mb mutant showed that the protein is stable with no observed changes in the distal site. The heme iron center is clearly accessible from the solvent, through the hole created by the absence of the E7 side chain (Fig. 3, top-right panel). The results for Trp-E7, on the other hand, were more surprising. The Trp-E7 side chain was drawn inward toward the distal site, as opposed to its initial outward conformation (Fig. 3, bottom panel, and Fig. 4). We observed a similar inward rotation of the hydrophobic side chain in the case of another mutant Phe-E7 Mb (data not shown). We should note that the new configuration adopted by Trp-E7 was previously observed in the crystal structures of the α Trp-E7 mutant subunit of human recombinant deoxyHbA containing wild-type β subunits (Fig. 4, right panel) (Protein Data Bank code 3NMM) (12). Both of the observed Trp-E7 conformations result in steric hindrance of the E7 pathway to the heme, as opposed to both Ala-E7 and the open His-E7 conformation (Fig. 3).
TABLE 2.
Experimentally reported ligand entry rates of oxygen migration into Sperm whale myoglobin at pH 7.0
The bimolecular rate constant for ligand entry, k′entry, was calculated as k′O2/Fgeminate (8). The errors for wild-type (WT) myoglobin were estimated from averages of multiple geminate analyses (>10) (8).
| Protein | k′O2 (μm−1 s−1) | k′entry (μm−1 s−1) |
|---|---|---|
| wt | 16 ± 3 | 34 ± 7 |
| Ala-E7 | 53 | 410 |
| Trp-E7 | 6,2 | 8.6 |
FIGURE 3.

Solvent-accessible surface contour of the E7 pathway. The surface of wt Mb (top left panel) is shown with His-E7 in the open conformation with the closed His-E7 conformation superimposed in a stick representation for reference. Surface representations of mutants Ala-E7 Mb (top right panel), Trp-E7 in the outward conformation (bottom left panel) and Trp-E7 in the inward conformation (bottom right panel) are also shown. Clear tunnels for ligand migration can be seen in the open His-E7 conformation (top left) and in the Ala-E7 (top right) mutant, both of which can be occupied by water molecules (shown as sticks). The heme group (yellow), iron atom (green), and nearby residues (cyan) are depicted in each panel.
FIGURE 4.

Conformational switch of Trp-E7 from the outward conformation (left panel) to the inward conformation (right panel). Trp-E7, Phe-CD1, Thr-E10, and Val-E11 are depicted in cyan. In both panels, the E7 pathway is blocked by the bulky Trp side chain (see Fig. 3). Position of Trp-E7 in the crystal structure of the Trp-E7 mutant α subunit of recombinant human hemoglobin with wild-type β subunit was superimposed in violet (Protein Data Bank code 3NMM) (12).
Oxygen Migration through the Conformation-dependent WT E7 Pathway
To characterize the oxygen entry process thermodynamically, we computed free energy profiles corresponding to the most populated conformation for each His-E7 state: the open conformation observed for HIP and HID and the closed conformation observed for HIE (Fig. 5). To compute each profile, we used multiple near-equilibrium perturbations and Jarzynski's equality (49, 50) to take dioxygen in the solvent, placed just outside the protein, and bring it to the bound state at 1.8 Å from the iron. The free energy was set to a value of 0 at 10 Å, where the oxygen molecule is fully solvated. The iron-O2 interaction was modeled using a Morse potential to avoid spurious steric repulsion when the oxygen is within bonding distance of the iron. This strategy has been successfully applied to the determination of ligand binding to heme proteins by our group and others (51, 54–56).
FIGURE 5.

Free energy profiles for ligand migration through the E7 pathway for the closed-HIE (black), open-HIP (red), and open-HID (green) conformations of the His-E7 in the wild-type Mb. The free energy was set to a value of 0 at 10 Å, where the oxygen molecule is fully solvated. Error bars represent mean square error considering the bias inherent to the Jarzynski free energy estimator for a small number of independent simulations (n = 40) (52).
The profiles for the open conformation (found in HIP and HID forms) present little or no barrier for oxygen entry into the E7 channel, which is characterized by a significant free energy well at ∼5 Å from the heme iron atom (Fig. 5). This well (∼3 kcal/mol) corresponds to an apolar cavity just above the edge of the porphyrin ring, but still away from the iron-O2 bond formation region. The ligand in this free energy well is in contact with apolar portions of the Thr-E10, His-E7 and Val-E11 side chains (Fig. 6). From this well, the oxygen does have to overcome a small barrier to be oriented just above the iron center for bond formation. The origin of this small barrier is the presence of Val-E11 (Fig. 6, right panel). Surprisingly, not even the closed conformation exhibits a significant barrier for oxygen entry into the E7 channel free energy well from the solvent (Fig. 5, black curve). As in the open conformation, there is only a small barrier to reach the iron, which in this case is due to the presence of Val-E11 and His-E7 (Fig. 6, left panel). The only notable difference between the closed and the open conformations is the depth of the free energy well, which is shallower in the closed conformation (∼1.5 versus 3 kcal/mol). In the closed conformation of the His-E7 side chain, the imidazole ring occupies part of apolar channel that corresponds to the free energy well, sterically restricting O2 in this pocket (Fig. 6, left panel).
FIGURE 6.

Schematic representation of the apolar channel site corresponding to the free energy well for oxygen migration, which is ∼5 Å from the iron in wt Mb. In the left panel, His-E7 is in the closed conformation, perturbing the well, whereas in the right panel, it is open, leaving the well unrestricted for oxygen entry. Heme group (yellow) with iron atom (green) and relevant E7, E10 and E11 residues (cyan) are shown explicitly. The arrow indicates the small lateral movement that the His-E7 side chain and the E helix undergo to allow oxygen to migrate toward the iron in the closed conformation.
Oxygen Entry through E7 Pathway in Trp-E7 and Ala-E7 Mb Mutants
The free energy profiles for Ala-E7 and Trp-E7 Mb provide an internal control of the methodology as well as a deeper understanding of the microscopic basis of the different ligand entry rate constants listed in Table 2. As expected, the profile for the Ala-E7 mutant exhibits absolutely no barrier for oxygen entry through the E7 channel. Surprisingly, the same free energy well, ∼5 Å from the iron atom, is found in the channel of Ala-E7 Mb and is considerably deeper than in the wild type case, ∼4 kcal/mol more favorable than bulk solvent (Fig. 7, magenta curve). The much smaller Ala side chain creates a larger and completely apolar cavity above the edge of the heme ring in which the oxygen molecule is more readily accommodated.
FIGURE 7.

Free energy profile of ligand migration through the E7 pathway for mutant proteins: Ala-E7 Mb (magenta curve), Trp-E7 Mb in the outward conformation as found in the crystal structure (blue curve) and Trp-E7 Mb in the inward conformation adopted during the simulation (orange curve). The free energy was set to a value of 0 at 10 Å, where the oxygen molecule is fully solvated. Error bars represent mean square error considering the bias inherent to the Jarzynski free energy estimator for a small number of independent simulations (n = 40) (52). The free energy profile for WT HIE (dashed black curve) and HID (dashed green curve) from Fig. 5 are included as a reference.
As described in the last section, the Trp-E7 Mb mutant was observed to sample two conformations in our simulations, including the one found in the Mb crystal structure (outward), and the one similar to that observed in the Trp-E7 α subunit of Hb mutant hybrid (inward). The resulting free energy profile for the outward but still blocked conformation of Trp-E7 shows a large ∼3 kcal/mol barrier at 8 Å from the iron center (Fig. 7, blue curve). This barrier is directly related to the presence of the bulky Trp-E7 hindering access to the heme group (Figs. 3 and 4). In this case, the free energy well, which is 5 Å from the iron atom, is completely blocked. The inward conformation of Trp-E7 presents an even higher (∼6 kcal/mol) barrier, which in this case is only 3 Å from the heme because the indole side chain has rotated into the distal pocket completely hindering access to the iron atom (Fig. 7, orange curve). The location of both barriers (outward and inward) corresponds to the shifted position of the Trp-E7 side chain in each case: at 8 Å from the iron center in the outward conformation and at 3 Å from the iron center in the inward conformation. The differences in heights are due to the mobility of the Trp-E7, which has more flexibility in the outward conformation, allowing the oxygen to circumvent the bulky side chain more easily than when it is in the inward conformation.
DISCUSSION
The goal of the present study was to understand the relationship between the rate of ligand entry into Mb and the His-E7 side chain, specifically the E7 protonation, tautomerization, and conformation, as well as its replacement with larger and smaller functional groups. Simulation of the limiting case mutants Trp-E7 and Ala-E7 Mbs provided internal tests for the validity of our methodology and a clearer understanding of the ligand entry process. The pH-dependent conformational change of the His-E7 side chain and the open conformation of the HID tautomer highlights the contribution of the hydrogen bond between the δ proton of the imidazole group and the nearby Asp-60 carbonyl, which stabilizes the open conformation. Our theoretical results are in agreement with experimental crystallographic studies showing an increase in the open conformation upon lowering the pH. These simulations also underscore the importance of the amphipathic nature of the histidine residue, which allows it to adopt the open conformation, even in a neutral tautomeric state (HID) or, alternatively, serve as an interface between the hydrophobic site and solvent molecules in the HIE tautomeric state.
The Impact of the Apolar Free Energy Well in the E7 Channel
The most significant, and novel characteristic seen in the free energy profiles is the presence of a well located near the heme at ∼5 Å from the iron. The well depth, relative to the free energy of O2 in water, increases in the order: Trp-E7 (no distinguishable well) < His-E7-closed (∼−1.5 kcal/mol) < His-E7-open (∼−3 kcal/mol) < Ala-E7 (∼−4 kcal/mol). The measured bimolecular rate constants for ligand entry, k′entry, shown in Table 2 follow the opposite trend: Ala-E7 Mb > His-E7-open (low pH) Mb > His-E7-closed (neutral pH) Mb > Trp-E7 Mb. Thus, the depth of the free energy well is directly related to the ligand entry rate. The deeper free energy well can enhance the rate of ligand entry given that solvated oxygen crosses no large barrier to reach the apolar cavity or from the cavity to the distal site itself. One possible mechanistic interpretation involves pre-concentration of molecular oxygen (or other small ligands) in the apolar cavity, which would be more favorable when His-E7 is in the open conformation or replaced with smaller amino acids. In this interpretation, the overall bimolecular rate of entry into the distal pocket is given by the first order rate of ligand movement from the apolar cavity into the distal site times the fraction of O2 molecules in the well. The latter fraction is very small and would be determined by [O2]/(Kd,cavity + [O2]) ≈ [O2]/Kd,cavity, where Kd,cavity is the equilibrium dissociation constant for O2 binding in the cavity. The approximation is reasonable because Kd,cavity is a very large number on the order of 0.1 to 10 m(57), whereas [O2] in water equilibrated with 1 atm of air is ∼0.00025 m. When the free energy well is made larger by the His-E7 to Ala mutation, Kd,cavity will decrease, increasing the bimolecular rate of entry.
In a previous work studying oxygen uptake in the α chain of an Antarctic fish Trematomus bernacchi hemoglobin, using a similar methodological approach, we also found that the closed conformation of His-E7 presents a very small free energy barrier to oxygen entry and that a free energy well at 5 Å is more prominent in the open conformation (38). Thus, although more exhaustive studies are needed, the conclusions from our simulations for ligand movement through the E7 channel seem to be valid for other globins with histidine in the E7 position as well.
The E7 Channel and Alternative Pathways
As mentioned in the Introduction, multiple pathways that do not involve the His-E7 gate have been proposed for ligand entry and exit in Mb. Such alternative migration pathways have been observed in other globins, such as in the Mycobacterium tuberculosis truncated Hb N (58–60) and in the Cerebratulus lacteus neuronal mini-globin (18), in which very clear apolar tunnels appear to be the main routes for ligand migration and not the E7 channel. In mammalian Mbs and Hbs, however, extensive experimental evidence has pointed to the E7 channel as the principal avenue for ligand migration (8, 11, 12, 18). Our calculated free energy profiles for both the open and closed His-E7 conformational states also lend considerable support to the E7 channel being a principal ligand migration route in Mb.
Comparison with Other Computational Studies
Previous simulation results of Mb have been interpreted to either support or discard the histidine gate hypothesis, based on the observed dynamics of His-E7. Bossa et al. (61) and Maragliano et al. (29) report the suspected opening of the His-E7 supporting the histidine gate hypothesis. On the other hand, some studies in both Hb and Mb report no opening motion of the His-E7 and raise doubt about the histidine gate hypothesis(32, 62). We believe that the difference between the results that lead to opposing conclusions may depend only on the tautomeric form of His-E7 used. The first set of authors used HID (mostly open in our simulations) and the second set used either HIE tautomer (exclusively closed) or did not comment on the protonation state. By considering all three forms of His-E7, we could differentiate between the conformational preference of each neutral tautomer (HID and HIE), which may reconcile some of the seemingly contradictory results above. The same exhaustive approach was used in earlier works by Jewsbury et al. (53) and more recently by Rabenstein et al. 7) leading to similar conclusions.
Regarding the ligand migration through the E7 channel, Cohen et al. report a free energy well at ∼5–6 Å from the iron center (30), which may be the same that we identify in the HIP/HID profiles. A high barrier for ligand exit through the E7 channel was later reported by Maragliano et al. (29), where the HID state was used, supporting our computed free energy profile. The counterintuitive result where ligand escaped through the E7 pathway when the histidine is in the closed conformation has recently been observed for Hb, although it was interpreted as evidence against the histidine gate hypothesis (32). By considering free energy profiles for all the protonation/conformational states of His-E7, we show that although ligand entry may occur in the closed His-E7 conformation, the presence of a deeper free energy well in the open conformation enhances the uptake of oxygen, lending new support for the histidine gate hypothesis.
Conclusions
The natural image of a gate controlling the entry to a given place is that of a steric barrier when the gate is closed and then the absence of one when the gate is open. Our analysis showed little or no barrier for oxygen entry even in the closed His-E7 conformation. The opening of His-E7 does not significantly lower a barrier; rather, it vacates a hydrophobic cavity and results in a free energy well for small apolar ligands such as molecular oxygen. This analysis suggests that movement to the open conformation creates a favorable space for the entering ligand. Thus, the His-E7 conformation influences the thermodynamics of ligand migration through this pathway by creating a lower free energy pathway rather than altering a steric barrier.
The driving force for ligand uptake into this cavity is the hydrophobic effect, in which solvent water expels the apolar diatomic ligand toward the apolar cavity, which is more pronounced by outward movement of the imidazole side chain.
Supplementary Material
Acknowledgment
Supercomputing time was granted by the Large Allocations Resource Committee (TG-MCA05T010) and by supercomputing center of INQUIMAE - FCEyN/UBA.
This work was supported in part by the University of Buenos Aires, ANPCyT (PICT-25667), Consejo Nacional de Investigaciones Científicas y Técnicas, and the EU FP7 Program (Project NOStress).

This article contains supplemental Movie 1.
- Hb
- hemoglobin
- Mb
- myoglobin
- MD
- molecular dynamics.
REFERENCES
- 1. Perutz M. F., Rossman M. G., Cullis A. F., Muirhead H., Will G., North A. C. (1960) Structure of haemoglobin: a three-dimensional Fourier synthesis at 5.5-A. resolution, obtained by x-ray analysis. Nature 185, 416–422 [DOI] [PubMed] [Google Scholar]
- 2. Kendrew J. C., Dickerson R. E., Strandberg B. E., Hart R. G., Davies D. R., Phillips D. C., Shore V. C. (1960) Structure of myoglobin: A three-dimensional Fourier synthesis at 2 A2 resolution. Nature 185, 422–427 [DOI] [PubMed] [Google Scholar]
- 3. Perutz M. F., Mathews F. S. (1966) An x-ray study of azide methaemoglobin. J. Mol. Biol. 21, 199–202 [DOI] [PubMed] [Google Scholar]
- 4. Frauenfelder H., McMahon B. H., Fenimore P. W. (2003) Myoglobin: the hydrogen atom of biology and a paradigm of complexity. Proc. Natl. Acad. Sci. U.S.A. 100, 8615–8617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Elber R. (2010) Ligand diffusion in globins: Simulations versus Experiment. Curr. Opin. Struct. Biol. 20, 162–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang F., Phillips G. N. (1996) Crystal structures of CO-, deoxy- and met-myoglobins at various pH values. J. Mol. Biol. 256, 762–774 [DOI] [PubMed] [Google Scholar]
- 7. Rabenstein B., Knapp E. W. (2001) Calculated pH-dependent population and protonation of carbon-monoxy-myoglobin conformers. Biophys. J. 80, 1141–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Scott E. E., Gibson Q. H., Olson J. S. (2001) Mapping the pathways for O2 entry into and exit from myoglobin. J. Biol. Chem. 276, 5177–5188 [DOI] [PubMed] [Google Scholar]
- 9. Milani M., Pesce A., Nardini M., Ouellet H., Ouellet Y., Dewilde S., Bocedi A., Ascenzi P., Guertin M., Moens L., Friedman J. M., Wittenberg J. B., Bolognesi M. (2005) Structural bases for heme binding and diatomic ligand recognition in truncated hemoglobins. J. Inorg. Biochem. 99, 97–109 [DOI] [PubMed] [Google Scholar]
- 10. Ouellet H., Milani M., LaBarre M., Bolognesi M., Couture M., Guertin M. (2007) The roles of Tyr(CD1) and Trp(G8) in Mycobacterium tuberculosis truncated hemoglobin O in ligand binding and on the heme distal site architecture. Biochemistry 46, 11440–11450 [DOI] [PubMed] [Google Scholar]
- 11. Olson J. S., Soman J., Phillips G. N. (2007) Ligand pathways in myoglobin: a review of Trp cavity mutations. IUBMB Life 59, 552–562 [DOI] [PubMed] [Google Scholar]
- 12. Birukou I., Soman J., Olson J. S. (2011) Blocking the gate to ligand entry in human hemoglobin. J. Biol. Chem. 286, 10515–10529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tian W. D., Sage J. T., Champion P. M. (1993) Investigations of ligand association and dissociation rates in the “open” and “closed” states of myoglobin. J. Mol. Biol. 233, 155–166 [DOI] [PubMed] [Google Scholar]
- 14. Esquerra R. M., Jensen R. A., Bhaskaran S., Pillsbury M. L., Mendoza J. L., Lintner B. W., Kliger D. S., Goldbeck R. A. (2008) The pH dependence of heme pocket hydration and ligand rebinding kinetics in photodissociated carbonmonoxymyoglobin. J. Biol. Chem. 283, 14165–14175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Coletta M., Ascenzi P., Traylor T. G., Brunori M. (1985) Kinetics of carbon monoxide binding to monomeric hemoproteins. Role of the proximal histidine. J. Biol. Chem. 260, 4151–4155 [PubMed] [Google Scholar]
- 16. Smith R. D., Blouin G. C., Johnson K. A., Phillips G. N., Jr., Olson J. S. (2010) Straight-chain alkyl isocyanides open the distal histidine gate in crystal structures of myoglobin. Biochemistry 49, 4977–4986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blouin G. C., Schweers R. L., Olson J. S. (2010) Alkyl isocyanides serve as transition state analogues for ligand entry and exit in myoglobin. Biochemistry 49, 4987–4997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Salter M. D., Blouin G. C., Soman J., Singleton E. W., Dewilde S., Moens L., Pesce A., Nardini M., Bolognesi M., Olson J. S. (2012) Determination of ligand pathways in globins: apolar tunnels versus polar gates. J. Biol. Chem. 287, 33163–33178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schmidt M., Nienhaus K., Pahl R., Krasselt A., Anderson S., Parak F., Nienhaus G. U., Srajer V. (2005) Ligand migration pathway and protein dynamics in myoglobin: A time-resolved crystallographic study on L29W MbCO. Proc. Natl. Acad. Sci. U.S.A. 102, 11704–11709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Elber R., Karplus M. (1990) Enhanced sampling in molecular dynamics: use of the time-dependent Hartree approximation for a simulation of carbon monoxide diffusion through myoglobin. J. Am. Chem. Soc. 112, 9161–9175 [Google Scholar]
- 21. Brunori M., Gibson Q. H. (2001) Cavities and packing defects in the structural dynamics of myoglobin. EMBO Rep. 2, 674–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bourgeois D., Vallone B., Schotte F., Arcovito A., Miele A. E., Sciara G., Wulff M., Anfinrud P., Brunori M. (2003) Complex landscape of protein structural dynamics unveiled by nanosecond Laue crystallography. Proc. Natl. Acad. Sci. U.S.A. 100, 8704–8709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tomita A., Sato T., Ichiyanagi K., Nozawa S., Ichikawa H., Chollet M., Kawai F., Park S. Y., Tsuduki T., Yamato T., Koshihara S. Y., Adachi S. (2009) Visualizing breathing motion of internal cavities in concert with ligand migration in myoglobin. Proc. Natl. Acad. Sci. U.S.A. 106, 2612–2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hummer G., Schotte F., Anfinrud P. A. (2004) Unveiling functional protein motions with picosecond x-ray crystallography and molecular dynamics simulations. Proc. Natl. Acad. Sci. U.S.A. 101, 15330–15334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lim M., Jackson T. A., Anfinrud P. A. (1997) Ultrafast rotation and trapping of carbon monoxide dissociated from myoglobin. Nat. Struct. Biol. 4, 209–214 [DOI] [PubMed] [Google Scholar]
- 26. Bossa C., Amadei A., Daidone I., Anselmi M., Vallone B., Brunori M., Di Nola A. (2005) Molecular dynamics simulation of sperm whale myoglobin: effects of mutations and trapped CO on the structure and dynamics of cavities. Biophys. J. 89, 465–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ceccarelli M., Anedda R., Casu M., Ruggerone P. (2008) CO escape from myoglobin with metadynamics simulations. Proteins 71, 1231–1236 [DOI] [PubMed] [Google Scholar]
- 28. Ostermann A., Waschipky R., Parak F. G., Nienhaus G. U. (2000) Ligand binding and conformational motions in myoglobin. Nature 404, 205–208 [DOI] [PubMed] [Google Scholar]
- 29. Maragliano L., Cottone G., Ciccotti G., Vanden-Eijnden E. (2010) Mapping the network of pathways of CO diffusion in myoglobin. J. Am. Chem. Soc. 132, 1010–1017 [DOI] [PubMed] [Google Scholar]
- 30. Cohen J., Arkhipov A., Braun R., Schulten K. (2006) Imaging the migration pathways for O2, CO, NO, and Xe inside myoglobin. Biophys. J. 91, 1844–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cohen J., Olsen K. W., Schulten K. (2008) Finding gas migration pathways in proteins using implicit ligand sampling. Methods Enzymol. 437, 439–457 [DOI] [PubMed] [Google Scholar]
- 32. Shadrina M. S., English A. M., Peslherbe G. H. (2012) Effective simulations of gas diffusion through kinetically accessible tunnels in multisubunit proteins: O2 pathways and escape routes in T-state deoxyhemoglobin. J. Am. Chem. Soc. 134, 11177–11184 [DOI] [PubMed] [Google Scholar]
- 33. Scott E. E., Gibson Q. H. (1997) Ligand migration in sperm whale myoglobin. Biochemistry 36, 11909–11917 [DOI] [PubMed] [Google Scholar]
- 34. Ruscio J. Z., Kumar D., Shukla M., Prisant M. G., Murali T. M., Onufriev A. V. (2008) Atomic level computational identification of ligand migration pathways between solvent and binding site in myoglobin. Proc. Natl. Acad. Sci. U.S.A. 105, 9204–9209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gordon J. C., Myers J. B., Folta T., Shoja V., Heath L. S., Onufriev A. (2005) H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 33, W368–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C. (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bayly C. I., Cieplak P., Cornell W., Kollman P. A. (1993) A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 97, 10269–10280 [Google Scholar]
- 38. Boechi L., Martì M. A., Vergara A., Sica F., Mazzarella L., Estrin D. A., Merlino A. (2011) Protonation of histidine 55 affects the oxygen access to heme in the alpha chain of the hemoglobin from the Antarctic fish Trematomus bernacchii. IUBMB Life 63, 175–182 [DOI] [PubMed] [Google Scholar]
- 39. Martí M. A., González Lebrero M. C., Roitberg A. E., Estrin D. A. (2008) Bond or cage effect: how nitrophorins transport and release nitric oxide. J. Am. Chem. Soc. 130, 1611–1618 [DOI] [PubMed] [Google Scholar]
- 40. Martí M. A., Bidon-Chanal A., Crespo A., Yeh S. R., Guallar V., Luque F. J., Estrin D. A. (2008) Mechanism of product release in NO detoxification from Mycobacterium tuberculosis truncated hemoglobin N. J. Am. Chem. Soc. 130, 1688–1693 [DOI] [PubMed] [Google Scholar]
- 41. Capece L., Marti M. A., Crespo A., Doctorovich F., Estrin D. A. (2006) Heme protein oxygen affinity regulation exerted by proximal effects. J. Am. Chem. Soc. 128, 12455–12461 [DOI] [PubMed] [Google Scholar]
- 42. Boechi L., Mañez P. A., Luque F. J., Marti M. A., Estrin D. A. (2010) Unraveling the molecular basis for ligand binding in truncated hemoglobins: the trHbO Bacillus subtilis case. Proteins 78, 962–970 [DOI] [PubMed] [Google Scholar]
- 43. Boechi L., Martí M. A., Milani M., Bolognesi M., Luque F. J., Estrin D. A. (2008) Structural determinants of ligand migration in Mycobacterium tuberculosis truncated hemoglobin O. Proteins 73, 372–379 [DOI] [PubMed] [Google Scholar]
- 44. Pesce A., Nardini M., Dewilde S., Capece L., Martí M. A., Congia S., Salter M. D., Blouin G. C., Estrin D. A., Ascenzi P., Moens L., Bolognesi M., Olson J. S. (2011) Ligand migration in the apolar tunnel of Cerebratulus lacteus mini-hemoglobin. J. Biol. Chem. 286, 5347–5358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bidon-Chanal A., Martí M. A., Crespo A., Milani M., Orozco M., Bolognesi M., Luque F. J., Estrin D. A. (2006) Ligand-induced dynamical regulation of NO conversion in Mycobacterium tuberculosis truncated hemoglobin-N. Proteins 64, 457–464 [DOI] [PubMed] [Google Scholar]
- 46. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–936 [Google Scholar]
- 47. Ryckaert J. P., Ciccotti G., Berendsen H. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 [Google Scholar]
- 48. Berendsen H. J. C., Postma J. P. M., Van Gunsteren W. F., DiNola A., Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 [Google Scholar]
- 49. Jarzynski C. (1997) Nonequilibrium Equality for Free Energy Differences. Phys. Rev. Lett. 78, 2690–2693 [Google Scholar]
- 50. Xiong H., Crespo A., Marti M., Estrin D., Roitberg A. E. (2006) Free Energy Calculations with Non-Equilibrium Methods: Applications of the Jarzynski Relationship. Theor. Chem. Acc. 116, 338–346 [Google Scholar]
- 51. Meuwly M., Becker O. M., Stote R., Karplus M. (2002) NO rebinding to myoglobin: a reactive molecular dynamics study. Biophys. Chem. 98, 183–207 [DOI] [PubMed] [Google Scholar]
- 52. Gore J., Ritort F., Bustamante C. (2003) Bias and error in estimates of equilibrium free-energy differences from nonequilibrium measurements. Proc. Natl. Acad. Sci. U.S.A. 100, 12564–12569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jewsbury P., Kitagawa T. (1994) The distal residue-CO interaction in carbonmonoxy myoglobins: a molecular dynamics study of two distal histidine tautomers. Biophys. J. 67, 2236–2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. De Biase P. M., Paggi D. A., Doctorovich F., Hildebrandt P., Estrin D. A., Murgida D. H., Marti M. A. (2009) Molecular basis for the electric field modulation of cytochrome c structure and function. J. Am. Chem. Soc. 131, 16248–16256 [DOI] [PubMed] [Google Scholar]
- 55. Nadra A. D., Martí M. A., Pesce A., Bolognesi M., Estrin D. A. (2008) Exploring the molecular basis of heme coordination in human neuroglobin. Proteins 71, 695–705 [DOI] [PubMed] [Google Scholar]
- 56. Capece L., Marti M. A., Bidon-Chanal A., Nadra A., Luque F. J., Estrin D. A. (2009) High pressure reveals structural determinants for globin hexacoordination: neuroglobin and myoglobin cases. Proteins 75, 885–894 [DOI] [PubMed] [Google Scholar]
- 57. Birukou I., Maillett D. H., Birukova A., Olson J. S. (2011) Modulating distal cavities in the α and β subunits of human HbA reveals the primary ligand migration pathway. Biochemistry 50, 7361–7374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wittenberg J. B., Bolognesi M., Wittenberg B. A., Guertin M. (2002) Truncated hemoglobins: a new family of hemoglobins widely distributed in bacteria, unicellular eukaryotes, and plants. J. Biol. Chem. 277, 871–874 [DOI] [PubMed] [Google Scholar]
- 59. Milani M., Pesce A., Ouellet Y., Ascenzi P., Guertin M., Bolognesi M. (2001) Mycobacterium tuberculosis hemoglobin N displays a protein tunnel suited for O2 diffusion to the heme. EMBO J. 20, 3902–3909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cazade P. A., Meuwly M. (2012) Oxygen migration pathways in NO-bound truncated hemoglobin. Chemphyschem 13, 4276–4286 [DOI] [PubMed] [Google Scholar]
- 61. Bossa C., Anselmi M., Roccatano D., Amadei A., Vallone B., Brunori M., Di Nola A. (2004) Extended molecular dynamics simulation of the carbon monoxide migration in sperm whale myoglobin. Biophys. J. 86, 3855–3862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cohen J., Schulten K. (2007) O2 migration pathways are not conserved across proteins of a similar fold. Biophys. J. 93, 3591–3600 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.