

Abstract
Background
Celery is an increasing popular vegetable species, but limited transcriptome and genomic data hinder the research to it. In addition, a lack of celery molecular markers limits the process of molecular genetic breeding. High-throughput transcriptome sequencing is an efficient method to generate a large transcriptome sequence dataset for gene discovery, molecular marker development and marker-assisted selection breeding.
Principal Findings
Celery transcriptomes from four tissues were sequenced using Illumina paired-end sequencing technology. De novo assembling was performed to generate a collection of 42,280 unigenes (average length of 502.6 bp) that represent the first transcriptome of the species. 78.43% and 48.93% of the unigenes had significant similarity with proteins in the National Center for Biotechnology Information (NCBI) non-redundant protein database (Nr) and Swiss-Prot database respectively, and 10,473 (24.77%) unigenes were assigned to Clusters of Orthologous Groups (COG). 21,126 (49.97%) unigenes harboring Interpro domains were annotated, in which 15,409 (36.45%) were assigned to Gene Ontology(GO) categories. Additionally, 7,478 unigenes were mapped onto 228 pathways using the Kyoto Encyclopedia of Genes and Genomes Pathway database (KEGG). Large numbers of simple sequence repeats (SSRs) were indentified, and then the rate of successful amplication and polymorphism were investigated among 31 celery accessions.
Conclusions
This study demonstrates the feasibility of generating a large scale of sequence information by Illumina paired-end sequencing and efficient assembling. Our results provide a valuable resource for celery research. The developed molecular markers are the foundation of further genetic linkage analysis and gene localization, and they will be essential to accelerate the process of breeding.
Introduction
Apium graveolens L. (family Apiaceae), originated from the Mediterranean basin, is a biennial species grown worldwide. There are three distinct taxonomic varieties, var. dulce (celery), var. rapaceum (celeriac), and var. secalinum (smallage) [1]. Celery is the most widely grown variety and it was spread to China in the Han Dynasty (the second century B. C.). After a long period of domestication, cultivars with slender petiole known as Chinese celery or local celery came into being [2]. Celery is rich in a variety of nutrients like vitamins, minerals and proteins and it has many pharmacological efficacies, which make it an increasing popular vegetable to consumers [3], [4]. In China, celery growing area has been increased from 0.54 million hectare in 2004 to 0.57 million hectare in 2006 according to the data statistics, almost accounting for 3% of the total vegetable planting area throughout the country. So it is essential to breed new celery varieties and enrich commercial species.
Molecular marker assisted selection has become a more and more important auxiliary means for conventional breeding. However, only a few molecular markers have been indentified in celery. After the development of isozyme markers and morphological markers [5], [6], Huestis et al [7] initially developed 21 restriction fragment-length polymorphism (RFLP) markers. Yang and Quiros [8] first used random amplified polymorphic DNA (RAPD) markers in celery to distinguish different cultivars. Domblides [9] did the similar research using RAPD markers. Then amplified fragment length polymorphism (AFLP) markers were used in celery for cultivar classification and genetic diversity analysis [10], [11]. In recent decades, simple sequence repeats (SSRs) as a marker system have been widely used. However, the lack of expressed sequence tags (ESTs) and genomic sequences prevents the development of SSR markers in celery. So far only 11 SSR markers have been reported [12]. Wang et al [13] used the 11 SSR makers and 60 sequence-related amplified polymorphism (SRAP) primer combinations to study the genetic diversity of 68 celery accessions.
During the last decades, large amounts of transcriptomic and genomic sequences have been available in many model organisms. For Apium graveolens, only about 2,000 ESTs were deposited in GenBank database. Therefore, extensive genomic or transcriptomic sequences are badly needed for Apium graveolens, which can be used for new genes discovery, molecular markers development, gene localization, and comparative genomics and so on. Given that the celery is a diploid with a large genome (3×109 bp) and a high degree of heterozygosity, it is infeasible to consider whole genome sequencing because of the high costs and time-consuming. Fortunately, the advent of transcriptome sequencing provides an alternative to the whole genome sequencing. Transcriptome sequences exclude non-coding DNA, thus the sequences obtained must contain a high content of functional information [14], [15] and are beneficial to reveal the molecular mechanism of functional genes [16].
A number of next-generation sequencing (NGS) technologies such as Roche/454, Illumina and AB SOLiD have recently become available. These technologies are capable of generating high-throughput reads at a relatively low cost and are being utilized for research in many areas like de novo sequencing, genome re-sequencing and whole genome or transcriptome analysis. In the past few years, the 454 pyrosequencing has been preferred for non-model organisms [17]–[20] due to its longer read length. With shorter read length, Illumina and SOLID platforms were confined to re-sequencing applications which rely on a reference sequence. Recently, with increased read length by Illumina technology improvement and development of new computational tools, short reads can be assembled and used for transcriptome analysis. De novo assemblies using sequence reads from Illumina technology have been reported in many non-model organisms without a reference sequence [21]–[23]. NGS technologies prove to be efficient, inexpensive and reliable for genome and transcriptome sequencing, and suitable for non-model organisms such as celery.
In this study, we sampled from different celery tissues (roots, stems, petioles and leaves) and used Illumina paired-end sequencing technology to generate a large-scale EST dataset and develop a set of EST-SSRs. This study is the first try to characterize the complete transcriptome of celery by analyzing large-scale transcriptome sequences. These sequences will serve as a valuable resource for novel gene discovery, development of molecular markers, gene mapping and comparative genomics.
Materials and Methods
Plant Material
Celery line “Q 111” was grown at the experimental station of China Agricultural University, Beijing, China. Samples were collected from roots, stems, petioles and leaves, frozen immediately in liquid nitrogen and stored at −80°C until use.
RNA Extraction and Library Preparation for Transcriptome Analysis
Total RNA was extracted using the Trizol reagent according to the manufacture’s instructions (Invitrogen). The total RNA concentration was quantified using UV spectrophotometry, and quality of total RNA is checked by electrophoresis in 1% agarose gel. Equal volumes of RNA from each of the four tissues were pooled. Paired-end libraries with approximate average insert lengths of 200 base pairs were synthesized using the Genomic Sample Prep kit (Illumina, San Diego, CA) according to manufacturer’s instructions. Prior to cluster generation, library concentration and size were assayed using the Agilent DNA1000 kit (Agilent, USA) on a 2100 Bioanalyzer (Agilent, USA). Libraries were sequenced as 100-mer×2 on a Hi-Seq 2000 equipped with a paired-end module at the MininGene Biotechnology Co. Ltd (Beijing, China).
Illumina Reads Processing and Assembly
A Perl script was written to remove low quality sequences (reads with a base quality less than 20). Then the high quality reads were assembled by Trinity [24] with default settings except K-mer value to construct unique consensus sequences.
Gene Annotations and Classifications
Functional annotations were performed by sequence comparison with public databases. All unigenes were compared with the NCBI non-redundant protein database (http://www.ncbi.nlm.nih.gov/), Swiss-Prot database (http://www.expasy.ch/sprot), the Clusters of Orthologous Groups database (http://www.ncbi.nlm.nih.gov/COG/) using BLASTx [25] with an E-value of less than 1e−5, 1e−10 and 1e−10 respectively. InterPro domains [26] were annotated by InterProScan [27] Release 16.0 and functional assignments were mapped onto Gene Ontology (GO) (http://www.geneontology.org/). Then we made GO classification and drew GO tree using WEGO (http://wego.genomics.org.cn/cgi-bin/wego/index.pl). Pathway assignments were carried out based on the KEGG database (http://www.genome.jp/kegg). Unigenes were first compared with KEGG database using BLASTx with an E-value of less than 1e−10, and then a Perl script was developed to retrieve KO (KEGG Orthology) information from BLASTx results and we established pathway correlation between unigenes and database.
EST-SSR Detection and Primer Design
Potential SSR markers were detected among the 42,280 unigenes using the MISA tool [28] (http://pjrc.ipk-gatersleben.de/misa/). We searched for SSRs with motifs ranging from mono- to hexa-nucleotides in size. The minimum of repeat units were set as follows: ten repeat units for mono-nucleotide, six for di-nucleotides and five for tri-, tetra-, penta- and hexa-nucelotides. Mono-nucleotide repeats were discarded given that it was difficult to distinguish genuine mono-nucleotide repeats from polyadenylation products and some mono-nucleotide repeats were generated by base mismatch or sequencing errors. Primer pairs were designed using Primer3 (http://fokker.wi.mit.edu/primer3/) with default parameters and 80 randomly selected primer pairs (Table S1) were synthesized (Beijing Sunbiotech co., Ltd) for polymorphism detection in celery.
Survey of EST-SSR Polymorphism
Polymorphism was validated among 31 celery accessions (Table S2). Genomic DNA was extracted from celery tender leaves using a modified version of the cetyltrimethyl ammonium bromide (CTAB) method [29]. PCR amplifications were conducted in a final volume of 10 µL containing 3.5 µL 2×Taq PCR MasterMix (Beijing Biomed co., Ltd), 4.5 µLddH2O, 0.5 µL of each primer (5 µM) and 1 µL of template (aprox. 20 ng/µL). PCR was performed as follows: denaturation at 94°C for 5 min, followed by 38 cycles of 30s at 94°C, 30s at Tm (annealing temperature), 1 min at 72°C and a final step at 72°C for 10 min. PCR products were firstly detected by agarose gel elctrophoresis and the products possessing single band or only a few bands were subjected to 7% polyacrylamide gel to separate alleles. With regard to those had no bands or multiple bands, we optimized the PCR condition to get better products for separation of alleles. PCR products were mixed with a volume of loading buffer and then denatured at 95°C for 10 min before being loaded on the polyacrylamide gel.
Data Collection and Cluster Analysis
The presence or absence of each single band was coded by 1 or 0 respectively. Scored data from polymorphic loci were used to calculate the polymorphism information content (PIC) according to the formula of PIC = 1–∑pi2 (pi is the frequency of ith allele for each locus) [30]. Observed heterozygosity (Ho) and expected heterozygosity (He) were calculated using the Popgene software version 1.31 [31]. A genetic similarity matrix was constructed using the NTSYSpc 2.1 software [32]. UPGMA (un-weighted pair group method with arithmetic mean) cluster analysis was performed to develop a dendrogram.
Results
Illumina Paired-end Sequencing and de novo Assembly
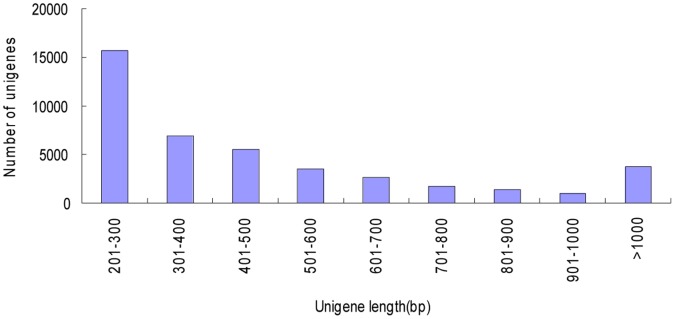
The paired-end sequencing yielded 2×101-bp reads from either end of the cDNA fragment. In this study, a total of 27,154,728 raw sequencing reads were generated from a 200 bp insert library, encompassing 2.7 Gb of sequence data. After stringent quality assessment and data filtering, reads with Q20 bases (those with a base quality greater than 20) were selected as high quality reads for further analysis. 25,915,104 (89.33%) reads were deemed high quality reads, of which 621,320 (2.4%) were ribosomal. The sequences have been deposited in DDBJ Sequence Read Archive (DRA, http://trace.ddbj.nig.ac.jp/dra/) with accession number DRA000903. We used the Trinity method with optimized k-mer length of 31 for de novo assembly. Finally all short sequences were assembled into 42,280 unigenes with an average length of 502.6 bp and a median length of 604 bp. There were 22,262,653 (85.91%) reads assembled into transcripts. The number of reads aligned to each unigene ranges form 1 to 92,345, with an average number of 228. The majority of the reads were in the range of 200–400 bp, which accounted for 53.46%. There were 15,906 unigenes (37.62%) in the length range of 401 to 1000 bp and 3,769 unigenes (8.91%) with length more than 1000 bp (Figure 1).
Figure 1. Length distribution of the celery unigenes de novo assembled from 42280 ESTs.
Annotation of all Non-redundant Unigenes
A sequence similarity search was conducted against the Nr database (E-value<1e−5) and Swiss-Prot database (E-value<1e−10) using the BLASTx algorithm, with the outcome that 33,160 (78.43%) and 20,686 (48.93%) unigenes showed homology with sequences in the Nr and Swiss-Prot database respectively. Our results showed that more than 80% of unigenes over 400 bp in length had BLAST matches against Nr database, whereas only 65.34% of unigenes shorter than 300 bp did (Figure 2). The same tendency was observed in the BLAST against Swiss-Prot database. We also made analysis of E-value and similarity distributions of the top hits in the Nr database. There were 35.92% and 35.49% of the sequences showing significant homology (E-value<1e−50) and high similarity (greater than 80%), respectively (Figure 3A and 3C). For E-value and similarity distributions of the top hits in the Swiss-Prot database, the percentages were 32.87% and 28.71% (Figure 3B and 3D).
Figure 2. Comparison of unigene length between hit and no hit unigenes.

Longer contigs were more likely to have BLASTx homologs in protein database.
Figure 3. Characteristics of similarity search of unigenes against Nr and Swiss-Prot databases.

(A) E-value distribution of BLAST hits for each unigene with a cutoff E-value of 1E-5 in the Nr database. (B) E-value distribution of BLAST hits for each unigene with a cutoff E-value of 1E-5 in the Swiss-Prot database. (C) Similarity distribution of the top BLAST hits for each unigene in Nr database. (D) Similarity distribution of the top BLAST hits for each unigene in Swiss-Prot database.
The celery unigenes were also compared with all the 25,276 Apiaceae ESTs in NCBI and 58,751 carrot transcriptome sequences [33] with the E-value of less than 1e−10, and with the result that 19,878 (47.02%) and 2,886 (6.83%) unigenes had matches against carrot transcripts and Apiaceae ESTs, respectively. Similarly, 19,872 carrot transcripts and 2,881 Apiaceae ESTs had significant matches to the celery transcripts when they were respectively compared to the celery transcripts.
In addition, we made an ORF prediction analysis. ORF was predicted using getORF from EMBOSS package and most unigenes (86.94%) were identified to have an ORF (Table S3). Among those with ORF, 31,352 (85.30%) unigenes had annotations against at least one of the following databases: Nr, SwissProt and KEGG database. They are probably genes encoding specific proteins, while unigenes without hits (14.70%) may represent new genes or errors in the assembly procedure.
Functional Classification by GO
Gene Ontology (GO) provides ontologies of defined terms representing gene product properties, and it has ontologies that describe gene products in terms of their associated biological processes, cellular components and molecular functions. In this study, 15,409 unigenes could be assigned to one or more ontologies and we assigned each unigene to a set of GO Slims. A summary with unigenes classified to each GO slim term is shown in Figure 4. Totally, 5,535 unigenes were grouped under cellular components, 13,934 unigenes under molecular functions and 11,210 unigenes under biological processes. Metabolic process (7,908 unigenes, 70.54%) and cellular process (7,024 unigenes, 62.66%) were the most highly represented groups under the biological process category. Genes involved in other important biological processes such as biological regulation, stimulus response, reproduction and developmental process were also identified. Furthermore, we also found that a relatively large numbers of unigenes (11.57%) were involved in the metabolism of pigmentation, which may play a role in the petiole color formation. For the cellular components category, cell and cell part were the most highly represented groups. Regarding molecular functions, binding and catalytic activity represented the majorities of the category with a large number of ligases, transferases, hydrolases, oxidoreductases annotated.
Figure 4. Gene Ontology classifications of assembled unigenes.

The unigenes are summarized into three main categories: biological process, cellular location, and molecular function. In total, 15,409 unigenes with BLASTx matches were assigned to gene ontologies.
Functional Classification by COG
All unigenes were subjected to a search against the COG database for functional prediction and classification. 9,332 sequences were assigned to COG classifications (Figure 5). Among the 24 COG categories, the cluster for general function prediction only (17.38%) was the largest group, followed by posttranslational modification, protein turnover and chaperones (9.14%), translation, ribosomal structure and biogenesis (8.82%) and carbohydrate transport and metabolism (8.36%).
Figure 5. Clusters of orthologous groups (COG) classification.

In total, 10,473 sequences were grouped into 24 COG classifications.
Functional Classification by KEGG
Unigenes were compared with KEGG using BLASTx with an E-value of less than 1e−10 and the corresponding pathways were established. Out of the 42,280 unigenes, 30,704 (72.62%) were annotated and 9,982 (32.51%) were assigned to KEGG pathways. Among the 9,982 unigenes, 4,921 were assigned to the metabolic pathways, composing the largest group of the five categories classified by KEGG (Figure 6B). The KEGG metabolic pathways (Figure 6A) mainly contained carbohydrate metabolism, energy metabolism, amino acid metabolism, lipid metabolism and glycan biosynthesis and nucleotide metabolism. In addition to the unigenes assigned to the metabolism pathways, 4,128 unigenes were sorted to the genetic information processing involving folding, sorting, degradation, replication and repair, transcription and translation. Additionally, there were 1,265 and 1,149 unigenes classified to cellular processes and environmental information processing respectively.
Figure 6. Pathway assignment based on KEGG.

(A) Classification based on metabolism categories; (B) Categories classified by KEGG.
Genes Potentially Controlling Petiole Traits and Bolting
There exist green and yellow petioles in celery, and green petiole color has been reported to be dominant to yellow [34], [35]. Yellowing mutation is a common phenomenon in the nature and the celery with yellow petiole is considered as a mutant of the celery with green petiole. Similar phenomenons have been reported in organism like cucumber [36]–[38], pepper [39] and carrot [40], [41]. Yellowing mutation plays an important role in studies of plant photosynthesis mechanism, Chlorophyll biosynthetic pathway, genetic control mechanism and genetic breeding. It has been reported that the content of chlorophyll in yellowing plants is dramatically lower than that in normal plants [34], [35], [36], [38], [41]. The celery unigenes contain genes potentially controlling petiole color (Table S4). And we found 85 unigenes participated in chlorophyll metabolism pathways. Combined with the GO annotations, there were 8 unigenes involved in chlorophyll biosynthetic process and 4 in chlorophyll catabolic process, and annotations of these unigenes were consistent with Nr or Swiss-Prot database.
According to petiole organizational structure, celery can be divided into hollow and solid types. Collenchyma, presented underneath the petiole epidermis, is the main mechanical organization of celery petiole with the function of supporting the whole plant. Cellulose and pectin are the main components of collenchyma cells walls which are uneven thickened, and the both may be correlated with the formation of hollow or solid petioles. About 100 homologs involved in metabolism of cellulose and pectin were found in our transcripts (Table S5).
Early bolting, being a great obstacle in production, dramatically affects the commercial quality of celery. The unigenes include orthologs of genes involved in flowering (Table S6) such as ELF8 (EARLY FLOWERING 8), ELF7 (EARLY FLOWERING 7), FCA (flowering time control), FLC (FLOWERING LOCUS C) [42], RKF3 (RECEPTOR-LIKE KINASE IN FLOWERS 3) [43], TM6 and AP3 (flowering-related B-class MADS-box protein) [44], TFL2 (TERMINAL FLOWER 2) [45] and PIE1 (PHOTOPERIOD-INDEPENDENT EARLY FLOWERING 1) [46]. In addition, the date of bolting and flowering can be regulated by plant hormones and environmental conditions and it has been reported that gibberellins (GA) can facilitates date of bolting [47], [48]. We detected some orthologs of genes including gibberellin 2-oxidase (GA2ox1, GA2ox2) and gibberellin receptor GID1 which were involved in signal transduction pathways [49].
EST-SSR Discovery: Distribution and Frequencies
All of the 42,280 unigenes generated in this study were used to mine potential microsatellites using MISA software. A total of 2,997 SSRs were identified in 2,601 unigenes. Of all the SSR containing unigenes, 339 sequences contained more than one SSR and 472 SSRs were present in compound form (Table 1). And we found 1 SSR per 10 Kb on average in this study, which was significantly different from the frequency in coffee (1 SSR per 1.5 Kb) [50] and in cotton (1 SSR per 20 Kb) [51]. Among the 2,997 SSRs identified, the di-nucleotide repeat motifs were the most abundant types (43.98%), followed by mono- (32.93%), tri- (21.62%), tetra- (0.97%), hexa- (0.13%) and penta-nucleotide (0.37%) repeat motifs. The frequencies of EST-SSRs with different numbers of tandem repeats were summarized in Table 2. SSRs with six tandem repeats (22.62%) were the most common, followed by ten tandem repeats (18.75%), five tandem repeats (12.98%), and seven tandem repeats (12.95%). Given that the mono-nucleotide repeats may not be accurate because of the sequencing errors and assembly mistakes, mono-nucleotide repeats were not used for further analysis. The most common type of di-nucleotide was GA/TC which accounted for 29.55% of the repeats, followed by AG/CT (20.15%) and CA/TG (5.57%). Among the tri-nucleotide repeats, the TGA/TCA (2.74%) and CCA/TGG (2.39%) were the most frequent motifs (Figure 7).
Table 1. Summary of SSR searching results.
| Item | Number |
| Total number of unigenes examined | 42,280 |
| Total size of examined unigenes (bp) | 21,250,238 |
| Number of SSR-containing unigenes | 2,578 |
| Total number of identified SSRs | 2,997 |
| Avarage number of SSRs per 1 K | 0.1 |
| Number of unigenes containing 1 SSR | 2,239 |
| Number of unigenes containing more than 1 SSR | 339 |
| Number of SSRs present in compound formation | 478 |
Table 2. Distribution of identified SSRs using the MISA software.
| Motif | Repeat Numbers | total | % | |||||||
| 5 | 6 | 7 | 8 | 9 | 10 | 11 | >11 | |||
| Mono- | 0 | 0 | 0 | 0 | 0 | 451 | 194 | 342 | 987 | 32.93 |
| Di- | 0 | 514 | 284 | 213 | 128 | 110 | 66 | 3 | 1,318 | 43.98 |
| Tri- | 356 | 157 | 102 | 25 | 5 | 0 | 0 | 3 | 648 | 21.62 |
| Tetra- | 23 | 4 | 2 | 0 | 0 | 0 | 0 | 0 | 29 | 0.97 |
| Penta- | 1 | 2 | 0 | 0 | 0 | 1 | 0 | 0 | 4 | 0.13 |
| Hexa- | 9 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 11 | 0.37 |
| Total | 389 | 678 | 388 | 238 | 134 | 562 | 260 | 348 | 2,997 | 100 |
| % | 12.98 | 22.62 | 12.95 | 7.94 | 4.47 | 18.75 | 8.68 | 11.61% | 100 | |
Figure 7. Frequency distribution of SSRs based on motif types.

The GA/TC di-nucleotide repeat motif was the most abundant motif detected.
Polymorphism Test of SSR Markers
31 celery accessions were used to evaluate the rate of amplication and polymorphism using 80 primer pairs randomly selected, of which 65 pairs (81.25%) successfully amplified fragments and 47 pairs (72.31%) produced PCR amplicons at the expected size. We also found 28 microsatellite loci showed allelic polymorphism (Table 3). The number of alleles per locus raged from two to four with an average of 2.68. The value of observed heterozygosity (Ho) varied from 0 to 0.92 (mean: 0.14), while the expected heterozygosity (He) varied form 0.06 to 0.74 (mean: 0.36). Polymorphism information content (PIC) values ranged form 0.06 to 0.72 with an average of 0.35.
Table 3. Characteristics of the primer pairs possessing polymorphic SSR loci texted in 31 different accessions of A. graveolens.
| Primer | SSRs | No.of alleles | Ho | He | PIC |
| QC10 | (GAAA)5 | 3 | 0.00 | 0.12 | 0.12 |
| QC 12 | (AACT)5 | 2 | 0.00 | 0.16 | 0.15 |
| QC 24 | (CT)6 | 2 | 0.34 | 0.46 | 0.45 |
| QC 28 | (GAG)8 | 4 | 0.06 | 0.56 | 0.55 |
| QC 34 | (ATT)5 | 2 | 0.09 | 0.24 | 0.24 |
| QC 41 | (TC)6 | 4 | 0.31 | 0.74 | 0.72 |
| QC 43 | (AT)6 | 3 | 0.03 | 0.15 | 0.15 |
| QC 46 | (TG)6 | 3 | 0.41 | 0.54 | 0.53 |
| QC 47 | (AG)6 | 3 | 0.10 | 0.55 | 0.54 |
| QC 48 | (TC)6 | 2 | 0.13 | 0.12 | 0.12 |
| QC 49 | (AG)6 | 2 | 0.00 | 0.18 | 0.17 |
| QC 51 | (TTG)6 | 3 | 0.92 | 0.58 | 0.57 |
| QC 52 | (GCA)5 | 4 | 0.00 | 0.59 | 0.58 |
| QC 53 | (GCT)6 | 3 | 0.03 | 0.51 | 0.51 |
| QC 55 | (TCA)5 | 3 | 0.03 | 0.56 | 0.55 |
| QC 56-1 | (ACC)6 | 2 | 0.00 | 0.18 | 0.18 |
| QC 56-2 | (ACC)6 | 3 | 0.04 | 0.23 | 0.23 |
| QC 57 | (TCA)5 | 2 | 0.00 | 0.06 | 0.06 |
| QC 62 | (TGA)6 | 2 | 0.00 | 0.06 | 0.06 |
| QC 63 | (GTG)6 | 2 | 0.20 | 0.50 | 0.49 |
| QC 67 | (GGT)7 | 2 | 0.00 | 0.06 | 0.06 |
| QC 70 | (TCA)5 | 4 | 0.11 | 0.63 | 0.60 |
| QC 71 | (GGT)7 | 3 | 0.03 | 0.19 | 0.18 |
| QC 72 | (TCA)7 | 2 | 0.14 | 0.38 | 0.38 |
| QC 73 | (CAG)6 | 2 | 0.00 | 0.06 | 0.06 |
| QC 75 | (CAG)5 | 2 | 0.86 | 0.50 | 0.49 |
| QC 80 | (GT)6 | 3 | 0.07 | 0.52 | 0.51 |
| QC 86 | (AT)8 | 3 | 0.03 | 0.54 | 0.53 |
| Mean | 2.68 | 0.14 | 0.36 | 0.35 |
Ho: Observed heterozygosity; He: Expected heterozygosity; PIC: Polymorphism Information Content.
The polymorphic SSR markers were then used to perform genetic correlation analysis among the 31 different A. graveolens accessions, and dendrograms were constructed from the genetic similarity matrix (Figure 8). At the genetic distance of 0.6, cultivated and wild species were separated. Within the cultivated species, almost all lines of A. graveolens L. var. dulce formed a cluster, except that five lines (C111, C67, C114, C123, and C163) were classified into a group with A. graveolens L. var.rapaceum (C163). The lines belonging to A. graveolens L. var. dulce were further classified into local celery (cultivars developed through ancient introductions) and celery (modern cultivars introduced from Europe and America). And most local celery formed one group with the remaining scattered in the celery group.
Figure 8. Similarity relationships of 31 different accessions of A. graveolens based on 28 EST-SSR loci.

LC: local celery; C: celery.
Discussion
Illumina Paired-end Sequencing and Assembly
With the introduction of next-generation sequencing, transcriptome sequencing, which displays a great advantage over traditional Sanger sequencing by lowering the cost, increasing the throughput and eliminating complicated bacterial cloning, has become an important tool used in research. Roche GS FLX platform, due to its longer read length, has been widely used for de novo transcriptome sequencing in many organisms like pigeonpea [52], yellow lupin [53], lodgepole pine [18], [54] and so on. Illumina was initially limited to organisms with reference genomes available due to its shorter read length [55]–[58]. While in recent years, with the improvement of read length by paired-end sequencing and the development of bioinformatics and computational methods [24], [59]–[62], the relatively short reads can be effectively assembled and has been successfully used for non-model organisms [63]–[65]. In accordance with the previous reports, our results also proved that short reads from Illumina paired-end sequencing could be well assembled and used for transcriptome analysis, gene identification and marker development in celery. Here, approximately 13 million paired-end reads were generated from HiSeq 2000 and they were finally assembled to 42,280 unigenes with an average length of 502.6 bp. The unigenes were larger than that reported for Lycoris sprengeri and cauliflower transcriptomes using Genome Analyzer IIx platform [66], [67]. The average length was also longer than that reported for rubber tree, Polygonum cuspidate, Hypericum perforatum and safflower transcriptomes obtained using the same platform with this study but different assembly software (SOAPdenovo) from this study (Trinity) except the safflower transcriptome (Trinity) [68]–[71]. The different assembly softwares used probably led to the diversity of average unigene length. The sequence reads produced unigenes (mean = 502.6 bp) even longer than some organisms like Ziziphus Celata (mean = 408 bp) and Salvia miltiorrhiza (mean = 414 bp) sequenced by Roche 454 GS FLX platform [72], [73].
Quality of the de novo assembly was validated by comparison with known databases, functional annotation and marker validation by the PCR amplification. It turned out that a high percentage of unigenes could match to the databases and most of the primers could produce amplicons, indicating the high quality of our assembled unigenes. By far, this is the first report of a large scale of transcriptome sequencing and analysis on celery.
Functional Annotation and Classification of Unigenes
Due to the lack of a reference genome, it is kind of difficult to estimate the number of genes and predict the potential functions of the transcripts. So we made a BLAST analysis using the public protein databases to indentify genes in our collection indirectly. In our study, 78.28% and 48.93% of the celery transcripts had homologs in the Nr and Swiss-Prot protein database, respectively. The percentage of hits was positive correlation with the number of long sequences, which was confirmed by the fact that 41.05% of unigenes shorter than 200 bp had no BLAST matches while only 1.67% longer than 1,000 bp had no hits. We also observed the same correlation in other studies [18], [64], [65]. The unigenes without hits probably belonged to untranslated regions, non-coding RNA, short sequences not containing a protein domain or assembly mistakes. The results of sequence comparison with Apiaceae ESTs indicated that the unigenes we got might represent potential celery-specific genes, and they would be a great supplement to the current database. 47.02% of unigenes had matches to carrot transcripts, suggesting the possibility of comparative genomics studies.
Large numbers of unigenes were assigned to a great diversity of GO categories (Figure 4) and COG classifications (Figure 5). Most representative transcripts were mapped to specific pathways, such as metabolism, genetic information processing, environmental information processing and cellular processes using the KEGG database. On the other hand, we found unigenes involved in the metabolism of chlorophyll, cellulose and pectin. Orthologs related to bolting and flowering were also detected. These transcripts may be genes controlling specific agronomic traits and they will be useful for gene function study.
By this study, our results demonstrated that high-throughput transcriptome sequencing is an efficient, reliable and inexpensive tool for transcriptome analysis and marker discovery in non-model organisms. The large number of sequences generated in present study will provide foundation for our further studies like marker development and validation, gene mapping and molecular marker-assisted selection breeding.
EST-SSR Markers Identification
In the previous research on celery, molecular markers were mainly focused on RFLP, RAPD and AFLP markers. Nowadays, polymorphic SSR markers play an important role in genetic diversity research, linkage map construction, identification of varieties, comparative genomics and related analysis. In this study, we performed a general screen on the celery transcripts for the presence of microsatellites and analyzed the distribution and frequency of the markers. A search for repeats from mono- to hexa-nucleotide yielded a total of 2,997 SSRs in 2,601 unigenes, accounting for approximately 6.15% of the total unigenes. This percentage agrees with the range of 4%–11% in previous studies [33], [74].
While as a result of various SSR searching tools and criteria, it’s not surprising that the frequencies, types and distributions of the potential SSRs are sifinificantly distinct in different studies. As was shown in Table 2, di-nucleotide repeats were the most frequent SSR motifs type followed by tri-nucleotide repeats in this study, which was consistent with some organisms reported like sesame [65], rubber tree [68], [75] and red pepper [76] but inconsistent with cereal [77]. The most frequent motif types were greatly diverse in different studies. In this study, GA/TC (29.55%) and TGA/TCA (2.74%) were the most frequent motifs among the di-nucleotide and tri-nucleotide respectively.
EST-SSR Marker Polymorphism
In present study, a total of 80 primer pairs were designed and used for further assessment of the assembly quality. 65 primer pairs (81.25%) could successfully yield amplicons and 47 primer pairs amplified PCR products at the expected sizes. The others resulted in larger PCR products than expected, which may be due to the presence of introns, a lack of specificity or assembly errors. The failure of 15 primer pairs to produce amplicons may be due to the large introns or insertions, the location of the primer across splice sites, primer synthesis problems or poor sequences quality. Most of the celery EST-SSRs generated high quality amplicons, suggesting that the transcriptome sequencing were accurate and the assembled unigenes were of high quality. Currently there exist only hundreds of genetic markers in celery, so the sequences seem more valuable and can be put to good use in the future. Based on the SSR-containing sequences in our dataset, more PCR primers could be designed and used for germplasm polymorphism assessment, linkage map construction, gene mapping, and comparative genomics in celery.
SSR markers were subjected to polymorphism test and most of the polymorphic SSRs gave no more than 4 alleles, which was in agreement with the previous result by Wang [13]. While the average PIC value in this study (0.35) was lower than that in Wang’s report (0.527) using also SSR markers, which was probably due to a smaller sample size of A. gravolence accessions tested here (n = 31) than the previous one (n = 68). However, the average PIC value obtained here (0.35) was higher than that reported by Wang using SRAP markers (0.264), suggesting that SSR is a higher polymorphism marker.
All varieties were classified into two groups at the level of genetic similarity 0.6, which was in good accord with the plant taxonomy based on genetic relationship. Several lines of celery were assigned to a group with celeriac, which may be cause by hybridization between celery and celeriac. Given that local celery and celery may have a similar genetic background and existence of hybridization during the long period of domestication, the result that some local celery lines were scattered to the celery group was nothing out of the ordinary. Molecular markers like SSRs can distinguish varieties without morphological diversities, and they have been a useful tool in research like cultivar classification and genetic diversity analysis.
Concluding Remarks
In this work, a large EST dataset composed of 42,280 transcripts was achieved. The functional annotation and classification were beneficial for us to have a better understanding of celery genomic information. Based on the sequences, 2,997 SSRs were identified and characterized as potential molecular markers. 80 primer pairs were randomly selected to detect polymorphism among 31 celery accessions, and 65 pairs (81.25%) successfully amplified fragments with 27 pairs showing polymorphic loci.
Through this research, we obtained a large-scale transcriptome dataset using relatively short reads generated by paired-sequencing technology. This was the first try on celery, an organism lacking genomic resources. The dataset will be important for gene discovery in celery and for annotation of the celery genome. The indentified unigenes involved in specific pathways provided candidates for genes controlling petiole traits, bolting and flowering. The SSRs indentified here will supply an important resource for constructing genetic linkage maps, mapping interested genes and marker-assisted breeding in celery. On the other hand, celery along with carrot, coriander, and fennel belong to umbelliferous vegetables. So the resource will enhance comparative genomics studies within the family and plants related.
Supporting Information
Primer pairs validated in this study.
(XLS)
Apium graveolens L. germplasms for polymorphism validation with EST-SSRs.
(XLS)
Summary of ORF-containing unigenes. Unigene length and predicted position of ORFs were indicated for the whole unigene collection.
(XLS)
The main identified chlorophyll metabolism-related genes potentially controlling petiole color.
(XLS)
The identified genes involved in cellulose and pectin metabolism.
(XLS)
Genes potentially controlling bolting and flowering.
(XLS)
Acknowledgments
We thank to Professor Wen-Cai Yang for experiment guide and useful suggestions, to Xin Li, Chen Liu and Ping-Yong Wang for experiment assistance, to Ting Liu for critical reading of the manuscript, to Jing-Jing Yang for partial data processing, and to the rest of our lab mates for field sampling and DNA extraction.
Funding Statement
This work was supported by Chinese Universities Scientific Fund (No.2012QJ134). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Muminovic J, Melchinger AE, Lubberstedt T (2004) Prospects for celeriac (Apium graveolens var. rapaceum) improvement by using genetic resources of Apium, as determined by AFLP markers and morphological characterization. Plant Genetic Resources 2: 189–198. [Google Scholar]
- 2.Sampson S (2006) Chinese celery has robust taste. Toronto Star (Canada) ISSN:0319–0781.
- 3.Song XJ, Wang YT (2008) Research Progress in the Medicinal Function of Celery. Journal of Anhui Agricultural Sciences 36: 6360–6361, 6395.
- 4. TANG F, GUO J, ZHANG J, LI J, SU M (2007) Study on Hypotensive and Vasodilatory Effects of Celery Juice. Food Science 28: 322–325. [Google Scholar]
- 5. Arus P, Ortan T (1984) Inheritance patterns and linkage relationships of eight genes of celery. Journal of Heredity 75: 11–14. [Google Scholar]
- 6. Quiros CF, Douches D, D’Antonio V (1987) Inheritance of annual habit in celery: cosegregation with isozyme and anthocyanin markers. TAG Theoretical and Applied Genetics 74: 203–208. [DOI] [PubMed] [Google Scholar]
- 7. Huestis G, McGrath JM, Quiros CF (1993) Development of genetic markers in celery based on restriction fragment length polymorphisms. TAG Theoretical and Applied Genetics 85: 889–896. [DOI] [PubMed] [Google Scholar]
- 8. Yang X, Quiros C (1993) Identification and classification of celery cultivars with RAPD markers. TAG Theoretical and Applied Genetics 86: 205–212. [DOI] [PubMed] [Google Scholar]
- 9. Domblides A, Domblides H, Kharchenko V (2008) Discrimination between celery cultivars with the use of RAPD markers. Proceedings of the Latvian Academy of Sciences. Section B. Natural, Exact, and Applied Sciences 62: 219–222. [Google Scholar]
- 10. Li G, Quiros CF (2000) Use of amplified fragment length polymorphism markers for celery cultivar identification. HortScience 35: 726–728. [Google Scholar]
- 11. Ju J (2007) An Analysis of Celery Genetic Diversity by AFLP. Chinese Agricultural Science Bulletin 23: 120–123. [Google Scholar]
- 12. Acquadro A, Magurno F, Portis E, Lanteri S (2006) dbEST-derived microsatellite markers in celery (Apium graveolens L. var. dulce). Molecular Ecology Notes 6: 1080–1082. [Google Scholar]
- 13. Wang S, Yang W, Shen H (2011) Genetic diversity in Apium graveolens and related species revealed by SRAP and SSR markers. Scientia Horticulturae 129: 1–8. [Google Scholar]
- 14. Andersen JR, Lubberstedt T (2003) Functional markers in plants. Trends in plant science 8: 554–560. [DOI] [PubMed] [Google Scholar]
- 15. Morozova O, Marra MA (2008) Applications of next-generation sequencing technologies in functional genomics. Genomics 92: 255–264. [DOI] [PubMed] [Google Scholar]
- 16. Guo S, Zheng Y, Joung JG, Liu S, Zhang Z, et al. (2010) Transcriptome sequencing and comparative analysis of cucumber flowers with different sex types. BMC Genomics 11: 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vera JC, Wheat CW, Fescemyer HW, Frilander MJ, Crawford DL, et al. (2008) Rapid transcriptome characterization for a nonmodel organism using 454 pyrosequencing. Molecular ecology 17: 1636–1647. [DOI] [PubMed] [Google Scholar]
- 18. Parchman TL, Geist KS, Grahnen JA, Benkman CW, Buerkle CA (2012) Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery. BMC Genomics 11: 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang H, Zhang H, Wong YH, Voolstra C, Ravasi T, et al. (2010) Rapid transcriptome and proteome profiling of a non-model marine invertebrate, Bugula neritina. . Proteomics 10: 2972–2981. [DOI] [PubMed] [Google Scholar]
- 20. Shahin A, van Gurp T, Peters SA, Visser RGF, van Tuyl JM, et al. (2012) SNP markers retrieval for a non-model species: a practical approach. BMC Research Notes 5: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Feldmeyer B, Wheat CW, Krezdorn N, Rotter B, Pfenninger M (2011) Short read Illumina data for the de novo assembly of a non-model snail species transcriptome (Radix balthica, Basommatophora, Pulmonata), and a comparison of assembler performance. BMC Genomics 12: 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mizrachi E, Hefer CA, Ranik M, Joubert F, Myburg AA (2010) De novo assembled expressed gene catalog of a fast-growing Eucalyptus tree produced by Illumina mRNA-Seq. BMC Genomics 11: 681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Crawford JE, Guelbeogo WM, Sanou A, Traore A, Vernick KD, et al. (2010) De novo transcriptome sequencing in Anopheles funestus using Illumina RNA-seq technology. PLoS one 5: e14202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, et al. (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature biotechnology 29: 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Barrell D, et al. (2003) The InterPro Database, 2003 brings increased coverage and new features. Nucleic acids research 31: 315–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zdobnov EM, Apweiler R (2001) InterProScan-an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17: 847–848. [DOI] [PubMed] [Google Scholar]
- 28. Thiel T, Michalek W, Varshney RK, Graner A (2003) Exploiting EST database for the development and characterization of gen-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106: 411–422. [DOI] [PubMed] [Google Scholar]
- 29. Kabelka E, Franchino B, Francis D (2002) Two loci from Lycopersicon hirsutum LA407 confer resistance to strains of Clavibacter michiganensis subsp. michiganensis . Phytopathology 92: 504–510. [DOI] [PubMed] [Google Scholar]
- 30.Weir BS (1990) Genetic data analysis. Methods for discrete population genetic data. Sunderland, Massachusetts: Sinauer Associates, Inc. Publishers. 377 p.
- 31.Yeh FC, Yang R, Boyle T (1999) PopGene Version 1.31: Microsoft Window-based freeware for population genetic analysis. Available from http://www.ualberta.ca/~fyeh. University of Alberta and Center for International Forestry Research, Edmonton, Alberta, Canada.
- 32.Rohlf F J (2000). NTSYS-pc.Numerical Taxonomy and Multivariate Analysis System,Version 2.1. New York: Exeter Software.
- 33. Iorizzo M, Senalik DA, Grzebelus D, Bowman M, Cavagnaro PF, et al. (2011) De novo assembly and characterization of the carrot transcriptome reveals novel genes, new markers, and genetic diversity. BMC Genomics 12: 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Townsend G, Emerson R, Newhall A (1946) Resistance to Cercospora-apii in celery. Phytopathology 36: 980–982. [Google Scholar]
- 35. Wang S, Shen H, Wu X (2010) Studies on inheritance and phenotype of the yellow mutant of celery. Acta Agriculturae Boreali-Sinica 25: 119–122. [Google Scholar]
- 36. Yanmei G, Xingfang G, Chunzhen Z, Xiujuan F, Shengping Z, et al. (2003) Genetic mechanism of the cucumber leaf mutant. Acta Horticulturae Sinica 30: 409–412. [Google Scholar]
- 37. Xingfang G, Shengping Z, Xiurong C (2005) Inheritance and linkage relationships among the genes of leaf mutant and bitterness with other five major genes in cucumber. Acta Horticulturae Sinica 32: 108–110. [Google Scholar]
- 38. Miao H, GU X, ZHANG S, WANG X, FANG Z, et al. (2010) Changes of the Photosynthetic Pigment and Differential Expression of the Correlated Genes in a Chlorophyll-Deficient Cucumber Mutant (Cucumis sativus L.). Scientia Agricultura Sinica 43: 4027–4035. [Google Scholar]
- 39. MA Z, WANG H, WEI W, YAN Y (2011) Change of Photosynthetic Pigments and Enzyme Activities of Hot Pepper (Capsicum annuum) CMS Line Leaves with Yellow-green Marker. China Cucurbits and Vegetables 24: 20–24. [Google Scholar]
- 40. Nothnagel T, Straka P (2003) Inheritance and mapping of a yellow leaf mutant of carrot (Daucus carota). Plant breeding 122: 339–342. [Google Scholar]
- 41. Shen H, Cheng J, Han Q (2006) Studies on Inheritance and Phenotype of the Yellow Mutant of Carrot. Acta Horticulturae Sinica 33: 856–858. [Google Scholar]
- 42. Mouradov A, Cremer F, Coupland G (2002) Control of flowering time interacting pathways as a basis for diversity. The Plant Cell Online 14: S111–S130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Takahashi T, Mu JH, Gasch A, Chua NH (1998) Identification by PCR of receptor-like protein kinases from Arabidopsis flowers. Plant molecular biology 37: 587–596. [DOI] [PubMed] [Google Scholar]
- 44. Poupin MJ, Federici F, Medina C, Matus JT, Timmermann T, et al. (2007) Isolation of the three grape sub-lineages of B-class MADS-box TM6, PISTILLATA and APETALA3 genes which are differentially expressed during flower and fruit development. Gene 404: 10–24. [DOI] [PubMed] [Google Scholar]
- 45. Latrasse D, Germann S, Houba-Herin N, Dubois E, Bui-Prodhomme D, et al. (2011) Control of flowering and cell fate by LIF2, an RNA binding partner of the polycomb complex component LHP1 . PloS one 6: e16592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Noh YS, Amasino RM (2003) PIE1, an ISWI family gene, is required for FLC activation and floral repression in Arabidopsis . The Plant Cell Online 15: 1671–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mutasa-Gottgens ES, Qi A, Zhang W, Schulze-Buxloh G, Jennings A, et al. (2010) Bolting and flowering control in sugar beet: Relationships and effects of gibberellin, the bolting gene B and vernalization. AoB Plants 10.1093/aobpla/plq012. [DOI] [PMC free article] [PubMed]
- 48. Mutasa-Gottgens E, Qi A, Mathews A, Thomas S, Phillips A, et al. (2009) Modification of gibberellin signalling (metabolism & signal transduction) in sugar beet: analysis of potential targets for crop improvement. Transgenic research 18: 301–308. [DOI] [PubMed] [Google Scholar]
- 49. Hirano K, Ueguchi-Tanaka M, Matsuoka M (2008) GID1-mediated gibberellin signaling in plants. Trends in plant science 13: 192–199. [DOI] [PubMed] [Google Scholar]
- 50. Aggarwal RK, Hendre PS, Varshney RK, Bhat PR, Krishnakumar V, et al. (2007) Identification, characterization and utilization of EST-derived genic microsatellite markers for genome analyses of coffee and related species. TAG Theoretical and Applied Genetics 114: 359–372. [DOI] [PubMed] [Google Scholar]
- 51. Cardle L, Ramsay L, Milbourne D, Macaulay M, Marshall D, et al. (2000) Computational and experimental characterization of physically clustered simple sequence repeats in plants. Genetics 156: 847–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dutta S, Kumawat G, Singh BP, Gupta DK, Singh S, et al. (2011) Development of genic-SSR markers by deep transcriptome sequencing in pigeonpea [Cajanus cajan (L.) Millspaugh]. BMC plant biology 11: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Parra-Gonzalez LB, Aravena-Abarzua GA, Navarro-Navarro CS, Udall JA, Maugham PJ, et al. (2012) Yellow lupin (Lupinus luteus L.) transcriptome sequencing: molecular marker development and comparative studies. BMC genomics 13: 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lesser MR, Parchman TL, Buerkle C (2012) Cross-species transferability of SSR loci developed from transciptome sequencing in lodgepole pine. Molecular Ecology Resources 12: 448–455. [DOI] [PubMed] [Google Scholar]
- 55. Wang J, Wang W, Li R, Li Y, Tian G, et al. (2008) The diploid genome sequence of an Asian individual. Nature 456: 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, et al. (2008) A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 321: 956–960. [DOI] [PubMed] [Google Scholar]
- 57. Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, et al. (2008) The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lister R, O’Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, et al. (2008) Highly Integrated Single-Base Resolution Maps of the Epigenome in Arabidopsis. Cell 133: 523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome research 18: 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dohm JC, Lottaz C, Borodina T, Himmelbauer H (2007) SHARCGS, a fast and highly accurate short-read assembly algorithm for de novo genomic sequencing. Genome research 17: 1697–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Butler J, MacCallum I, Kleber M, Shlyakhter IA, Belmonte MK, et al. (2008) ALLPATHS: De novo assembly of whole-genome shotgun microreads. Genome research 18: 810–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Martin JA, Wang Z (2011) Next-generation transcriptome assembly. Nature Reviews Genetics 12: 671–682. [DOI] [PubMed] [Google Scholar]
- 63. Zhang J, Liang S, Duan J, Wang J, Chen S, et al. (2012) De novo assembly and Characterisation of the Transcriptome during seed development, and generation of genic-SSR markers in Peanut (Arachis hypogaea L.). BMC Genomics 13: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang Z, Fang B, Chen J, Zhang X, Luo Z, et al. (2010) De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweetpotato (Ipomoea batatas). BMC Genomics 11: 726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wei W, Qi X, Wang L, Zhang Y, Hua W, et al. (2011) Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genomics 12: 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chang L, Chen J, Xiao Y, Xia Y (2011) De novo characterization of Lycoris sprengeri transcriptome using Illumina GA II. African Journal of Biotechnology 10: 12147–12155. [Google Scholar]
- 67. Zhou X, Fei Z, Thannhauser TW, Li L (2011) Transcriptome analysis of ectopic chloroplast development in green curd cauliflower (Brassica oleracea L. var. botrytis). BMC Plant Biology 11: 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li D, Deng Z, Qin B, Liu X, Men Z (2012) De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genomics 13: 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hao DC, Ma P, Mu J, Chen SL, Xiao PG, et al. (2012) De novo characterization of the root transcriptome of a traditional Chinese medicinal plant Polygonum cuspidatum. Science China. Life sciences 55: 452–466. [DOI] [PubMed] [Google Scholar]
- 70. He M, Wang Y, Hua W, Zhang Y, Wang Z (2012) De Novo Sequencing of Hypericum perforatum Transcriptome to Identify Potential Genes Involved in the Biosynthesis of Active Metabolites. PloS one 7: e42081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Huang L, Yang X, Sun P, Tong W, Hu S (2012) The First Illumina-Based De Novo Transcriptome Sequencing and Analysis of Safflower Flowers. PloS one 7: e38653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Edwards CE, Parchman TL, Weekley CW (2012) Assembly, gene annotation and marker development using 454 floral transcriptome sequences in Ziziphus celata (Rhamnaceae), a highly endangered, Florida endemic plant. DNA research 19: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Li Y, Sun C, Luo H, Li X, Niu Y, et al. (2010) Transcriptome characterization for Salvia miltiorrhiza using 454 GS FLX. Acta pharmaceutica Sinica 45: 524–529. [PubMed] [Google Scholar]
- 74. Blanca J, Canizares J, Roig C, Ziarsolo P, Nuez F, et al. (2011) Transcriptome characterization and high throughput SSRs and SNPs discovery in Cucurbita pepo (Cucurbitaceae). BMC Genomics 12: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Triwitayakorn K, Chatkulkawin P, Kanjanawattanawong S, Sraphet S, Yoocha T, et al. (2011) Transcriptome sequencing of Hevea brasiliensis for development of microsatellite markers and construction of a genetic linkage map. DNA research 18: 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lu FH, Cho MC, Park YJ (2012) Transcriptome profiling and molecular marker discovery in red pepper, Capsicum annuum L. TF68. Molecular biology reports 39: 3327–3335. [DOI] [PubMed] [Google Scholar]
- 77. La Rota M, Kantety RV, Yu JK, Sorrells ME (2005) Nonrandom distribution and frequencies of genomic and EST-derived microsatellite markers in rice, wheat, and barley. BMC Genomics 6: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primer pairs validated in this study.
(XLS)
Apium graveolens L. germplasms for polymorphism validation with EST-SSRs.
(XLS)
Summary of ORF-containing unigenes. Unigene length and predicted position of ORFs were indicated for the whole unigene collection.
(XLS)
The main identified chlorophyll metabolism-related genes potentially controlling petiole color.
(XLS)
The identified genes involved in cellulose and pectin metabolism.
(XLS)
Genes potentially controlling bolting and flowering.
(XLS)