

Abstract
Inositol 1,4,5-trisphosphate receptors (IP3R) are intracellular Ca2+ channels. Most animal cells express mixtures of the three IP3R subtypes encoded by vertebrate genomes. Adenophostin A (AdA) is the most potent naturally occurring agonist of IP3R and it shares with IP3 the essential features of all IP3R agonists, namely structures equivalent to the 4,5-bisphosphate and 6-hydroxyl of IP3. The two essential phosphate groups contribute to closure of the clam-like IP3-binding core (IBC), and thereby IP3R activation, by binding to each of its sides (the α- and β-domains). Regulation of the three subtypes of IP3R by AdA and its analogues has not been examined in cells expressing defined homogenous populations of IP3R. We measured Ca2+ release evoked by synthetic adenophostin A (AdA) and its analogues in permeabilized DT40 cells devoid of native IP3R and stably expressing single subtypes of mammalian IP3R. The determinants of high-affinity binding of AdA and its analogues were indistinguishable for each IP3R subtype. The results are consistent with a cation-π interaction between the adenine of AdA and a conserved arginine within the IBC α-domain contributing to closure of the IBC. The two complementary contacts between AdA and the α-domain (cation-π interaction and 3″-phosphate) allow activation of IP3R by an analogue of AdA (3″-dephospho-AdA) that lacks a phosphate group equivalent to the essential 5-phosphate of IP3. These data provide the first structure-activity analyses of key AdA analogues using homogenous populations of all mammalian IP3R subtypes. They demonstrate that differences in the Ca2+ signals evoked by AdA analogues are unlikely to be due to selective regulation of IP3R subtypes.
Introduction
Inositol 1,4,5-trisphosphate receptors (IP3R) are intracellular Ca2+ channels that are expressed in almost all animal cells. They allow release of Ca2+ from intracellular stores in response to the many stimuli that activate phospholipase C [1], [2]. The genomes of vertebrates encode three closely related IP3R subtypes (IP3R1-3), and most cells from vertebrates express functional IP3R that are homo- or hetero-tetrameric assemblies of these IP3R subtypes and their splice variants [3]. The physiological significance of this IP3R diversity is poorly understood, and nor are there ligands that usefully discriminate between IP3R subtypes. It is, however, clear that activation of IP3R is initiated by binding of IP3 to the conserved IP3-binding core (IBC, residues 224-604 of IP3R1) of each IP3R subunit [4]. Mixed populations of IP3R in native cells make it difficult to define unambiguously the functional properties of each IP3R subtype. Stable heterologous expression of mammalian IP3R in the only vertebrate cell line engineered to lack all endogenous IP3R (DT40 KO cells) [5] provides an effective means of addressing this difficulty [6]. We previously used DT40 cells expressing homogeneous populations of each mammalian IP3R subtype to define structure-activity relationships for key endogenous and synthetic inositol phosphates [7]. Here, we extend the approach to examine the interactions of each IP3R subtype with adenophostin A (1, AdA) and its most important analogues [8] (Figure 1A).
Figure 1. Structures of the analogues of AdA used.

(A) Key moieties within IP3 and AdA are highlighted in matching colours to indicate their proposed structural equivalence. (B and C). The Ca2+ contents of the intracellular stores of populations of permeabilized DT40-IP3R1 cells are shown after addition of ATP to allow active Ca2+ uptake, and then addition of the indicated concentrations of IP3 or AdA with thapsigargin (1 µM) to inhibit further Ca2+ uptake. The traces, which are typical of those from all subsequent analyses, show the average response from 2 wells on a single plate. The results demonstrate that both IP3 and AdA evoke quantal Ca2+ release.
AdA, originally isolated from Penicillium brevicompactum [9], [10] and later synthesized [11], is a potent agonist of IP3R. It is also resistant to degradation by the enzymes that degrade IP3 via phosphorylation or dephosphorylation [10]. Although AdA is based on a glucose ring, rather than the inositol ring of IP3, its structure retains the key functional groups of IP3 that are known to be essential for IP3 activity at IP3R [12] (Figure 1A). Considerable evidence supports the original suggestion [10] that the essential 4,5-bisphosphate and 6-hydroxyl of IP3 are effectively mimicked by the 4″,3″-bisphosphate and 2″-hydroxyl of AdA (red highlights in Figure 1A). The interactions that allow AdA to bind to IP3R with about 10-fold greater affinity than IP3 have been more difficult to resolve. One view was that the 2′-phosphate of AdA is equivalent to the 1-phosphate of IP3 and, like the latter [13] (blue in Figure 1A), contributes to high-affinity binding to the IBC. The suggestion was that the 2′-phosphate of AdA forms a stronger interaction with the IBC than does the 1-phosphate of IP3. Our recent analyses have challenged this idea and instead suggest that a cation-π interaction between the adenine ring of AdA and a guanidinium side chain of an arginine residue within the α-domain of the IBC (R504 in IP3R1) may be a more important determinant of the increased affinity of AdA for IP3R [12].
The high-affinity and metabolic stability of AdA have generated considerable interest in both the synthesis of AdA analogues and their application to analyses of IP3R activation and associated changes in cytosolic Ca2+ signalling [12]. There has, however, been no systematic analysis of the activities of AdA or its analogues with defined populations of homogenous IP3R subtypes. The need for such analyses is particularly important in attempting to explain results in which Ca2+ signals evoked by IP3 differ from those evoked by AdA [14], [15], [16], [17], [18], [19], [20], [21], or where different analogues of AdA evoke different cellular responses [reviewed in 12,22]. Here we use DT40 cells in which all endogenous IP3R have been genetically inactivated [5] to stably express homogenous populations of mammalian IP3R subtypes and thereby define structure-activity relationships for AdA and its key analogues for each IP3R subtype.
Materials and Methods
Materials
Sources of most reagents were provided in a previous publication [7]. The structures of the ligands used and their abbreviations are shown in Figure 1A. IP3 was from Alexis Biochemicals (Nottingham, UK). AdA [23], imidophostin [24], ribophostin [25], furanophostin [26], manno-AdA and xylo-AdA [27], 3″-dephospho AdA and 4″-dephospho AdA [28], and 2′-dephospho AdA were synthesized, purified and characterized as previously described.
Measurement Ca2+ Release by IP3 Receptors
From quantitative analyses of western blots using antisera that selectively recognise each IP3R subtype or react equally with all three subtypes, we established that in the DT40 cells used, levels of IP3R expression (relative to IP3R3) were IP3R1 (71±8%, n = 3), IP3R2 (48±5%) and IP3R3 (100%) [7]. It is impracticable to achieve identical levels of IP3R expression for each cell line, and differences (albeit modest in our cell lines) may affect both the size of the IP3-sensitive Ca2+ pool and its sensitivity to IP3 [29]. The different levels of IP3R expression do not compromise the analyses reported here, which are entirely concerned with relative potencies of AdA analogues for each IP3R subtype (see below).
A comprehensive description of the methods used to measure free [Ca2+] within the endoplasmic reticulum of permeabilized DT40 cells was provided in preceding publications [7], [30]. Briefly, the endoplasmic reticulum of DT40 cells stably expressing each of the three mammalian IP3R subtypes was loaded with a low-affinity Ca2+ indicator (Mag fluo-4) [30]. After permeabilization of the plasma membrane with saponin (10 µg/mL, ∼4 min, 37°C), the permeabilized cells in cytosol-like medium (CLM) were distributed into 96-well plates at 20°C. Addition of MgATP (1.5 mM) then allowed active Ca2+ accumulation, which was monitored at intervals of ∼1 s using a FlexStation 3 fluorescence plate-reader (MDS Analytical Devices). CLM had the following composition: 140 mM KCl, 20 mM NaCl, 1 mM EGTA, 20 mM Pipes, pH 7, free [Ca2+] ∼220 nM (after addition of MgATP), and carbonyl cyanide 4-trifluoromethoxy-phenyl hydrazone (FCCP, 10 µM) to inhibit mitochondrial Ca2+ uptake. After 150 s, when the stores had loaded to steady-state with Ca2+, IP3, AdA or its analogues was added with thapsigargin (1 µM) to prevent further Ca2+ uptake, and after a further 30 s, the response was recorded. Agonist-evoked Ca2+ release was expressed as a fraction of that released by ionomycin (1 µM) [30]. All experiments were performed at 20°C.
Statistical Analysis
Concentration-effect relationships were fitted to Hill equations using GraphPad Prism (version 5.0) from which Hill coefficients (h), the fraction of the intracellular Ca2+ stores released by maximally effective concentrations of agonist, and pEC50 values (-log EC50) were calculated. For convenience some results are presented as EC50 values, but all statistical comparisons use pEC50 values. Within each experiment, the pEC50 for AdA was determined to allow paired comparisons with values obtained for each AdA analogue. These are reported as ΔpEC50, where:
We note that Table 1 reports pooled results from experiments collected over a considerable period, whereas ΔpEC50 values, like those shown in Table 2, compare only paired values. The latter provide the most robust means of comparing agonist potencies. Results are expressed as means ± SEM from n independent experiments, with each experiment performed in triplicate.
Table 1. Effects of AdA analogues on Ca2+ release by subtypes of IP3 receptor.
| IP3R1 | IP3R2 | IP3R3 | |||||||||||||
| EC50 | pEC50 | h | Ca2+release | n | EC50 | pEC50 | h | Ca2+release | n | EC50 | pEC50 | h | Ca2+release | n | |
| (1,4,5)IP3 | 87 | 7.06±0.05 | 0.99±0.05 | 75±1 | 31 | 145 | 6.84±0.06 | 1.29±0.09 | 61±2 | 34 | 417 | 6.38±0.05 | 1.26±0.07 | 64±2 | 30 |
| AdA | 8.3 | 8.08±0.09 | 1.17±0.09 | 72±3 | 10 | 18.2 | 7.74±0.06 | 1.79±0.21 | 56±2 | 13 | 33 | 7.48±0.09 | 1.13±0.07 | 61±2 | 14 |
| Imidophostin | 37 | 7.43±0.28 | 1.17±0.21 | 78±5 | 3 | 68 | 7.17±0.14 | 1.84±0.50 | 59±3 | 3 | 166 | 6.78±0.16 | 1.73±0.39 | 67±7 | 3 |
| Ribophostin | 40 | 7.40±0.29 | 1.34±0.16 | 77±4 | 3 | 102 | 6.99±0.11 | 1.60±0.50 | 61±2 | 3 | 295 | 6.53±0.21 | 1.42±0.08 | 68±4 | 3 |
| Furanophostin | 51 | 7.29±0.25 | 0.90±0.10 | 79±6 | 3 | 76 | 7.12±0.01 | 1.73±0.20 | 60±3 | 3 | 457 | 6.34±0.18 | 1.27±0.21 | 71±3 | 3 |
| Manno-AdA | 34 | 7.47±0.19 | 1.33±0.30 | 75±7 | 3 | 69 | 7.16±0.07 | 1.33±0.22 | 57±3 | 3 | 245 | 6.61±0.23 | 1.23±0.15 | 69±4 | 3 |
| Xylo-AdA | 5.9 | 8.23±0.17 | 1.27±0.27 | 73±7 | 3 | 7.9 | 8.10±0.10 | 1.52±0.40 | 52±6 | 3 | 29 | 7.54±0.12 | 1.58±0.29 | 64±9 | 3 |
| 2′-dephospho-AdA | 275 | 6.56±0.13 | 1.31±0.15 | 66±7 | 3 | 575 | 6.24±0.10 | 0.85±0.07 | 63±2 | 3 | 692 | 6.16±0.03 | 1.5±0.22 | 55±7 | 4 |
| 3″-dephospho-AdAc | ND | ND | ND | 15±6b | 7 | ND | ND | ND | 6±2b | 6 | ND | ND | ND | 7±7b | 5 |
| 4″-dephospho-AdA | Inactivea | Inactivea | Inactivea | ND | 6 | Inactivea | Inactivea | Inactivea | ND | 6 | Inactivea | Inactivea | Inactivea | ND | 5 |
The EC50 (nM), pEC50 (/M), Hill coefficient (h) and fraction (%) of the intracellular Ca2+ stores released by a maximally effective concentration of each analogue are shown for each IP3R subtype. All results (except EC50) are shown as means ± SEM from n independent experiments.
Inactive at 300 µM.
Ca2+ release evoked by 300 µM 3″-dephospho AdA.
Refer to Table 2 for relative potencies of 3″-dephospho AdA. ND, not determined.
Table 2. Relative potencies of AdA analogues at different IP3 receptor subtypes.
| IP3R1 | IP3R2 | IP3R3 | |
| IP3 | 1.02±0.02 | 0.9±0.30 | 1.1±0.30 |
| Imidophostin | 0.78±0.15 | 0.78±0.08 | 0.81±0.04 |
| Ribophostin | 0.82±0.18 | 0.96±0.20 | 1.06±0.07 |
| Furanophostin | 0.92±0.13 | 0.83±0.14 | 1.25±0.05 |
| Manno-AdA | 0.74±0.08 | 0.79±0.18 | 0.98±0.08 |
| Xylo-AdA | −0.01±0.07 | −0.3±0.27 | 0.05±0.08 |
| 2′-dephospho-AdA | 1.24±0.33 | 1.60±0.18 | 1.68±0.16 |
| 3″-dephospho-AdAa | 4.03±0.09 | 4.47±0.30 | 4.13±0.14 |
From paired comparisons with AdA, the potency (ΔpEC50) of the analogues relative to AdA is shown for each IP3R subtype. Results are means ± SEM, with n provided in Table 1. ND, not determined. aBecause the very low affinity of 3″-dephospho AdA for IP3R made it impracticable to stimulate cells with a maximally effective concentration, ‘ΔpEC50’ for 3″-dephospho AdA was estimated by comparing concentrations of it and AdA that evoked the same sub-maximal Ca2+ release.
Statistical comparisons used Student’s t-test or ANOVA followed by Bonferroni’s post hoc test, as appropriate, with P<0.05 considered significant. Because not all comparisons of the relative potencies of AdA and IP3 were paired, the SEM of this ΔpEC50 value was calculated from:
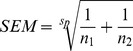 |
where, sp is the estimate of the population variance:
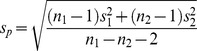 |
where, s1 and s2 are the sample standard deviations, and n1 and n2 are the sample sizes [31].
Results
Quantal Ca2+ Release Evoked by AdA and IP3
The kinetics of IP3-evoked Ca2+ release from intracellular stores are unexpectedly complex. It is widely observed that under conditions where Ca2+ uptake into the endoplasmic reticulum (ER) is inhibited, submaximally effective concentrations of IP3 rapidly release only a fraction of the IP3-sensitive Ca2+ stores [32]. Thereafter, there is either no, or a massively reduced, effect of IP3 on the rate of Ca2+ release. The mechanisms underlying this pattern of response, known as quantal Ca2+ release [33], remain unclear. It may require desensitization of IP3R as the Ca2+ content of the ER declines [34] or heterogeneity among IP3-senstive Ca2+ stores [35]. The results shown in Figures 1B and C confirm that the Ca2+ release evoked by submaximal concentrations of either IP3 or AdA from permeabilized DT40-IP3R1 cells is quantal. These observations provide the justification for all subsequent experiments in which the concentration-dependent effects of IP3 or AdA were measured 30 s after their addition (see Methods).
AdA is a Potent Agonist of All Three IP3 Receptor Subtypes
The results shown in Figure 2 and Tables 1 and 2 demonstrate that AdA is ∼10-times more potent than IP3 at each IP3R subtype, and for each subtype, maximally effective concentrations of IP3 and AdA release the same fraction of the intracellular Ca2+ stores. This is consistent with many analyses of IP3 and AdA in a variety of cell types using both functional and binding assays, in which AdA behaves as a full agonist with ∼10-fold greater affinity than IP3 [reviewed in 8]. Our results do, however, provide the first direct demonstration that AdA interacts similarly with all three IP3R subtypes. Subsequent experiments examine the interactions between key analogues of IP3 and AdA with each IP3R subtype.
Figure 2. AdA is a potent agonist of all three IP3 receptor subtypes.

(A) Concentration-dependent effects of AdA on Ca2+ release from the intracellular stores of cells expressing IP3R1, IP3R2 or IP3R3. All results are expressed as percentages of the Ca2+ release evoked by ionomycin. The same colour codes are used in all subsequent figures. (B) Comparison, for each IP3R subtype, of the Ca2+ release evoked by IP3 and AdA. Results are means ± SEM from the number of independent experiments given in Table 1. Here, and in many subsequent figures, some error bars are smaller than the symbols.
Trimming the Adenosine Moiety of AdA Reduces its Potency at All IP3 Receptor Subtypes
Systematic trimming of the adenosine moiety of AdA successively produces imidophostin (which lacks the pyrimidine ring of AdA), ribophostin (in which a methoxy group replaces the adenine moiety of AdA) and furanophostin (in which only the furanoid ring remains) (Figure 1A). Maximally effective concentrations of each of these analogues released the same fraction of the intracellular Ca2+ stores as AdA in cells expressing each of the three IP3R subtypes, and each analogue was ∼5-10-fold less potent than AdA (Figure 3, Tables 1 and 2). These results are consistent with previous analyses of IP3R in hepatocytes, which express predominantly IP3R2 [24], [36], with analyses of binding of ribophostin and furanophostin to an N-terminal fragment of IP3R1 [12], and with evidence from other analogues that trimming the adenosine moiety decreases affinity for cerebellar IP3R, which are largely IP3R1 [37].
Figure 3. Trimming the adenosine moiety of AdA reduces potency.

(A–F) Effects of imidophostin (A), ribophostin (C) and furanophostin (E) on Ca2+ release via each of the three IP3R subtypes, and the same analogues compared with AdA (B, D and F). Results are means ± S.E.M. from 3 independent experiments. (G) A cation-π interaction between the adenine of AdA and R504 within the α-domain of the IBC is proposed to stabilize AdA binding (left). Closure of the clam-like IBC is proposed to be mediated by interactions between the 3″-phosphate of AdA and the α-domain of the IBC (blue ribbon), and between the 4″-phosphate and the β-domain of the IBC (green ribbon). In 3″-dephospho AdA, a cation-π interaction between AdA and the IBC α-domain is proposed to be sufficient to allow some effective closure of the clam. R504 is conserved in all three mammalian IP3R subtypes (right).
These results are consistent with our earlier conclusion that the 10-fold greater affinity of AdA relative to IP3 requires the adenine moiety of AdA positioned to allow it to form a cation-π interaction with Arg-504 in the α-domain of the IBC of IP3R1, a residue that is conserved in all IP3R subtypes [8], [12] (Figure 3G). We suggest that this interaction of AdA with IP3R is likely to be similar for all IP3R subtypes.
Hydroxyl Moieties that are Important for IP3 Binding are Less Important for Binding of AdA
The 5″-CH2OH and 2″-OH substituents of the glucose ring of AdA are thought to mimic the 3-OH and 6-OH of IP3, respectively (Figure 1A). A structure equivalent to the 6-OH of IP3 is an essential feature of all inositol phosphate analogues that bind to IP3R [13], [38], [39] and inversion of its orientation from equatorial to axial reduces affinity by more than 100-fold at all IP3R subtypes [40]. It is therefore surprising, but consistent with previous analyses of native hepatic IP3R [36], that manno-AdA, which differs from AdA only in the orientation of its 2″-OH, should be only 5- to 10-fold less potent than AdA at each IP3R subtype (Figures 4A and B, Tables 1 and 2). Why, when the 6-OH of IP3 and 2″-OH of AdA seem to be analogous in the ligand structures, should these moieties make such different contributions to the interactions of IP3 and AdA with IP3R?
Figure 4. Hydroxyl groups within the glucose ring of AdA are unimportant.

(A–D) Effects of manno-AdA (A) and xylo-AdA (C) on Ca2+ release via each IP3R subtype, and the same analogues compared with AdA (B and D). Results are means ± S.E.M. from 3 independent experiments.
The 6-OH of IP3 interacts, through a water molecule, with a lysine residue (K569) in the IBC [41] and, by interacting with the adjacent 1-phosphate, it has also been proposed to influence the behaviour of the 4,5-bisphosphate moiety of IP3 [42]. The latter interaction is unlikely to contribute to AdA binding because the structures equivalent to the 6-OH (2″-OH of AdA) and the 1-phosphate of IP3 (2′-phosphate of AdA) are in different rings in AdA (Figure 1A). We suggest that the lesser importance in AdA of a structure equivalent to the essential 6-OH of IP3 comes from this hydroxyl mediating a relatively minor interaction with K569 in AdA, whereas for IP3 it contributes also to appropriately orienting the critical 4,5-bisphosphate moiety.
The 3-OH group, although less important than the 6-OH, is another feature of IP3 that contributes to high-affinity binding [43]. Our recent analyses of the functional effects of 3-deoxy-IP3 established that it was ∼40-fold less potent than IP3 at all three IP3R subtypes [7]. This is consistent with earlier work showing that 3-deoxy-IP3 and analogues with other modifications of the 3-position have reduced affinity for the three IP3R subtypes [40]. However, the equivalent modification of AdA, removal of its 5″-CH2OH to give xylo-AdA (Figure 1A), had no significant effect on its potency at any IP3R subtype (Figures 4C and D, Tables 1 and 2). This is consistent with a previous functional analysis of hepatic IP3R, where xylo-AdA was only marginally less potent than AdA (ΔpEC50 ∼0.28) [36]. Our results suggest that despite the apparent structural similarity between the 3-OH of IP3 and the 5″-CH2OH of AdA (Figure 1A), the two hydroxyl groups do not contribute similarly to ligand binding. Previous analyses of IP3 analogues suggested that replacing the 3-OH with the larger CH2OH moiety caused the affinity to decrease by no more than 7-fold [40]. A partial explanation for the lack of effect of removing the 5″-CH2OH of AdA may therefore be that this moiety is less readily accommodated than a hydroxyl group in the IBC. This would suggest that an analogue of AdA in which the 5″-CH2OH is replaced by 5″-OH might bind with increased affinity. We are unaware of such an analogue having been synthesized. The larger substituent at the 5″-position of AdA is, however, unlikely to provide the sole explanation for it making no discernible contribution to binding.
The 2′-phosphate of AdA is not a Super-optimal Mimic of the 1-phosphate of IP3
It has been suggested that the 2′-phosphate of AdA interacts with the IBC in a manner that allows it to behave as a super-optimal mimic of the 1-phosphate of IP3 [44], [45]. However, our recent study combining structure-activity analyses with mutagenesis of the binding site suggest that the 1-phosphate of IP3 is more important for binding than is the 2′-phosphate of AdA [12]. Removal of the 1-phosphate from IP3 (to give (4,5)IP2) caused its potency and affinity for IP3R1 to decrease by ∼100-fold [12], whereas removal of the 2′-phosphate from AdA (2′-dephospho AdA) causes a decrease in potency of ∼17-fold in IP3R1 (Figure 5) and ∼40-fold decreases in potency were obtained with 2′-dephospho AdA and IP3R2 and IP3R3 (Figure 5, Table 1 and 2). These results establish that for all three IP3R subtypes, the enhanced affinity of AdA is not due to its 2′-phosphate interacting more effectively than the 1-phosphate of IP3 with the IBC.
Figure 5. The 2′-phosphate of AdA is not the primary cause of its increased potency.

(A) Effects of 2′-dephospho AdA on Ca2+ release via each IP3R subtype. (B) The same analogue compared with AdA. Results are means ± S.E.M. from 3–4 independent experiments.
A Bisphosphate Moiety is not Essential for Activation of IP3 Receptors by AdA
All known active analogues of IP3 have structures equivalent to its 4,5-bisphosphate moiety [13]. Structures of the IBC with and without IP3 bound provide a rationale for this requirement by revealing that these two phosphate groups contact opposite sides (the α- and β-domains) of the clam-like IBC, closure of which initiates IP3R activation [4], [41]. Substantial evidence suggests that the 4″,3″-bisphosphate moiety of AdA mimics the critical 4,5-bisphosphate of IP3 [8] (Figure 1A).
4″-dephospho-AdA at concentrations up to 300 µM failed to evoke Ca2+ release via any IP3R subtype (Figure 6A). This is consistent with previous analyses by both functional and binding assays of IP3R1 [28], [46]. 3″-dephospho-AdA did, however, cause detectable Ca2+ release albeit with much reduced potency (Figure 6B). The synthetic route used to prepare 3″-dephospho-AdA makes it extremely unlikely that the activity could be due to minor contamination with AdA or related structures with a vicinal bisphosphate moiety. Maximal attainable concentrations of 3″-dephospho-AdA (300 µM) failed to release the entire IP3-sensitive Ca2+ store, but comparison of the concentrations required to achieve the same submaximal Ca2+ release suggests that 3″-dephospho-AdA is ∼10,000-fold less potent than AdA at all three IP3R subtypes. With such a massive reduction in potency the lesser sensitivity of DT40-IP3R3 cells to AdA means that even the highest practicable concentration of 3″-dephospho-AdA (300 µM) is close to the threshold for detecting Ca2+ release (Figure 6B).
Figure 6. Structures equivalent to the 4,5-bisphosphate of IP3 are not essential for AdA activity.

(A, B) Concentration-dependent effects on Ca2+ release via each IP3R subtype of 4″-dephospho AdA (A) and 3″-dephospho AdA (B) compared with AdA. Results are means ± SEM from n independent experiments (n is provided in Table 1). (C) Concentration-dependent effects of IP3 alone on Ca2+ release via IP3R1 or after pre-incubation (30 s) with 3″-dephospho AdA (30 µM), which itself evoked release of 21±5% of the intracellular Ca2+ stores. Results (C) are means ± SEM from 3 independent experiments.
The inability of high concentrations of 3″-dephospho-AdA to release the entire IP3-sensitive Ca2+ store is likely to be due solely to its reduced affinity rather than reduced efficacy. A concentration of 3″-dephospho-AdA (30 µM) that caused detectable Ca2+ release via IP3R1 (∼21±5%) had no effect on the sensitivity of the Ca2+ release evoked by a subsequent addition of IP3. The pEC50 was 7.00±0.02 and 7.04±0.06 (n = 3) for (1,4,5)IP3 alone and in the presence of 3″-dephospho-AdA, respectively (Figure 6C). A partial agonist would be expected to shift the sensitivity to higher concentrations of IP3. These results suggest that 3″-dephospho-AdA is a low-affinity full agonist of IP3R.
These results extend our previous analyses of IP3R1 by demonstrating that for all IP3R subtypes, the 4″-phosphate group of AdA is essential for activity, whereas the 3″-phosphate is important but not essential. 3″-dephospho-AdA is the only known agonist of IP3R to lack a structure equivalent to the 4,5-bisphosphate moiety of IP3.
Discussion
AdA is a high-affinity full agonist of IP3R that has been extensively used to explore the behaviour of IP3R [reviewed in8]. The activity of AdA has been confirmed in many cell types, but hitherto there has been no assessment of its activity in homogenous populations of IP3R subtypes. We have demonstrated that AdA is ∼10-fold more potent than IP3R at each IP3R subtype (Figure 2, Tables 1 and 2), and the structural determinants of its high-affinity interaction with IP3R are similar for all three IP3R subtypes. Contrary to an earlier suggestion that the 2′-phosphate of AdA mediates its enhanced affinity by forming a stronger interaction with the IBC than the analogous 1-phosphate of IP3, we find that the 1-phosphate makes a greater contribution to IP3 binding than does the 2′-phosphate of AdA (Figure 5) [12]. A more likely explanation for the enhanced affinity of AdA is a cation-π interaction between its adenine moiety and R504 within the α-subunit of the IBC (Figure 3G) [28]. That explanation is supported by results for each IP3R subtype showing that truncation of the adenosine moiety of AdA brings the potency of the resulting analogues (imidophostin, ribophostin and furanophostin) close to that of IP3 (Figure 3).
A key step in the initial activation of IP3R by IP3 appears to be closure of its clam-like IBC as the 4-phosphate of IP3 contacts one side of the clam (its β-domain) and the 5-phosphate contacts the other side (α-domain) [4]. That mechanism provides a satisfying explanation for the long-standing observation that all inositol phosphates that activate IP3R share this essential 4,5-bisphosphate moiety. AdA is different in that its 4″-phosphate (analogous to the 4-phosphate of IP3, Figure 1A) is essential, but 3″-dephospho-AdA retains activity at all three IP3R subtypes, albeit with very low affinity (Figure 6). We suggest that for AdA, the need for the bisphosphate moiety to cause closure of the IBC can be partially replaced for all IP3R subtypes by having an interaction between the adenine of AdA and the α-domain substitute for the interaction between the 3″-phosphate (analogous to the 5-phosphate of IP3) and the α-domain [28]. Finally, whereas the 6-OH and, to a lesser extent, the 3-OH of IP3 are important for IP3 binding, the equivalent structures within AdA play lesser roles.
Both store-operated Ca2+ entry, which is triggered by depletion of IP3-sensitive Ca2+ stores [47], and the spatial organization of subcellular Ca2+ signals have been reported to be differentially affected by IP3, AdA or its analogues [14], [16], [17], [19], [21], [22]. Our present results, which demonstrate that AdA structure-activity relationships are similar for all IP3R subtypes, suggest that different physiological effects of IP3, AdA or its analogues are more likely to result from differences in their affinities, kinetics or rates of degradation than from selective interactions with different IP3R subtypes.
Funding Statement
This work was supported by grants from the Wellcome Trust to CWT [085295], and BVLP and AMR [082837]. HS is supported by a research studentship from the Jameel Family Trust. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Foskett JK, White C, Cheung KH, Mak DO (2007) Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87: 593–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Taylor CW, Tovey SC (2010) IP3 receptors: toward understanding their activation. Cold Spring Harb Persp Biol 2: a004010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Taylor CW, Genazzani AA, Morris SA (1999) Expression of inositol trisphosphate receptors. Cell Calcium 26: 237–251. [DOI] [PubMed] [Google Scholar]
- 4. Seo M-D, Velamakanni S, Ishiyama N, Stathopulos PB, Rossi AM, et al. (2012) Structural and functional conservation of key domains in InsP3 and ryanodine receptors. Nature 483: 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sugawara H, Kurosaki M, Takata M, Kurosaki T (1997) Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. EMBO J 16: 3078–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Taylor CW, Rahman T, Tovey SC, Dedos SG, Taylor EJA, et al. (2009) IP3 receptors: some lessons from DT40 cells. Immunol Rev 231: 23–44. [DOI] [PubMed] [Google Scholar]
- 7.Saleem H, Tovey SC, Rahman T, Riley AM, Potter BVL, et al. (2012) Stimulation of inositol 1,4,5-trisphosphate (IP3) receptor subtypes by analogues of IP3. PLoS ONE In Press. [DOI] [PMC free article] [PubMed]
- 8. Rossi AM, Riley AM, Potter BVL, Taylor CW (2010) Adenophostins: high-affinity agonists of IP3 receptors. Curr Top Membr 66: 209–233. [DOI] [PubMed] [Google Scholar]
- 9. Takahashi M, Kagasaki T, Hosoya T, Takahashi S (1994) Adenophostins A and B: potent agonists of inositol-1,4,5-trisphosphate receptor produced by Penicillium brevicompactum. Taxonomy, fermentation, isolation, physico-chemical and biological properties. J Antibiot 46: 1643–1647. [DOI] [PubMed] [Google Scholar]
- 10. Takahashi M, Tanzawa K, Takahashi S (1994) Adenophostins, newly discovered metabolites of Penicillium brevicompactum, act as potent agonists of the inositol 1,4,5-trisphosphate receptor. J Biol Chem 269: 369–372. [PubMed] [Google Scholar]
- 11. Hotoda H, Takahashi M, Tanzawa K, Takahashi S, Kaneko M (1995) IP3 receptor ligand.1: Synthesis of adenophostin A. Tetrahedon Lett. 36: 5037–5040. [PubMed] [Google Scholar]
- 12. Rossi A, Sureshan KM, Riley AM, Potter BVL, Taylor CW (2010) Selective determinants of inositol 1,4,5-trisphosphate and adenophostin A interactions with type 1 inositol 1,4,5-trisphosphate receptors. Br J Pharmacol 161: 1070–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wilcox RA, Primrose WU, Nahorski SR, Challiss RAJ (1998) New developments in the molecular pharmacology of the myo-inositol 1,4,5-trisphosphate receptor. Trends Pharmacol Sci 19: 467–475. [DOI] [PubMed] [Google Scholar]
- 14. Marchant JS, Parker I (1998) Kinetics of elementary Ca2+ puffs evoked in Xenopus oocytes by different Ins(1,4,5)P 3 receptor agonists. Biochem J 334: 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mak D-O, McBride S, Foskett JK (2001) ATP-dependent adenophostin activation of inositol 1,4,5-trisphosphate receptor channel gating. Kinetic implications for the duration of calcium puffs in cells. J Gen Physiol 117: 299–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bird GSJ, Takahashi M, Tanzawa K, Putney JWJ (1999) Adenophostin A induces spatially restricted calcium signaling in Xenopus laevis occytes. J Biol Chem 274: 20643–20649. [DOI] [PubMed] [Google Scholar]
- 17. DeLisle S, Marksberry EW, Bonnett C, Jenkins DJ, Potter BVL, et al. (1997) Adenophostin A can stimulate Ca2+ influx without depleting the inositol 1,4,5-trisphosphate-sensitive Ca2+ stores in the Xenopus oocyte. J Biol Chem 272: 9956–9961. [DOI] [PubMed] [Google Scholar]
- 18. Gregory RB, Wilcox RA, Berven LA, van Straten NC, van der Marel GA, et al. (1999) Evidence for the involvement of a small subregion of the endoplasmic reticulum in the inositol trisphosphate receptor-induced activation of Ca2+ inflow in rat hepatocytes. Biochem J 341: 401–408. [PMC free article] [PubMed] [Google Scholar]
- 19. Hartzell HC, Machaca K, Hirayama Y (1997) Effects of adenophostin-A and inositol 1,4,5-trisphosphate on Cl- currents in Xenopus laevis oocytes. Mol Pharmacol 51: 683–692. [DOI] [PubMed] [Google Scholar]
- 20. Huang Y, Takahashi M, Tanzawa K, Putney JW Jr (1998) Effect of adenophostin A on Ca2+ entry and calcium release-activated calcium current (I crac) in rat basophilic leukemia cells. J Biol Chem 273: 31815–31821. [DOI] [PubMed] [Google Scholar]
- 21. Machaca K, Hartzell HC (1999) Adenophostin A and inositol 1,4,5-trisphosphate differentially activate Cl- currents in Xenopus oocytes because of disparate Ca2+ release kinetics. J Biol Chem 274: 4824–4831. [DOI] [PubMed] [Google Scholar]
- 22. Parekh AB, Riley AM, Potter BVL (2002) Adenophostin A and ribophostin, but not inositol 1,4,5-trisphosphate or manno-adenophostin, activate a Ca2+ release-activated Ca2+ current, I CRAC, in weak intracellular Ca2+ buffer. Biochem J 361: 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marwood RD, Correa V, Taylor CW, Potter BVL (2000) Synthesis of adenophostin A. Tetrahedron: Asymmetry. 11: 397–403. [Google Scholar]
- 24. Marwood RD, Jenkins DJ, Correa V, Taylor CW, Potter BVL (2000) Contribution of the adenine base to the activity of adenophostin A investigated using a base replacement strategy. J Med Chem 43: 4278–4287. [DOI] [PubMed] [Google Scholar]
- 25. Jenkins DJ, Marwood RD, Potter BVL (1997) A disaccharide polyphosphate mimic of 1d-myo-inositol 1,4,5-trisphosphate. Chem Comm 5: 449–450. [Google Scholar]
- 26. Marwood RD, Riley AM, Correa V, Taylor CW, Potter BVL (1999) Simplification of adenophostin A defines a minimal structure for potent glucopyranoside-based mimics of d-myo-inositol 1,4,5-trisphosphate. Bioorg Med Chem Lett 9: 453–458. [DOI] [PubMed] [Google Scholar]
- 27.Marwood RD, Riley AM, Jenkins DJ, Potter BVL (2000) Synthesis of adenophostin A and congeners modified at glucose. J Chem Soc Perkin Trans: 1935–1947.
- 28.Sureshan KM, Riley AM, Rossi AM, Tovey SC, Dedos SG, et al. (2009) Activation of IP3 receptors by synthetic bisphosphate ligands. Chem Comm: 1204–1206. [DOI] [PMC free article] [PubMed]
- 29. Yamazaki H, Chan J, Ikura M, Michikawa T, Mikoshiba K (2010) Tyr-167/Trp-168 in type1/3 inositol 1,4,5-trisphosphate receptor mediates functional coupling between ligand binding and channel opening. J Biol Chem 285: 36081–36091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tovey SC, Sun Y, Taylor CW (2006) Rapid functional assays of intracellular Ca2+ channels. Nature Prot 1: 259–263. [DOI] [PubMed] [Google Scholar]
- 31.Ott RL, Longnecker M (2010) An introduction to statistical methods and data analysis: Brooks/Cole, Cengage Learning. 1273.
- 32. Taylor CW, Potter BVL (1990) The size of inositol 1,4,5-trisphosphate-sensitive Ca2+ stores depends on inositol 1,4,5-trisphosphate concentration. Biochem J 266: 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Muallem S, Pandol SJ, Beeker TG (1989) Hormone-evoked calcium release from intracellular stores is a quantal process. J Biol Chem 264: 205–212. [PubMed] [Google Scholar]
- 34. Irvine RF (1990) “Quantal” Ca2+ release and the control of Ca2+ entry by inositol phosphates - a possible mechanism. FEBS Lett 262: 5–9. [DOI] [PubMed] [Google Scholar]
- 35. Hirose K, Iino M (1994) Heterogeneity of channel density in inositol-1,4,5-trisphosphate-sensitive Ca2+ stores. Nature 372: 791–794. [DOI] [PubMed] [Google Scholar]
- 36. Correa V, Riley AM, Shuto S, Horne G, Nerou EP, et al. (2001) Structural determinants of adenophostin A activity at inositol trisphosphate receptors. Mol Pharmacol 59: 1206–1215. [DOI] [PubMed] [Google Scholar]
- 37. Shuto S, Tatani K, Ueno Y, Matsuda A (1998) Synthesis of adenophostin analogues lacking the adenine moiety as novel potent IP3 receptor ligands: Some structural requirements for the significant activity of adenophostin A. J Org Chem. 63: 8815–8824. [Google Scholar]
- 38. Polokoff MA, Bencen GH, Vacca JP, se Solms SJ, Young SD, et al. (1988) Metabolism of synthetic inositol trisphosphate analogs. J Biol Chem 263: 11922–11927. [PubMed] [Google Scholar]
- 39. Safrany ST, Wojcikiewicz RJH, Strupish J, Nahorski SR, Dubreuil D, et al. (1991) Interaction of synthetic d-6-deoxy-myo-inositol 1,4,5-trisphosphate with the Ca2+-releasing d-myo-inositol 1,4,5-trisphosphate receptor, and the metabolic enzymes 5-phosphatase and 3-kinase. FEBS Lett 278: 252–256. [DOI] [PubMed] [Google Scholar]
- 40. Nerou EP, Riley AM, Potter BVL, Taylor CW (2001) Selective recognition of inositol phosphates by subtypes of inositol trisphosphate receptor. Biochem J 355: 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bosanac I, Alattia J-R, Mal TK, Chan J, Talarico S, et al. (2002) Structure of the inositol 1,4,5-trisphosphate receptor binding core in complex with its ligand. Nature 420: 696–700. [DOI] [PubMed] [Google Scholar]
- 42. Felemez M, Ballereau S, Schlewer G, Spiess B (2000) Inframolecular protonation process of 6-modified myo-inositol 1,4,5-tris(phosphates): substitution effects on the cooperativity between the phosphate groups. New J Chem 24: 631–638. [Google Scholar]
- 43. Wilcox RA, Challiss RAJ, Traynor JR, Fauq AH, Ognayanov VI, et al. (1994) Molecular recognition at the myo-inositol 1,4,5-trisphosphate receptor. 3-position substituted myo-inositol 1,4,5-trisphosphate analogues reveal the binding and Ca2+ release requirements for high affinity interaction with the myo-inositol 1,4,5-trisphosphate receptor. J Biol Chem 269: 26815–26821. [PubMed] [Google Scholar]
- 44. Takahashi S, Takeshi K, Takahashi M (1994) Adenophostins A and B: potent agonists of inositol-1,4,5-trisphosphate receptors produced by Penicillium brevicompactum. Structure elucidation. J Antibiot 47: 95–100. [DOI] [PubMed] [Google Scholar]
- 45. Hotoda H, Murayama K, Miyamoto S, Iwata Y, Takahashi M, et al. (1999) Molecular recognition of adenophostin, a very potent Ca2+ inducer, at the d-myo-inositol 1,4,5-trisphosphate receptor. Biochemistry 38: 9234–9241. [DOI] [PubMed] [Google Scholar]
- 46. Sureshan KM, Riley AM, Thomas MP, Tovey SC, Taylor CW, et al. (2012) Contribution of phosphates and adenine to the potency of adenophostins at the IP3 receptor: synthesis of all possible bisphosphates of adenophostin A. J Med Chem. 55: 1706–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Putney JW (2009) Capacitative calcium entry: from concept to molecules. Immunol Rev 231: 10–22. [DOI] [PubMed] [Google Scholar]