

Abstract
Methylmercury (MeHg) is a well known environmental pollutant that induces serious neuronal damage. Although MeHg readily crosses the blood-brain barrier, and should affect both neurons and glial cells, how it affects glia or neuron-to-glia interactions has received only limited attention. Here, we report that MeHg triggers ATP/P2Y1 receptor signals in astrocytes, thereby protecting neurons against MeHg via interleukin-6 (IL-6)-mediated pathways. MeHg increased several mRNAs in astrocytes, among which IL-6 was the highest. For this, ATP/P2Y1 receptor-mediated mechanisms were required because the IL-6 production was (i) inhibited by a P2Y1 receptor antagonist, MRS2179, (ii) abolished in astrocytes obtained from P2Y1 receptor-knockout mice, and (iii) mimicked by exogenously applied ATP. In addition, (iv) MeHg released ATP by exocytosis from astrocytes. As for the intracellular mechanisms responsible for IL-6 production, p38 MAP kinase was involved. MeHg-treated astrocyte-conditioned medium (ACM) showed neuro-protective effects against MeHg, which was blocked by anti-IL-6 antibody and was mimicked by the application of recombinant IL-6. As for the mechanism of neuro-protection by IL-6, an adenosine A1 receptor-mediated pathway in neurons seems to be involved. Taken together, when astrocytes sense MeHg, they release ATP that autostimulates P2Y1 receptors to upregulate IL-6, thereby leading to A1 receptor-mediated neuro-protection against MeHg.
Introduction
Methylmercury (MeHg), a well-known environmental pollutant, easily crosses the blood-brain barrier [1], [2] inducing several types of serious neuronal damage and disorders [3], [4], [5], [6]. Although most studies about MeHg-induced toxicity in the CNS have focused on its effects on neurons, MeHg, acting on a much higher number of glial cells, should affect their functions and viabilities. This is of great importance because it has become apparent that glial cells regulate a large variety of neuronal functions both in physiological and pathophysiological CNS [7]. However, the effects of MeHg on glial cells or neuron-to-glia interactions have received only limited attention.
Recently, it has become apparent that MeHg causes diverse responses in glial cells, i.e., it upregulates antioxidant genes [8], [9], while it rather inhibits the uptake of cysteine, a critical precursor of glutathione synthesis, leading to a decrease in antioxidants [10]. As one of the mechanisms of MeHg-induced neuronal loss is oxidative stress [11], [12], [13], [14], these glial responses by MeHg may greatly affect neuronal functions or viability. Inflammatory responses in glial cells are also involved in several types of neuronal damage. It has been reported that MeHg produces proinflammatory cytokines including interleukin-6 (IL-6) in glial cells [15], [16], [17]. In general, these cytokines facilitate inflammatory responses, leading to deterioration of the neuronal viability. However, we [18] and others [19] have already demonstrated that astrocytic IL-6 in response to various chemicals or insults protected neurons against oxidative neuronal death. However, the physiological or pathophysiological significance of the increased IL-6 in response to MeHg remains largely unknown, and even less is known about the mechanisms underlying MeHg-induced IL-6 in astrocytes.
Here, we demonstrate that MeHg upregulates several genes in astrocytes, among which IL-6 is the highest. And, as mentioned above, astrocytes protect neurons against MeHg by IL-6-mediated mechanisms. We also demonstrate that, when astrocytes sense MeHg, they release ATP that autostimulates P2Y1 receptors in astrocytes, thereby leading to IL-6 production via p38-mediated mechanisms. The released IL-6 appears to exhibit neuro-protection by upregulating adenosine A1 receptors in neurons.
Materials and Methods
Chemicals and Antibodies
Reagents were obtained from the following sources. Adenosine 5′-triphosphate (ATP), apyrase (grade III), bovine serum albumin (BSA), DPCPX, methylmercury (MeHg), MRS2179, (NH4)2S, Pb(NO3)2, suramin and Tris-maleate were purchased from Sigma Chemical (MO, USA). PD98059, SB203580, and SP600125 were purchased from Tocris bioscience (Bristol, UK). Recombinant rat IL-6 and anti IL-6 antibody were purchased from R&D Systems (MN, USA). Fura 2-acetoxymethyl ester (fura 2-AM) was purchased from Invitrogen (CA, USA). Polyclonal antibodies against total p38 and phosphorylated p38 were purchased from Cell Signaling Technology (MA, USA). Anti-MAP2 antibody was obtained from Chemicon (CA, USA). Anti-GFAP antibody was obtained from Millipore (MA, USA). Dextran T250 was purchased from Extrasynthase (Genay, France).
Cell Culture
All of the animals used in this study were obtained, housed, cared for and used in accordance with the guidelines of the University of Yamanashi. Every effort was made to minimize the number of experimental animals used and their suffering. The culture of cortical neurons was prepared as described [20] with minor modifications. In brief, cerebral cortices dissected from 17-day-old fetal Wistar rats were digested with papain (9 units/ml) dissolved in PBS containing 0.02% L-cysteine monohydrate, 0.5% glucose, and 0.02% BSA at 37°C for 15 min. After enzyme treatment, cells were plated on BD PureCoat Amine 96 well cell culture plates (Becton, Dickinson and Company, NJ, USA) at a density of 8×104 cells/well. The cells were maintained in DMEM supplemented with 1 mM glutamine, N1 supplement, 10 units/ml penicillin, and 10 µg/ml streptomycin under 5% CO2 at 37°C. The culture of cortical astrocytes was prepared as previously reported [20]. Cerebral cortices dissected from newborn Wistar rats were digested 0.1% Trypsin-EDTA at 37°C for 10 min. After enzyme treatment, the cells were dispersed by agitation through a pipette and plated in a flask. For purification of the astrocytes from the cortical cultures, the flask was shaken for 24 hr 7–10 days after seeding to remove detached cells. Then, astrocytes were subcultured in 6-well cell culture plates at a density of 2×105 cells/well, 96-well cell culture plates at a density of 7×103 cells/well, and LAB-TEK II chambered coverglass (Nalge Nunc International, NY, USA) at a density of 2.5×104 cells/well.
Mice
C57BL/6 mice (17-day-old fetal) were purchased from Japan SLC. P2Y1 knock-out mice (C57BL/6 background) have been developed as previously reported [21]. The cortical astrocytes from these mice were prepared as it is for the rat cortical astrocytes.
WST-1 Assay
Neuronal viability and astrocytic viability were estimated by WST-1 assay using a cell counting kit (Dojindo, Kumamoto, Japan). After incubation with MeHg for 20 or 44 hr, 1/10 volume of WST-1 solution was added to the cell culture medium and incubated for an additional 4 hr. The absorbance of supernatants was measured with a microplate reader at 450 nm as the test wavelength and at 630 nm as the reference wavelength.
DNA Microarray Analysis
For this experiment, astrocytes were exposed to 10 µM of MeHg for 2 hr. Converting total RNA (100 ng) to the targets for Affymetrix GeneChip DNA microarray hybridization was done according to the manufacturer’s instructions. The targets were hybridized onto a rat genome U34A GeneChip DNA microarray (Affymetrix, Santa Clara, CA) for 16–24 hr at 45°C. After hybridization, DNA microarrays were washed and stained on a Fluidics Station according to the protocol provided by Affymetrix. Afterward, the DNA microarrays were scanned, and then the images obtained were analyzed by GeneChip Operating System software (version 1.4; Affymetrix). The microarray data is available upon request.
Quantitative RT-PCR
Total RNA was isolated and purified from astrocytes and neurons using RNeasy (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Reverse transcription (RT)-PCR was performed using a one step primescript® RT-PCR Kit (Takara Bio Inc., Shiga, Japan) according to the manufacturer's protocol. The reaction mix contained 40 ng of total RNA, 200 nM primers, 100 nM TaqMan probe, TAKARA EX Taq® HS and PrimeScript™ RT enzyme Mix. RT-PCR amplification and real-time detection were performed using an Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems, CA, USA). The reverse transcription was performed at 42°C for 5 min followed by inactivation at 95°C for 10 s. The temperature profile consisted of 40 cycles of denaturation at 95°C for 5 s, and annealing/extension at 60°C for 34 s. The sequence of the primers and probe for rat IL-6 were as follows: the TaqMan probe, 5′-CAGAATTGCCATTGCACAACTCTTTTCTCA-3′; the forward primer, 5′-CAGTGCATCATCGCTGTTCA-3′; and the reverse primer, 5′-CATATGTTCTCAGGGAGATCTTGGA-3′. The sequence of the primers and probe for rodent A1 receptor were as follows: the TaqMan probe, 5′-CGAGTCAAGATCCCTCTCCGGTACAAGA-3′; the forward primer, 5′-TCATCCTCACCCAGAGCTCC-3′; and the reverse primer, 5′-ATGGGTGTCAGGCCTACCAC-3′. Primers and the Taquman probe for GAPDH were obtained from Rodent GAPDH Control Reagents (Applied Biosystems). Mouse IL-6 expression was estimated using the probe set (Mm0046190-m1) from Applied Biosystems (Foster City, CA).
Enzyme-linked Immunosorbent Assay of IL-6
The MeHg-induced IL-6 production from astrocytes was measured using a Quantikine® rat IL-6 immunoassay kit (R&D Systems, MN, USA). Astrocytes were incubated with MeHg (1 or 3 µM) in serum-free medium for 12 or 24 hr and the supernatants were collected. The assay was performed according to the manufacturer’s instructions. All standards and samples were measured with a microplate reader at a wavelength of 450 nm.
Ca2+-imaging
Changes in intracellular Ca2+ were measured by the fura 2 method with minor modifications [22]. In brief, the culture medium was replaced with balanced salt solution (BSS) of the following composition (in mM): NaCl 150, KCl 5.0, CaCl2 1.8, MgCl2 1.2, HEPES 25, and d-glucose 10 (pH 7.4). Cells were loaded with fura 2 by incubation with 10 µM fura 2-acetoxymethyl ester (fura 2-AM) at room temperature (RT) in BSS for 45 min. After loading, the samples were mounted on a microscope (ECLIPSE TE2000-U, Nikon,Tokyo, Japan) equipped with a 75-W xenon lamp and band-pass filters of 340 and 380 nm wavelengths for measurement of the Ca2+-dependent signals (F340 and F380 nm). Image data were recorded by a CCD camera (ORCA-ER, Hamamatsu Photonics, Shizuoka, Japan). For evaluation, we used the ratio of F340/F380.
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde for 30 min at RT and they were incubated with the primary antibodies (anti-GFAP antibody at 1∶2000; and anti-MAP2 antibody at 1∶500) in a Can Get Signal A (TOYOBO, Osaka, Japan) for 24 hr at 4°C. Then, the cells were further incubated with Alexa 488- or Alexa 546-conjugated second antibodies (1∶2000) for 1 hr at RT. Fluorescent images were obtained by a laser scanning confocal microscope FV-1000 (Olympus, Tokyo, Japan).
Measurement of Extracellular ATP
The extracellular ATP concentration of the MeHg-treated astrocytes was determined with an ATP bioluminescence assay kit CLS II (Roche Applied Science, Mannheim, Germany). Astrocytes were incubated with MeHg (1 or 3 µM) in serum-free medium and the supernatants were collected and boiled at 95°C for 10 min. Equal volumes of luciferin/luciferase reagents and samples (100 µl each) were mixed a few times by gentle pipetting. All standards and samples were measured with a Lumat LB9501 tube luminometer (Berthold, Wildbad, Germany). The ATP concentrations were calculated from the intensities of a series of standard ATP.
Western Blotting
Cells were lysed and the lysates were electrophoresed with 10% SDS-PAGE gels and transferred to PVDF membranes. The membranes were blocked for 1 hr in Tris-buffered saline containing 0.1% Tween-20 and 5% BSA at RT and were incubated with primary antibodies (1∶5000) over night at 4°C. Membranes were then incubated with horseradish peroxidase-conjugated 2nd antibodies (1∶20000) for 1 hr at RT. Protein bands were visualized by rinsing the membrane with supersignal west pico chemiluminescence substrate (Thermo scientific, PA, USA). Images were obtained using LAS-4000 (Fujifilm, Tokyo, Japan).
Enzyme Histochemistry
Enzyme histochemistry for ecto-ATPases activity has been performed on the basis of a previous report [23]. Briefly, cells were fixed with 4% paraformaldehyde for 30 min at RT and preincubated for 30 min at RT with Tris-maleate-sucrose buffer (250 mM sucrose, 50 mM Tris-maleate, pH 7.4) containing 2 mM CaCl2. The enzyme reaction was performed in a reaction buffer (2 mM Pb(NO3)2, 5 mM MnCl2, 2 mM CaCl2, 50 mM Tris-maleate (pH7.4), 250 mM sucrose, 3% dextran T250) with 1 mM of ATP as a substrate. After 1 hr reaction at RT, cells were washed with H2O and the ecto-ATPase activity was visualized by 0.5% (v/v) of (NH4)2S.
Statistics
Data were expressed as means ± SEM. Student’s t-test was used for comparison of two groups. One way analysis of variance (ANOVA) followed by Tukey test was applied for multiple comparisons. The differences were considered to be significant when the P value was less than 5%.
Results
MeHg Upregulates IL-6 Expression in Astrocytes
We first performed transcriptome analysis in cultured astrocytes stimulated with MeHg (10 µM) using DNA microarray (Table 1). MeHg changed the expressions of a large number of genes including those of cytokines and chaperones in astrocytes. Among them, interleukin-6 (IL-6) mRNA showed the most remarkable increase (638 fold), and we confirmed its upregulation using quantitative RT-PCR. The increase in IL-6 mRNA expression was concentration-dependent over a concentration range of from 0.1 to 3 µM with 2 hr-exposure (0.1 µM, 2.6±0.7; 1.0 µM, 6.1±0.7; 3.0 µM, 30.0±7.2 fold increase vs. control, n = 3) (Fig. 1A). The low concentration of MeHg (0.1 µM) never increased IL-6 mRNA expression at any exposure time tested (1–12 hr) (1 hr, 1.3±0.1; 2 hr, 2.6±0.7; 6 hr, 1.7±0.2; 12 hr, 1.2±0.2 fold increase vs. control, n = 3). The increase in the IL-6 mRNA level was transient and reached the maximal level at 2 hr after the exposure with 1 µM (1 hr, 2.5±0.6; 2 hr, 6.1±0.7; 6 hr, 5.1±2.0; 12 hr, 1.2±0.1 fold increase vs. control, n = 3) and 3 µM (1 hr, 4.3±1.1; 2 hr, 29.9±7.2; 6 hr, 5.1±1.6; 12 hr, 1.9±0.2 fold increase vs. control, n = 3) of MeHg. ELISA analysis of the supernatants showed that MeHg (1 and 3 µM, 24 hr) increased IL-6 derived from astrocytes (1 µM MeHg, 51.5±11.4 pg/ml; 3 µM MeHg, 80.9±18.6 pg/ml, n = 4) (Fig. 1B). With 12-hr exposure, a lower level of IL-6 release was observed (1 µM MeHg, 8.7±5.0 pg/ml; 3 µM MeHg, 30.5±16.9 pg/ml, n = 4). Without MeHg stimulation, no detectable level of IL-6 was observed (n.d.).
Table 1. A list of top 5 genes upregulated in astrocytes by MeHg (10 µM, 2 hr).
Figure 1. MeHg-induced IL-6 mRNA upregulation and protein release from astrocytes.

(A) Effect of MeHg on IL-6 mRNA expression in astrocytes. MeHg (1–3 µM) transiently increased IL-6 expression and the induction peak was observed at 2 hr exposure. Low concentration of MeHg (0.1 µM) had no effect on IL-6 expression. *P<0.05 and **P<0.01 vs. control. (B) MeHg-induced IL-6 protein production to the supernatant from astrocytes. MeHg (1 or 3 µM, 12 or 24 hr) induced IL-6 production. The 12-hr exposure exhibited a lower level of IL-6 release than that with 24-hr exposure of MeHg.
Activation of P2Y1 Receptor and Subsequent p38 Phosphorylation Mediate IL-6 Expression in Astrocytes
The mechanisms underlying the MeHg-evoked IL-6 production were investigated. Since astrocytes release or leak ATP that functions as a gliotransmitter or inflammatory mediator in response to various environmental changes, we firstly focused on ATP/P2 receptor-mediated signals. The MeHg (3 µM, 2 hr)-evoked increase in IL-6 mRNA was significantly suppressed by the broad P2 receptor antagonist suramin (100 µM) (MeHg/suramin, 58.7±8.1% of MeHg, n = 8) (Fig. 2A). The selective P2Y1 receptor antagonist MRS2179 (10 µM) also inhibited the IL-6 mRNA upregulation to a similar extent (MeHg/MRS2179, 64.0±4.0% of MeHg, n = 9), suggesting the predominant involvement of P2Y1 receptors in the MeHg-evoked IL-6 production. In addition, MeHg (3 µM, 2 hr) failed to increase IL-6 mRNA in P2Y1R KO astrocytes (WT, 5.8±0.7; P2Y1R KO, 1.0±0.7 fold increase vs. control, n = 3) (Fig. 2B), confirming that P2Y1 receptors are necessary in this process. As for intracellular signaling mechanisms, it is known that ATP activates mitogen-activated protein kinases (MAPKs) including ERK1/2, JNK, and p38 in glial cells via several types of P2 receptors [24], [25], [26], [27]. PD98059 (10 µM), an inhibitor of mitogen-activated protein kinase kinase, the upstream activator of ERK [28] and SP600125 (10 µM), an inhibitor of SAPK/JNK [29], exhibited no effect on the IL-6 expression (MeHg/PD98059, 78.3±37.5% of MeHg, n = 4; MeHg/SP600125, 127.5±19.5% of MeHg, n = 6) (Fig. 2C). In contrast, a p38 inhibitor, SB203580 (10 µM) [30], significantly suppressed the MeHg-mediated increase in IL-6 mRNA (MeHg/SB203580, 54.7±13.9% of MeHg, n = 6). Western blotting analysis revealed that MeHg (3 µM, 30 min) induced p38 phosphorylation, which was strongly blocked by suramin (control, 100.0±8.5; MeHg, 210.0±36.3; MeHg/suramin, 114.0±19.7% of control, n = 4) (Fig. 2D). The phosphorylation of p38 in astrocytes was mimicked by ATP (100 µM, 30 min) (246.3±58.0% of control, n = 4). Overall, the data suggest that the activation of P2Y1 receptors and subsequent phosphorylation of p38 MAPK pathway should be involved in the MeHg-evoked IL-6 production.
Figure 2. IL-6 upregulation by MeHg is mediated by P2Y1 receptors followed by p38 activation.
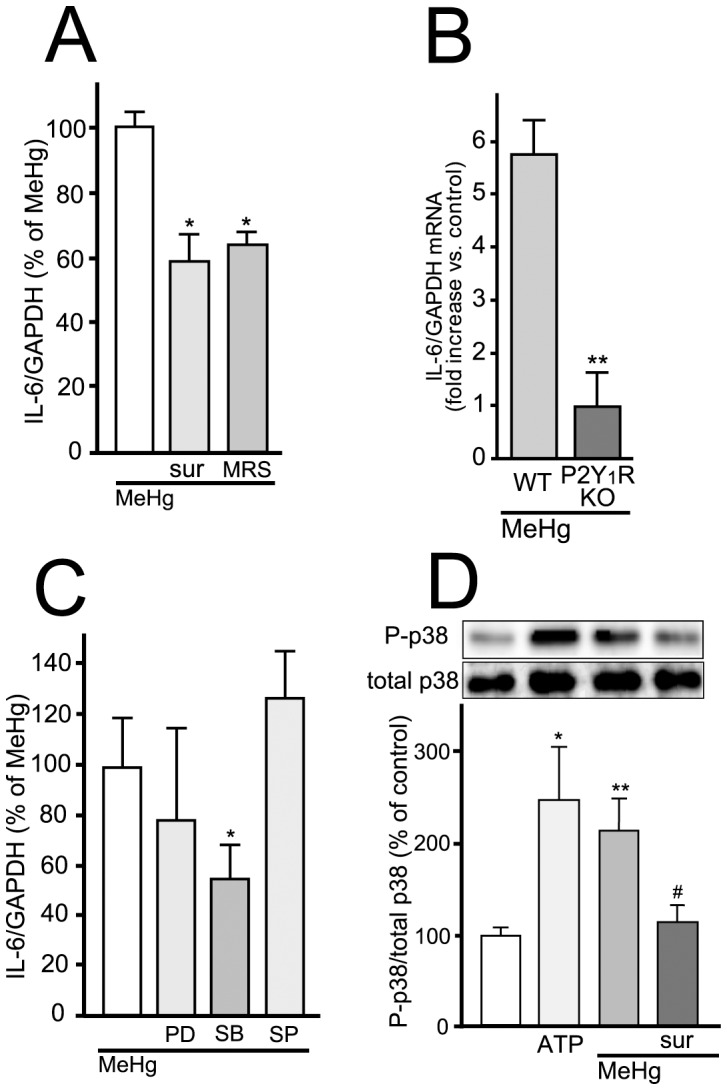
(A) P2Y1 receptor blockade suppresses IL-6 mRNA expression induced by MeHg. MeHg (3 µM, 2 hr)-increased IL-6 mRNA expression was inhibited by either suramin (sur, 100 µM) or MRS2179 (MRS, 10 µM). *P<0.05 vs. MeHg. (B) P2Y1 receptor mediates MeHg-induced IL-6 mRNA expression. P2Y1R KO astrocytes exhibited no increase in IL-6 mRNA with MeHg (3 µM, 2 hr). **P<0.01 vs. WT. (C) Contribution of p38 in MeHg-induced IL-6 mRNA expression. The IL-6 mRNA expression evoked by MeHg (3 µM, 2 hr) was inhibited by SB203580 (SB, 10 µM) but not by PD98059 (PD, 10 µM) or SP600125 (SP, 10 µM). *P<0.05 vs. MeHg. (D) Downstream signaling molecule of P2Y1 receptor is p38. MeHg (3 µM, 30 min)-induced p38 phosphorylation was inhibited by suramin (sur, 100 µM). ATP (100 µM) also induced p38 phosphorylation. *P<0.05, **P<0.01 vs. control, #P<0.05 vs. MeHg.
MeHg Evoked Release of ATP in Astrocytes
Next we examined whether MeHg elicits the release of ATP from astrocytes. To measure the release of ATP, we used a luciferin-luciferase based chemiluminescence assay. MeHg (1 and 3 µM for 15 min) increased the extracellular ATP level to 194.6±43.2% (1 µM MeHg) and 358.5±87.1 (3 µM MeHg) % of the pre-stimulated basal control level (77.0±18.0 pM; control, 100±22.9%, n = 10) (Fig. 3A). We analyzed the MeHg (3 µM)-evoked time-course of ATP release and found that it was transient, i.e., it was initiated at 15 min and peaked at 1 to 3 hr, and then was back to non-stimulated basal level in 6 hr (basal ATP level, 0.50±0.03 nM; control, 100.0±5.6%; 5 min, 110±3.2%; 15 min, 140.1±1.0%; 1 hr, 255.1±2.4%; 3 hr, 296.0±51.6%; 6 hr, 101.6±0.9%; 12 hr, 82.3±0.6%; 24 hr, 134.14±25.5%, n = 5) (Fig. 3B). It has been reported that ATP is released from astrocytes via several pathways including exocytosis and diffusion through ATP-permeable plasma membrane channels such as connexin hemichannels, pannexin channels, Maxi-anion channels or P2X7 receptors [31], [32], [33], [34], [35], [36], [37]. Using several pharmacological inhibitors, we found that neither Gd3+ (50 µM, a maxi-anion channel blocker) [33], nor carbenoxolone (CBX, 100 µM, an inhibitor of connexin hemi-channel, pannexin channel and P2X7 receptor) [36], [38], [39] showed an inhibitory effect on the MeHg (3 µM, 15 min)-evoked ATP release from astrocytes (MeHg, 169.0±9.6%; MeHg/Gd3+, 234.8±1.0%; MeHg/CBX, 156.3±1.9% of control, n = 4) (Fig. 3C). In contrast to these inhibitors, both Botulinum toxin A (BoNT) (5 units/ml, 24 hr pretreatment), a toxin that cleaves SNAPs [40], [41], thereby preventing exocytosis, and the intracellular Ca2+ chelator BAPTA-AM (10 µM) significantly suppressed the MeHg-induced ATP release from astrocytes (MeHg/BoNT, 112.8±0.6%; MeHg/BAPTA, 104.0±0.5% of control, n = 4) (Fig. 3C).
Figure 3. Exocytotic ATP release and Ca2+ oscillation in astrocytes evoked by MeHg.

(A) MeHg increased the extracellular ATP level of astrocytes. The effect of MeHg on the ATP release was concentration-dependent. MeHg was applied to the cell 15 min before the ATP measurement. *P<0.05 and **P<0.01 vs. control. (B) The time-course of extracellular ATP level of MeHg-treated astrocytes. MeHg (3 µM) increased extracellular ATP level of astrocytes, which was transient and peaked at 3 hr. (C) MeHg induces exocytotic ATP release from astrocytes. BoNT (5 units/ml, 24 hr pretreatment) and BAPTA-AM (10 µM) abolished MeHg (3 µM, 15 min)-induced ATP release from astrocytes. Gd3+(50 µM) rather enhanced ATP release, and CBX (100 µM) had no effect. **P<0.01 vs. MeHg. (D) The time-course of MeHg-induced changes in the frequency of Ca2+ oscillation. MeHg increased frequency of Ca2+ oscillation in astrocytes, which was transient and peaked at 1 hr. (E) MeHg increases the frequency of spontaneous calcium oscillations in astrocytes via enhancing ATP release. The number of Ca2+ oscillations was significantly increased by MeHg (3 µM, 30 min) and the increase was abolished by apyrase (Apy, 20 units/ml). **P<0.01 vs. MeHg. (F) Exocytotic pathway contributes to the MeHg-increased Ca2+ oscillation. BoNT (5 units/ml, 24 hr pretreatment) reduced the number of Ca2+ oscillations. **P<0.01 vs. MeHg. (G) Inhibitory effect of P2Y1 receptor antagonist on the Ca2+ oscillation frequency increased by MeHg. Ca2+ oscillation frequency was dramatically increased with MeHg (3 µM), and the increase was blocked by MRS2179 (MRS, 10 µM). **P<0.01 vs. MeHg. (H) P2Y1R is essential for the Ca2+ oscillation evoked by MeHg. WT astrocytes exhibited enhanced Ca2+ oscillation by MeHg (3 µM) but not in P2Y1R KO astrocytes. *P<0.05, **P<0.01 vs. control (WT), ##P<0.01 vs. MeHg (WT).
We also evaluated the release of ATP using another indicator, i.e., Ca2+ oscillation in astrocytes, because released ATP auto-stimulates P2 receptors to increase the frequency of Ca2+ oscillations or increase the distance of Ca2+ waves in astrocytes [20], [42], [43]. The time-course of changes in frequency of Ca2+ oscillation was also transient. After the addition of MeHg (3 µM), the frequency of spontaneous Ca2+ oscillations increased with time and peaked at 1 hr followed by decrease (control, 0.06±0.02; 5 min, 0.09±0.03; 15 min, 0.24±0.03; 1 hr, 0.36±0.05; 3 hr, 0.14±0.03; 6 hr, 0.12±0.02; 12 hr, 0.13±0.03 times/cell/5 min, n = 40) (Fig. 3D). The MeHg (3 µM, 30 min)-increased Ca2+ oscillation was completely suppressed by the nucleotide-degrading enzyme apyrase (20 units/ml) (control, 0.16±0.06 times/cell/5 min; MeHg, 1.02±0.15 times/cell/5 min; apyrase/MeHg, 0.01±0.01 times/cell/5 min, n = 100) (Fig. 3E). Similar to the ATP release, the frequency of Ca2+ oscillations was reduced by BoNT (5 units/ml, 24 hr pretreatment) (control, 0.13±0.05 times/cell/5 min; MeHg, 2.04±0.15 times/cell/5 min; MeHg/BoNT, 1.04±0.20 times/cell/5 min, n = 100) (Fig. 3F). Because many reports have shown that the P2Y1 receptor is essential in the ATP-mediated astrocytic Ca2+ signaling [42], [43], [44], the contribution of P2Y1 receptors to the MeHg-evoked Ca2+ oscillation was tested using the selective P2Y1 receptor antagonist MRS2179 [45] or P2Y1R KO mice astrocytes. The MeHg-evoked increase in Ca2+ oscillation was significantly suppressed by MRS2179 (10 µM) (MeHg/MRS2179, 0.02±0.01 times/cell/5 min, n = 100) (Fig. 3G) and not observed in astrocytes from P2Y1R KO mice (WT/control, 0.21±0.05 times/cell/5 min; P2Y1R KO/control, 0.13±0.04 times/cell/5 min; WT/MeHg, 2.03±0.15 times/cell/5 min; P2Y1R KO/MeHg, 0.36±0.06 times/cell/5 min, n = 200) (Fig. 3H). P2Y1R KO astrocytes exhibited normal Ca2+ responses to exogenously applied UTP (100 µM), a P2Y2/4 receptor agonist, but not to the P2Y1 agonist 2MeSADP (1 µM) (Fig. S1).
Astrocyte-derived IL-6 Protects Neurons against MeHg
We [18] and others [19], [46], [47], [48], [49], [50], [51], [52] have already reported that IL-6 had neuro-protective effects against several types of insults. Using immunocytochemical analysis and WST-1 assay, we evaluated whether IL-6 showed neuro-protection against MeHg. As shown in Fig. 4B, healthy cortical neurons exhibited clear cell bodies and extended dendrites when stained with anti-MAP2 antibody (Fig. 4B, MAP2-control). After MeHg treatment (3 µM, 48 hr), the anti-MAP2 signals were dramatically changed into signals with only cell body-like structures and fragmented or bead-like process structures (Fig. 4B, MAP2-MeHg). In contrast to neurons, anti-GFAP signals exhibited no significant changes with or without MeHg (Fig. 4B, GFAP). WST-1 assay revealed that MeHg (48 hr) decreased neuronal viability in a concentration-dependent manner over a range of from 0.01 to 3 µM (control, 100±5.0%; 0.01 µM, 84.9±4.3%; 0.1 µM, 75.2±4.3%; 1 µM, 50.0±3.2%; 3 µM, 19.9±3.2%) (Fig. 4C, gray columns), whereas astrocytes showed no decrease in cell viability (control, 100±2.4%; 0.01 µM, 96.6±2.9%; 0.1 µM, 96.7±1.8%; 1 µM, 100.6±4.1%; 3 µM, 103.9±4.6%) (Fig. 4C, white columns). Recombinant IL-6 protein (100 pg/ml, 24 hr pretreatment) suppressed the MeHg-induced morphological changes in neurons (Fig. 4D, MeHg+IL-6). IL-6 itself had no significant morphological effect on neurons (Fig. 4D, IL-6). Recombinant IL-6 protein also restored the MeHg (1 or 3 µM, 48 hr)-reduced neuronal viability (MeHg 1 µM, 35.5±3.9%; MeHg 3 µM, 16.4±2.4%; MeHg 1 µM/IL-6, 49.7±4.2%; MeHg 3 µM/IL-6, 46.7±4.0%, n = 20) (Fig. 4E). IL-6 itself had no significant effect on neuronal cell viability (93.2±3.3%, n = 14). When IL-6 was added to the neuronal culture 12 hr after MeHg treatment, it did not show neuro-protection (data not shown). As it is known that neuronal culture contains small portion of astrocytes, we estimated glial contamination in the culture of cortical neurons by immunocytochemical analysis. In our culture condition, almost all cells were positive to MAP2 (98.8±1.2%, n = 780), and remained 1.2±0.2% of cells were positive to GFAP, suggesting that glial contamination could be negligible.
Figure 4. Neuro-protective effect of recombinant IL-6.

(A) The experimental schedule. Recombinant IL-6 (100 pg/ml) was pretreated to the cell culture 24 hr before MeHg exposure (48 hr). Cells were then fixed for immunostaining or used for WST-1 assay. (B) MeHg-induced morphological changes in neurons. MAP2 signals in the processes were disrupted by MeHg (3 µM), whereas the GFAP signals exhibited no differences with or without MeHg. Scale bar: 50 µm. (C) MeHg-decreased the neuronal viability. Neuronal viability was significantly reduced by MeHg in a concentration-dependent fashion at a concentration range of from 0.01 to 3 µM (gray columns). MeHg did not affect the astrocytic viability (white columns). *P<0.05, **P<0.01 vs. control. (D) Recombinant IL-6 restored the MeHg-induced morphological changes in neurons. The collapsed MAP2 signals in neuronal processes were recovered in the presence of recombinant IL-6 (100 ng/ml). IL-6 did not change the neuronal morphology in controls. Scale bar: 50 µm. (E) IL-6 protects neurons against MeHg. Recombinant IL-6 (100 ng/ml) significantly restored the MeHg (1 or 3 µM)-reduced neuronal viability. **P<0.01 vs. MeHg.
We then examined the neuro-protective effect of astrocyte-conditioned medium (ACM, MeHg-treated astrocytes conditioned medium). As shown in figure 5A, astrocytes were incubated with MeHg (1 or 3 µM, 24 hr) and ACM was transferred into neuronal culture and neurons were further incubated in the either presence or absence of anti-IL-6 antibody (100 ng/ml) for 48 hr. ACM restored the MeHg-decreased neuronal viability (1 µM MeHg, 41.8±3.0%; 1 µM MeHg/ACM, 70.6±2.9%; 3 µM MeHg, 18.9±2.0%; 3 µM MeHg/ACM, 52.3±4.0%, n = 30) (Fig. 5B). The protective effects of ACM were abolished by IL-6 antibody (1 µM MeHg/ACM/IL-6 Ab, 47.4±4.3%; 3 µM MeHg/ACM/IL-6 Ab, 35.5±3.1%, n = 15), indicating that the ACM-mediated neuro-protection is dependent on IL-6. IL-6 antibody itself showed no direct effect on neuronal cell viability (97.7±9.8%; n = 5). When IL-6 was added to neurons 12 hr after MeHg, it no longer showed neuro-protection (1 µM of MeHg, 57.0±6.4%; MeHg/IL-6, 64.6±8.0%, n = 6, P>0.05), suggesting that an effective time window seems to be present for the IL-6- or ACM-mediated neuro-protection.
Figure 5. Neuro-protection by astrocyte-derived IL-6.

(A) The experimental schedule. MeHg (1 or 3 µM) was treated to the astrocyte culture for 24 h and the ACM was transferred into the neuronal culture. The ACM-treated neuronal cultures were further incubated for 48 h with or without IL-6 antibody. (B) ACM restored the reduction in neuronal viability evoked by MeHg (1 or 3 µM, 48 hr). The ACM-induced neuro-protection against MeHg was significantly reduced by IL-6 antibody (100 ng/ml). **P<0.01 vs. MeHg.
Neuro-protective Action of IL-6 is Mediated by Neuronal A1 Receptor
The neuro-protection by recombinant IL-6 was significantly inhibited by 1 µM cycloheximide (CHX), an inhibitor of protein synthesis (MeHg, 65.7±7.6%; MeHg/IL-6, 108.3±4.5%; MeHg/IL-6/CHX, 86.1±4.2%, n = 5) (Fig. 6A). CHX (1 µM, added to the culture 24 hr after IL-6 treatment) alone had no effect on neuronal cell viability (107.2±2.2%, n = 5). Thus, it appears that IL-6 would newly synthesize neuro-protective molecules that would account for the neuro-protection against MeHg. IL-6 has been reported to increase A1 receptor expression thereby inducing neuro-protection against cytotoxicity [53], [54]. We found that recombinant IL-6 (200 pg/ml, 2 hr) significantly increased adenosine A1 receptor mRNA in cortical neurons (control, 100±6.7%; IL-6, 173.1±23.9%, n = 7) (Fig. 6B). We further tested whether the IL-6-mediated A1 receptor upregulation contributes to excitability of cortical neurons. Firstly, we investigated whether pre-treatment with IL-6 might affect basal level of [Ca2+]i in the cortical neurons, because this can reflect a baseline activity of synaptic transmission. Thus, when excitatory synaptic transmission is inhibited by either TTX, antagonists of glutamate receptors, or agonists of presynaptic auto-receptors such as A1 receptors, the basal [Ca2+]i is decreased [20], [21]. As shown in Fig. 6C (i), the basal [Ca2+]i in IL-6-treated neurons (100 pg/ml, 24 hr) was lower than that in control neurons, which was restored by an A1 receptor antagonist DPCPX (1 µM, 5 min) (F340/F380: no treatment, 0.72±0.06; IL-6, 0.56±0.01; IL-6/DPCPX, 0.74±0.05, n = 99). Secondly, we tested whether the glutamate-evoked responses were affected by the IL-6-treatment. The glutamate (10 µM)-evoked increase in [Ca2+]i in IL-6-treated neurons was smaller than that in control neurons(F340/F380: no treatment, 3.2±0.2; IL-6, 2.2±0.2, n = 99), which was also restored by 1 µM DPCPX (IL-6/DPCPX, 3.0±0.2, n = 99) (Fig. 6C (ii) (iii)). DPCPX alone (1 µM) never affected the neuronal viability (control, 100.0±2.9%; DPCPX, 95.6±6.0%, n = 5). All these findings suggest that IL-6 could regulate excitability of cortical neurons by increasing both expression and function of adenosine A1 receptors, which might contribute to neuro-protection against MeHg.
Figure 6. Adenosine A1 receptor mediates neuro-protection against MeHg via suppressing excitatory neurotransmission.

(A) Newly synthesized proteins participate in the IL-6-mediated neuro-protection by recombinant IL-6 (100 pg/ml) was suppressed by CHX (1 µM). **P<0.01 vs. MeHg/IL-6. (B) Upregulation of adenosine A1 receptor mRNA by recombinant IL-6 (200 pg/ml, 2 hr) in the cortical neurons. *P<0.05 vs. no treatment. (C) Changes in [Ca2+]i in control and IL-6-treated cortical neurons, showing effect of A1 receptors. (i) The basal [Ca2+]i level in IL-6-treated (100 pg/ml, 24 hr) neurons was significantly lower than that in control neurons. This decrease was restored by DPCPX (1 µM). *P<0.05 vs. IL-6 alone. (ii) Representative traces of the glutamate-evoked increases in [Ca2+]i in non-treated control (left), IL-6-treated (100 pg/ml, 24 hr) (right) and IL-6-treated neurons in the presence of 1 µM DPCPX. Glutamate (10 µM) was added to the neurons for 10 s. Bold line in each panel showed averaged changes in [Ca2+]i in neurons, which was summarized in (iii). The glutamate-evoked increase in [Ca2+]i in IL-6-treated neurons was significantly lower than that in control neurons, which was restored by DPCPX. **P<0.01 vs. Glu/IL-6. (D) A1 receptor-mediated neuro-protection by IL-6. The protective effect of IL-6 (100 pg/ml) was suppressed by DPCPX (1 µM). **P<0.01 vs. MeHg/IL-6. (E) ATP-induced neuro-protection is mediated by A1 receptor. Exogenously applied ATP (100 µM) restored the MeHg (1 µM, 48 hr)-reduced neuronal viability, and this effect was blocked by DPCPX (1 µM). *P<0.05, **P<0.01 vs. MeHg/ATP. (F) The MeHg-evoked increase in activity of ecto-ATPases in astrocytes. Activity of ecto-ATPases was analyzed by an enzyme histochemical assay. When stimulated with MeHg (3 µM), the activity (shown as brown signals) was increased, which peaked at around 6 to 12 hr. Scale bar, 200 µm.
Next we asked whether the increased A1 receptors in cortical neurons are related to neuro-protection against MeHg. The protective effect of IL-6 (100 pg/ml, 24 hr) against MeHg (1 µM) was abolished by DPCPX (1 µM) (MeHg, 45.3±2.6%; MeHg/IL-6, 63.1±4.9%; MeHg/IL-6/DPCPX, 45.4±4.5%, n = 18) (Fig. 6D). Similarly, simultaneous application of ATP (100 µM) with MeHg also exhibited a neuro-protective effect, which was blocked by DPCPX (1 µM) (MeHg, 45.3±2.6%; MeHg/ATP, 58.5±4.7%; MeHg/ATP/DPCPX, 43.2±3.1%, n = 18) (Fig. 6E). As tonic A1 receptor activation would require an increased level of extracellular adenosine, we then performed an enzyme histochemistry for evaluating activity of ecto-ATPases in astrocytes. As shown in figure 6F, MeHg (3 µM) increased the activity of ecto-ATPases (shown as brown signals), which peaked around 6–12 hr. The MeHg-evoked increase in extracellular ATP was observed prior to the increase in ecto-ATPases activity (i.e. peaked at 3 hr) (Fig. 3B). These findings strongly suggest that the MeHg-treated ACM should contain higher levels of adenosine.
Discussion
MeHg easily passes the blood-brain barrier and causes serious damage in the CNS. Unlike neurons, its effect on glial cells has received only limited attention. In the present study, we demonstrated that, when exposed to MeHg, astrocytes exhibited neuro-protection against MeHg, in which ATP/P2Y1 receptor-mediated signals and subsequent IL-6 production in astrocytes have a pivotal role.
MeHg significantly increased extracellular ATP level of astrocytes. Although it has been described that MeHg induces astrocytic swelling [55], [56], we did not observe significant morphological changes or injured structures in astrocytes when evaluated by immunocytochemical analysis using anti-GFAP antibody. The increase in the extracellular ATP level did not seem to be due to leakage by cell damage because MeHg never decreased the astrocyte viability (Fig. 4). Although astrocytes release ATP by multiple mechanisms, exocytosis might be one of them because both BoNT and BAPTA-AM reduced the ATP release, while neither CBX nor Gd3+ inhibited the release (Fig. 3). However, the finding that the inhibition of the Ca2+ oscillation by BoNT was incomplete suggests that SNARE-independent pathways for ATP release might also be involved (Fig. 3F). With regard to ATP exocytosis, a recent report has demonstrated that lysosomes mediate at least part of the exocytotic ATP release in astrocytes [37]. In addition to lysosomes, vesicular nucleotide transporter (VNUT) has been reported to mediate exocytotic ATP release [57]. We must await further studies to clarify the involvement of lysosomes and/or VNUT-vesicles in the MeHg-evoked ATP release by astrocytes or the initial target molecule(s) of MeHg. However, our present data clearly showed that, when astrocytes sense MeHg, they release ATP in part by exocytosis. This initial ATP release should be a key response because subsequent events, e.g., IL-6 production or neuro-protection by astrocytes, were dependent on ATP/P2Y1 receptors.
In the transcriptome analysis, we found that MeHg upregulated several genes, among which IL-6 mRNA showed the most remarkable increase (table 1). The MeHg-evoked upregulation of IL-6 mRNA peaked at 2 hr and IL-6 protein production was observed at 12 or 24 hr (Fig. 1A and 1B). Such time lag between IL-6 mRNA and protein expression can be also seen in other reports of various stimuli-induced IL-6 expression/production in astrocytes [58], [59], [60], [61]. It would take longer time for the de novo synthesis or release of IL-6 after its mRNA upregulation in astrocytes, but we must await further investigation to clarify it since we did not check the IL-6 release at earlier (<12 hr) or later time points (>24 hr). The MeHg-evoked IL-6 production was dependent on the activation of P2Y1 receptors in astrocytes, because the IL-6 production was (i) reduced by the antagonists to P2Y1 receptors, (ii) was not observed in P2Y1R KO astrocytes, and (iii) was mimicked by exogenously applied ATP. As for the downstream signaling of P2Y1 receptors, we focused on MAPKs. Of the three MAPK members (i.e. ERK1/2, JNK, and p38), only the p38 inhibitor SB203580 suppressed the MeHg-induced IL-6 mRNA upregulation (Fig. 2C). In addition, the phosphorylation of p38 by MeHg was blocked by the P2 receptor antagonist. ATP itself also evoked p38 phosphorylation (Fig. 2D). All these findings suggest that MeHg triggered the exocytosis of ATP, which in turn autostimulates P2Y1 receptors, thereby leading to p38-mediated IL-6 production in astrocytes. Many reports have shown that p38 is required for the induction of IL-6 in astrocytes, together with a variety of stimuli including prostaglandin E2 [62], co-stimulation with IL-6 and IL-17 [60], thromboxane A2 [63], oncostatin M [64], and ICAM-1 ligation [65]. The p38 activation might be a common key pathway for IL-6 expression in astrocytes. The p38 inhibitor and P2 receptor antagonists (i.e. suramin and MRS2179) exhibited lesser extent of inhibitory effects than those by P2Y1R KO astrocytes. This discrepancy might be due to their lower concentrations because previous studies have shown that 10 µM of SB203580 does not show complete blockade for stimuli-induced p38 phosphorylation in astrocytes [66], [67], [68], [69], [70]. Similarly, we previously showed that neither suramin (100 µM) nor MRS2179 (1 µM) completely suppressed the ATP (100 µM)-evoked Ca2+ transient (i.e. about 70% suppression) [26].
Since IL-6 is a cytokine with major regulating effects on the inflammatory response, in general an elevation in the proinflammatory cytokine IL-6 is considered to have a damaging effect on neurons. We and others, however, reported that IL-6 could protect neurons against a variety of damages including trauma, ischemia, excitotoxicity and oxidative stress [18], [19], [46], [47], [48], [49], [50], [51], [52]. In the present study, astrocytes showed neuro-protection against MeHg via IL-6-mediated mechanisms because ACM-induced neuro-protection was IL-6 dependent (Fig. 5) and exogenously applied recombinant IL-6 protein mimicked the neuro-protection (Fig. 4). One possible mechanism of the IL-6-mediated neuro-protection is that IL-6 stimulates the induction of neuro-protective molecules. We showed that the neuro-protection by IL-6 disappeared in the presence of a protein synthesis inhibitor CHX (Fig. 6A), suggesting that de novo synthesis of certain neuro-protective molecules appears to be required. Recent reports by Biber et al. demonstrated that IL-6 inhibits the glutamate-induced excitototoxicity of cortical neurons requires adenosine A1 receptor functions in neurons [53], [54]. The IL-6 increased both mRNA and proteins of adenosine A1 receptors in the neurons, and the protection by IL-6 disappeared in the presence of CHX [54]. In the present study, we also found that IL-6 increases A1 receptor mRNA expression in cortical neurons (Fig. 6B). IL-6 increased not only mRNA but also A1 receptor-mediated tonic inhibition on an excitatory neurotransmitter (Fig. 6C). Supporting these results, the IL-6-induced neuro-protection against MeHg was suppressed by the A1 receptor antagonist DPCPX (Fig. 6D). All these findings may support the idea that one of the neuro-protective molecules induced by ACM or IL-6 would be adenosine A1 receptors.
However, anti-IL-6 antibody could not abolish the effect of ACM, indicating the involvement of IL-6-independent neuro-protective mechanisms. We considered that the astrocyte-derived ATP itself might function as another neuro-protective molecule because, without ACM, the exogenously applied ATP alone showed neuro-protection (Fig. 6E). Interestingly, this protection by ATP was also inhibited by the antagonist to adenosine A1 receptor DPCPX (Fig. 6E). Astrocytic ATP either as ATP [20] or metabolized into adenosine by ecto-nucleotidases [71], [72], inhibits excess excitatory synaptic transmission, leading to inhibition of excitatory neuronal death. Our time-lapse analysis of extracellular ATP level and enzyme histochemistry have shown that MeHg gradually increased ATP release from astrocytes followed by an increase in ATPase activity (Fig. 3B and 6F). Under this condition, extracellular adenosine would increase and activate neuronal A1 receptor.
Since it takes 24 hr for IL-6 to protect neurons against MeHg (Fig. 4), the delayed production of IL-6 (Fig. 1) in astrocytes might be problematic to the neuro-protection, because in general both neurons and astrocytes would be simultaneously exposed to MeHg in situ. However, astrocytes could release ATP in response to MeHg as early as 15 min after MeHg (Fig. 3A), and the released ATP or its metabolite adenosine directly protected neurons against MeHg (Fig. 6), suggesting that astrocytes should show the IL-6-independent neuro-protection even in the early stage. Thus, the astrocyte-mediated neuro-protection shown in the present study could work in situ. Furthermore, we previously showed that activation of P2Y1 receptors in astrocytes increased tolerance against oxidative stress by the upregulation of various oxidoreductase genes [26], [73]. Therefore, the astrocytic ATP release and activation of P2Y1 receptors appear to be key events that trigger multiple neuro-protective responses.
Taken together, as summarized in Figure 7, when astrocytes are exposed to MeHg, they exocytose ATP and show a neuro-protective phenotype. The released ATP functions as an autocrine to stimulate P2Y1 receptors, thereby leading to the protection of neurons against MeHg via IL-6-mediated pathways. The IL-6 increases neuronal A1 receptor expression and function. The released ATP, being metabolized into adenosine, may also function as a paracrine to exert neuro-protection via suppressing excitatory neurotransmission.
Figure 7. A schematic diagram, illustrating mechanisms underlying astrocyte-mediated neuro-protection against MeHg.

MeHg stimulates exocytosis of astrocytic ATP that functions as both (a) autocrine and (b) paracrine signals to reveal neuro-protection, i.e., (a) the released ATP as an autocrine signal, autostimulates P2Y1 receptors to induce IL-6 that upregulates neuronal adenosine A1 receptors, (b) the released ATP from astrocytes being degraded into adenosine, stimulates neuronal adenosine A1 receptors and suppresses neuronal excitability as a paracrine signal, thereby leading to further inhibition of neuronal excitability. As for mechanisms for IL-6 synthesis and release, an increase in [Ca2+]i in astrocytes mediated by P2Y1 receptors, and subsequent p38 phosphorylation were involved (insert).
Supporting Information
Differences in Ca2+ responses to 2MeSADP and UTP in WT and P2Y1R KO mice. (A) Typical Ca2+ responses to the P2Y1R agonist 2methyl-thio-ADP (2MeSADP) (1 µM) and the P2Y2/4 receptor agonist UTP (100 µM) in control astrocytes obtained from WT mice (upper traces) and those from P2Y1R KO mice (lower traces). Although UTP evoked [Ca2+]i elevations in both WT and P2Y1R KO astrocytes, 2MeSADP failed to produce the [Ca2+]i increse in P2Y1R KO astrocytes, which was summarized in B.
(TIF)
Acknowledgments
We thank Dr. Shigetomi, Ms. Hirayama and Mr. Komatsu for fruitful discussion.
Funding Statement
This work was supported by Study (Group) of the Health Effects of Heavy Metals Organized by Ministry of the Environment, Japan (SK). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Clarkson TW (1997) The toxicology of mercury. Crit Rev Clin Lab Sci 34: 369–403. [DOI] [PubMed] [Google Scholar]
- 2. Lapham LW, Cernichiari E, Cox C, Myers GJ, Baggs RB, et al. (1995) An analysis of autopsy brain tissue from infants prenatally exposed to methymercury. Neurotoxicology 16: 689–704. [PubMed] [Google Scholar]
- 3. Ceccatelli S, Dare E, Moors M (2010) Methylmercury-induced neurotoxicity and apoptosis. Chem Biol Interact 188: 301–308. [DOI] [PubMed] [Google Scholar]
- 4. Clarkson TW, Magos L, Myers GJ (2003) The toxicology of mercury–current exposures and clinical manifestations. N Engl J Med 349: 1731–1737. [DOI] [PubMed] [Google Scholar]
- 5. Farina M, Rocha JB, Aschner M (2011) Mechanisms of methylmercury-induced neurotoxicity: evidence from experimental studies. Life Sci 89: 555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yuan Y (2012) Methylmercury: a potential environmental risk factor contributing to epileptogenesis. Neurotoxicology 33: 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haydon PG (2001) GLIA: listening and talking to the synapse. Nat Rev Neurosci 2: 185–193. [DOI] [PubMed] [Google Scholar]
- 8. Ni M, Li X, Yin Z, Sidoryk-Wegrzynowicz M, Jiang H, et al. (2011) Comparative study on the response of rat primary astrocytes and microglia to methylmercury toxicity. Glia 59: 810–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang L, Jiang H, Yin Z, Aschner M, Cai J (2009) Methylmercury toxicity and Nrf2-dependent detoxification in astrocytes. Toxicol Sci 107: 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Allen JW, Shanker G, Tan KH, Aschner M (2002) The consequences of methylmercury exposure on interactive functions between astrocytes and neurons. Neurotoxicology 23: 755–759. [DOI] [PubMed] [Google Scholar]
- 11. Ali SF, LeBel CP, Bondy SC (1992) Reactive oxygen species formation as a biomarker of methylmercury and trimethyltin neurotoxicity. Neurotoxicology 13: 637–648. [PubMed] [Google Scholar]
- 12. Gasso S, Cristofol RM, Selema G, Rosa R, Rodriguez-Farre E, et al. (2001) Antioxidant compounds and Ca(2+) pathway blockers differentially protect against methylmercury and mercuric chloride neurotoxicity. J Neurosci Res 66: 135–145. [DOI] [PubMed] [Google Scholar]
- 13. Mundy WR, Freudenrich TM (2000) Sensitivity of immature neurons in culture to metal-induced changes in reactive oxygen species and intracellular free calcium. Neurotoxicology 21: 1135–1144. [PubMed] [Google Scholar]
- 14. Yee S, Choi BH (1996) Oxidative stress in neurotoxic effects of methylmercury poisoning. Neurotoxicology 17: 17–26. [PubMed] [Google Scholar]
- 15. Chang JY (2007) Methylmercury causes glial IL-6 release. Neurosci Lett 416: 217–220. [DOI] [PubMed] [Google Scholar]
- 16. Chang JY (2011) Methylmercury-induced IL-6 release requires phospholipase C activities. Neurosci Lett 496: 152–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chang JY, Tsai PF (2009) IL-6 release from mouse glia caused by MeHg requires cytosolic phospholipase A2 activation. Neurosci Lett 461: 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fujishita K, Ozawa T, Shibata K, Tanabe S, Sato Y, et al. (2009) Grape seed extract acting on astrocytes reveals neuronal protection against oxidative stress via interleukin-6-mediated mechanisms. Cell Mol Neurobiol 29: 1121–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Suzuki S, Tanaka K, Suzuki N (2009) Ambivalent aspects of interleukin-6 in cerebral ischemia: inflammatory versus neurotrophic aspects. J Cereb Blood Flow Metab 29: 464–479. [DOI] [PubMed] [Google Scholar]
- 20. Koizumi S, Fujishita K, Tsuda M, Shigemoto-Mogami Y, Inoue K (2003) Dynamic inhibition of excitatory synaptic transmission by astrocyte-derived ATP in hippocampal cultures. Proc Natl Acad Sci U S A 100: 11023–11028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leon C, Hechler B, Freund M, Eckly A, Vial C, et al. (1999) Defective platelet aggregation and increased resistance to thrombosis in purinergic P2Y(1) receptor-null mice. J Clin Invest 104: 1731–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koizumi S, Inoue K (1997) Inhibition by ATP of calcium oscillations in rat cultured hippocampal neurones. Br J Pharmacol 122: 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Braun N, Zhu Y, Krieglstein J, Culmsee C, Zimmermann H (1998) Upregulation of the enzyme chain hydrolyzing extracellular ATP after transient forebrain ischemia in the rat. J Neurosci 18: 4891–4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Neary JT, Kang Y, Bu Y, Yu E, Akong K, et al. (1999) Mitogenic signaling by ATP/P2Y purinergic receptors in astrocytes: involvement of a calcium-independent protein kinase C, extracellular signal-regulated protein kinase pathway distinct from the phosphatidylinositol-specific phospholipase C/calcium pathway. J Neurosci 19: 4211–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Panenka W, Jijon H, Herx LM, Armstrong JN, Feighan D, et al. (2001) P2X7-like receptor activation in astrocytes increases chemokine monocyte chemoattractant protein-1 expression via mitogen-activated protein kinase. J Neurosci 21: 7135–7142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shinozaki Y, Koizumi S, Ishida S, Sawada J, Ohno Y, et al. (2005) Cytoprotection against oxidative stress-induced damage of astrocytes by extracellular ATP via P2Y1 receptors. Glia 49: 288–300. [DOI] [PubMed] [Google Scholar]
- 27. Tran MD, Neary JT (2006) Purinergic signaling induces thrombospondin-1 expression in astrocytes. Proc Natl Acad Sci U S A 103: 9321–9326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR (1995) PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem 270: 27489–27494. [DOI] [PubMed] [Google Scholar]
- 29. Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, et al. (2001) SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A 98: 13681–13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McLaughlin MM, Kumar S, McDonnell PC, Van Horn S, Lee JC, et al. (1996) Identification of mitogen-activated protein (MAP) kinase-activated protein kinase-3, a novel substrate of CSBP p38 MAP kinase. J Biol Chem 271: 8488–8492. [DOI] [PubMed] [Google Scholar]
- 31. Arcuino G, Lin JH, Takano T, Liu C, Jiang L, et al. (2002) Intercellular calcium signaling mediated by point-source burst release of ATP. Proc Natl Acad Sci U S A 99: 9840–9845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kang J, Kang N, Lovatt D, Torres A, Zhao Z, et al. (2008) Connexin 43 hemichannels are permeable to ATP. J Neurosci 28: 4702–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu HT, Toychiev AH, Takahashi N, Sabirov RZ, Okada Y (2008) Maxi-anion channel as a candidate pathway for osmosensitive ATP release from mouse astrocytes in primary culture. Cell Res 18: 558–565. [DOI] [PubMed] [Google Scholar]
- 34. North RA, Verkhratsky A (2006) Purinergic transmission in the central nervous system. Pflugers Arch 452: 479–485. [DOI] [PubMed] [Google Scholar]
- 35. Pankratov Y, Lalo U, Verkhratsky A, North RA (2006) Vesicular release of ATP at central synapses. Pflugers Arch 452: 589–597. [DOI] [PubMed] [Google Scholar]
- 36. Suadicani SO, Brosnan CF, Scemes E (2006) P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2+ signaling. J Neurosci 26: 1378–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang Z, Chen G, Zhou W, Song A, Xu T, et al. (2007) Regulated ATP release from astrocytes through lysosome exocytosis. Nat Cell Biol 9: 945–953. [DOI] [PubMed] [Google Scholar]
- 38. Bruzzone R, Barbe MT, Jakob NJ, Monyer H (2005) Pharmacological properties of homomeric and heteromeric pannexin hemichannels expressed in Xenopus oocytes. J Neurochem 92: 1033–1043. [DOI] [PubMed] [Google Scholar]
- 39. Locovei S, Bao L, Dahl G (2006) Pannexin 1 in erythrocytes: function without a gap. Proc Natl Acad Sci U S A 103: 7655–7659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Binz T, Blasi J, Yamasaki S, Baumeister A, Link E, et al. (1994) Proteolysis of SNAP-25 by types E and A botulinal neurotoxins. J Biol Chem 269: 1617–1620. [PubMed] [Google Scholar]
- 41. Blasi J, Chapman ER, Link E, Binz T, Yamasaki S, et al. (1993) Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature 365: 160–163. [DOI] [PubMed] [Google Scholar]
- 42. Bowser DN, Khakh BS (2004) ATP excites interneurons and astrocytes to increase synaptic inhibition in neuronal networks. J Neurosci 24: 8606–8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bowser DN, Khakh BS (2007) Vesicular ATP is the predominant cause of intercellular calcium waves in astrocytes. J Gen Physiol 129: 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gallagher CJ, Salter MW (2003) Differential properties of astrocyte calcium waves mediated by P2Y1 and P2Y2 receptors. J Neurosci 23: 6728–6739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Moro S, Guo D, Camaioni E, Boyer JL, Harden TK, et al. (1998) Human P2Y1 receptor: molecular modeling and site-directed mutagenesis as tools to identify agonist and antagonist recognition sites. J Med Chem 41: 1456–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Carlson NG, Wieggel WA, Chen J, Bacchi A, Rogers SW, et al. (1999) Inflammatory cytokines IL-1 alpha, IL-1 beta, IL-6, and TNF-alpha impart neuroprotection to an excitotoxin through distinct pathways. J Immunol 163: 3963–3968. [PubMed] [Google Scholar]
- 47. Fujita T, Tozaki-Saitoh H, Inoue K (2009) P2Y1 receptor signaling enhances neuroprotection by astrocytes against oxidative stress via IL-6 release in hippocampal cultures. Glia 57: 244–257. [DOI] [PubMed] [Google Scholar]
- 48. Hirota H, Kiyama H, Kishimoto T, Taga T (1996) Accelerated Nerve Regeneration in Mice by upregulated expression of interleukin (IL) 6 and IL-6 receptor after trauma. J Exp Med 183: 2627–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Loddick SA, Turnbull AV, Rothwell NJ (1998) Cerebral interleukin-6 is neuroprotective during permanent focal cerebral ischemia in the rat. J Cereb Blood Flow Metab 18: 176–179. [DOI] [PubMed] [Google Scholar]
- 50. Peng YP, Qiu YH, Lu JH, Wang JJ (2005) Interleukin-6 protects cultured cerebellar granule neurons against glutamate-induced neurotoxicity. Neurosci Lett 374: 192–196. [DOI] [PubMed] [Google Scholar]
- 51. Penkowa M, Giralt M, Carrasco J, Hadberg H, Hidalgo J (2000) Impaired inflammatory response and increased oxidative stress and neurodegeneration after brain injury in interleukin-6-deficient mice. Glia 32: 271–285. [DOI] [PubMed] [Google Scholar]
- 52. Yamada K, Umegaki H, Maezawa I, Iguchi A, Kameyama T, et al. (1997) Possible involvement of catalase in the protective effect of interleukin-6 against 6-hydroxydopamine toxicity in PC12 cells. Brain Res Bull 43: 573–577. [DOI] [PubMed] [Google Scholar]
- 53. Biber K, Lubrich B, Fiebich BL, Boddeke HW, van Calker D (2001) Interleukin-6 enhances expression of adenosine A(1) receptor mRNA and signaling in cultured rat cortical astrocytes and brain slices. Neuropsychopharmacology 24: 86–96. [DOI] [PubMed] [Google Scholar]
- 54. Biber K, Pinto-Duarte A, Wittendorp MC, Dolga AM, Fernandes CC, et al. (2008) Interleukin-6 upregulates neuronal adenosine A1 receptors: implications for neuromodulation and neuroprotection. Neuropsychopharmacology 33: 2237–2250. [DOI] [PubMed] [Google Scholar]
- 55. Aschner M (1996) Astrocytes as modulators of mercury-induced neurotoxicity. Neurotoxicology 17: 663–669. [PubMed] [Google Scholar]
- 56. Aschner M (2000) Astrocytic swelling, phospholipase A2, glutathione and glutamate: interactions in methylmercury-induced neurotoxicity. Cell Mol Biol (Noisy-le-grand) 46: 843–854. [PubMed] [Google Scholar]
- 57. Sawada K, Echigo N, Juge N, Miyaji T, Otsuka M, et al. (2008) Identification of a vesicular nucleotide transporter. Proc Natl Acad Sci U S A 105: 5683–5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. DeForge LE, Remick DG (1991) Kinetics of TNF, IL-6, and IL-8 gene expression in LPS-stimulated human whole blood. Biochem Biophys Res Commun 174: 18–24. [DOI] [PubMed] [Google Scholar]
- 59. Lu-Kuo JM, Austen KF, Katz HR (1996) Post-transcriptional stabilization by interleukin-1beta of interleukin-6 mRNA induced by c-kit ligand and interleukin-10 in mouse bone marrow-derived mast cells. J Biol Chem 271: 22169–22174. [DOI] [PubMed] [Google Scholar]
- 60. Ma X, Reynolds SL, Baker BJ, Li X, Benveniste EN, et al. (2010) IL-17 enhancement of the IL-6 signaling cascade in astrocytes. J Immunol 184: 4898–4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Oberbach A, Schlichting N, Bluher M, Kovacs P, Till H, et al. (2010) Palmitate induced IL-6 and MCP-1 expression in human bladder smooth muscle cells provides a link between diabetes and urinary tract infections. PLoS One 5: e10882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fiebich BL, Schleicher S, Spleiss O, Czygan M, Hull M (2001) Mechanisms of prostaglandin E2-induced interleukin-6 release in astrocytes: possible involvement of EP4-like receptors, p38 mitogen-activated protein kinase and protein kinase C. J Neurochem. 79: 950–958. [DOI] [PubMed] [Google Scholar]
- 63. Obara Y, Kurose H, Nakahata N (2005) Thromboxane A2 promotes interleukin-6 biosynthesis mediated by an activation of cyclic AMP-response element-binding protein in 1321N1 human astrocytoma cells. Mol Pharmacol 68: 670–679. [DOI] [PubMed] [Google Scholar]
- 64. Van Wagoner NJ, Choi C, Repovic P, Benveniste EN (2000) Oncostatin M regulation of interleukin-6 expression in astrocytes: biphasic regulation involving the mitogen-activated protein kinases ERK1/2 and p38. J Neurochem 75: 563–575. [DOI] [PubMed] [Google Scholar]
- 65. Lee SJ, Drabik K, Van Wagoner NJ, Lee S, Choi C, et al. (2000) ICAM-1-induced expression of proinflammatory cytokines in astrocytes: involvement of extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways. J Immunol 165: 4658–4666. [DOI] [PubMed] [Google Scholar]
- 66. Bhat NR, Zhang P, Lee JC, Hogan EL (1998) Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-alpha gene expression in endotoxin-stimulated primary glial cultures. J Neurosci 18: 1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Harris JE, Green JA, Elkington PT, Friedland JS (2007) Monocytes infected with Mycobacterium tuberculosis regulate MAP kinase-dependent astrocyte MMP-9 secretion. J Leukoc Biol 81: 548–556. [DOI] [PubMed] [Google Scholar]
- 68. Hua LL, Zhao ML, Cosenza M, Kim MO, Huang H, et al. (2002) Role of mitogen-activated protein kinases in inducible nitric oxide synthase and TNFalpha expression in human fetal astrocytes. J Neuroimmunol 126: 180–189. [DOI] [PubMed] [Google Scholar]
- 69. Sheng WS, Hu S, Nettles AR, Lokensgard JR, Vercellotti GM, et al. (2010) Hemin inhibits NO production by IL-1beta-stimulated human astrocytes through induction of heme oxygenase-1 and reduction of p38 MAPK activation. J Neuroinflammation 7: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhu D, Tan KS, Zhang X, Sun AY, Sun GY, et al. (2005) Hydrogen peroxide alters membrane and cytoskeleton properties and increases intercellular connections in astrocytes. J Cell Sci 118: 3695–3703. [DOI] [PubMed] [Google Scholar]
- 71. Fellin T, Halassa MM, Terunuma M, Succol F, Takano H, et al. (2009) Endogenous nonneuronal modulators of synaptic transmission control cortical slow oscillations in vivo. Proc Natl Acad Sci U S A 106: 15037–15042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, et al. (2003) ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron 40: 971–982. [DOI] [PubMed] [Google Scholar]
- 73. Shinozaki Y, Koizumi S, Ohno Y, Nagao T, Inoue K (2006) Extracellular ATP counteracts the ERK1/2-mediated death-promoting signaling cascades in astrocytes. Glia 54: 606–618. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Differences in Ca2+ responses to 2MeSADP and UTP in WT and P2Y1R KO mice. (A) Typical Ca2+ responses to the P2Y1R agonist 2methyl-thio-ADP (2MeSADP) (1 µM) and the P2Y2/4 receptor agonist UTP (100 µM) in control astrocytes obtained from WT mice (upper traces) and those from P2Y1R KO mice (lower traces). Although UTP evoked [Ca2+]i elevations in both WT and P2Y1R KO astrocytes, 2MeSADP failed to produce the [Ca2+]i increse in P2Y1R KO astrocytes, which was summarized in B.
(TIF)