

ABSTRACT
Herpesvirus entry requires the viral glycoprotein triad of gB and gH/gL to carry out fusion between the virion envelope and a cellular membrane in order to release the nucleocapsid into the target cell. Herpes simplex virus (HSV) also requires glycoprotein gD to initiate the fusion cascade by binding a cell receptor such as nectin 1 or herpesvirus entry mediator (HVEM). While the structure of gB is that of a class III fusion protein, gH/gL has no features that resemble other viral fusion proteins. Instead, it is suggested that gH/gL acts as a regulator of gB. The crystal structure of HSV-2 gH/gL was obtained with a functional protein that had a deletion of 28 residues at the gH N terminus (gHΔ48/gL). Unexplainably, monoclonal antibodies (MAbs) with virus-neutralizing activity map to these residues. To reconcile these two disparate observations, we studied the ability of gHΔ48/gL to regulate fusion. Here, we show that the protein induces low (constitutive) levels of fusion by gB in the absence of gD and/or receptor. However, when gD and receptor are present, this mutant functions as well as does wild-type (wt) gH/gL for fusion. We propose that gHΔ48/gL has an intermediate structure on the pathway leading to full regulatory activation. We suggest that a key step in the pathway of fusion is the conversion of gH/gL to an activated state by receptor-bound gD; this activated gH/gL resembles gHΔ48/gL.
IMPORTANCE
Herpes simplex viruses (HSVs) cause many human diseases, from mild cold sores to lethal neonatal herpes. As an enveloped virus, HSV must fuse its membrane with a host membrane in order for replication to take place. The virus uses four glycoproteins for this process, gD, gB, and gH/gL, and either of two cell receptors, herpesvirus entry mediator (HVEM) and nectin 1. Although the virus can enter the cell by direct fusion at the plasma membrane or via endocytosis, the same four glycoproteins are involved. The absence of any of these proteins abolishes the entry process. Here, we show that a mutant form of gH/gL, gHΔ48/gL, can induce fusion of gB-expressing cells in the absence of gD and a gD receptor. Our study supports the concept that gB is the HSV fusogen and its activity is regulated by gH/gL.
Introduction
Herpesviruses enter cells by fusing their envelopes with host cell membranes. Unlike most enveloped viruses, which use a single fusion protein (1, 2), herpesviruses in general use gB and the gH/gL heterodimer as the major components of the fusion machine (3–6). Herpesviruses also employ additional accessory glycoproteins required for cell tropism (e.g., UL128-131 of cytomegalovirus [CMV]) (7) or to trigger the fusion machinery for virus entry (e.g., herpes simplex virus [HSV] gD) (5).
Glycoproteins gB, gD, gH, and gL of HSV mediate membrane fusion events required for both entry and virus-induced cell fusion. Deletion of any of these four glycoproteins results in mutant virions that cannot penetrate host cells (8). Moreover, all four glycoproteins and a receptor (either nectin 1 or herpesvirus entry mediator [HVEM]) are both necessary and sufficient to induce cell fusion (5, 9, 10). Ultrastructural and biochemical studies revealed that fusion occurs in a series of highly regulated steps that begin with binding of gD to receptor and end with fusion caused by conformational changes to gB (11–16). First (5), structures of gD bound to nectin 1 or HVEM revealed that C-terminal residues of the gD ectodomain must be displaced to allow binding of either receptor (12, 14, 17). Second, this activated form of gD then interacts with gH/gL (18), which in turn interacts with gB (10, 19), triggering this class III fusion protein (16) to carry out virus-cell or cell-cell fusion. Although these broad steps are supported by biochemical data (18, 20), significant details of the cascade are still unknown. Among the unanswered questions is what effect the activation of gD by receptor has on the structure of gH/gL that allows it to trigger gB into a fusogenic state.
Previously, we proposed a model for how the four essential entry proteins work to initiate virus entry and cell fusion (18). Binding of gD to its receptors causes conformational changes to gD. Altered gD then interacts with and activates the regulatory activity of gH/gL, which in turn upregulates the fusogenic activity of gB. The structures of gB that have been solved (16, 21) are generally agreed to represent its postfusion form. Changes to gB which allow it to go from a hypothetical prefusion form to ones that lead to membrane fusion are currently not known, nor are there any structural data that explain how gH/gL activates gB to carry out this process. Importantly, according to our model, gB is the sole fusion protein of HSV. Key evidence that gH/gL is not a fusion protein that acts as a membrane-bound cofusogen with gB was the observation that gH/gL and gB are functional even when the two proteins are in trans (18, 22). Moreover, we showed that cell fusion can be initiated by adding a combination of soluble forms (non-membrane bound) of gD and gH/gL to nectin-bearing cells that express gB (18).
Here, we provide evidence that receptor-activated gD induces structural changes to gH/gL and in particular to the N terminus of gH and the C terminus of gL that activate the heterodimer. We show that a membrane-bound form of gH/gL lacking the first 28 residues of the mature protein (gHΔ48/gL) triggers low but reproducible levels of cell-cell fusion in the absence of gD or a receptor, an activity which we call constitutive fusion. We conclude that (i) gB, as the fusion protein, is the only glycoprotein required to be anchored in the membrane and (ii) fusion occurs sequentially, whereby receptor-activated gD alters the structure of gH/gL and this form in turn activates gB.
RESULTS
Soluble and cell-bound gH2Δ29/gL2 and gH2Δ48/gL2 trigger fusion of cells expressing gB, gD, and nectin 1 receptor.
This report describes newly discovered properties of two previously derived type 2 gH mutants (gH2Δ29 and gH2Δ48) and a type 1 gH deletion, gH1Δ48 (Fig 1) (23). All mutants retain the gH signal sequence and lack either the first 10 amino acids (residues 19 to 28 for gH2Δ29) or the first 29 amino acids (residues 19 to 47 for gH1Δ48 and gH2Δ48) of mature gH (Fig. 1). However, the ability of these mutants to promote cell-cell fusion or virus entry (23) contrasted with the fact that epitopes of two virus-neutralizing gH2-specific monoclonal antibodies (MAbs), CHL17 and CHL32, map within the deleted residues of the gH2 N terminus (24). This apparent disparity warranted a closer look at the gH2 N terminus and its role in gH2/gL2 function.
FIG 1 .

Crystal structure and diagrams of constructs used. (A) Crystal structure of gHΔ48/gL. Domain organization as published. Residues 19 to 47 of gH, which are missing from gHΔ48/gL, and the unresolved gL residues 204 to 224 are shown as dashed lines (green and aqua, respectively). (B) Diagrams of gH and gL constructs used in this study. The epitopes of monoclonal antibodies used are indicated in the same color scheme as in panel A. For gH, CHL2 (white; MAR 116), CHL25 (pink; 73 to 92), 52S (dark blue; MAR 536), and LP11 (red; MARs 86, 168, and 329). For gL, CHL34 (purple; 146 to 165), CΔ48L3 (black; 173 to 183), CHL26 (brown; 195 to 208), and CHL18 (aqua; 209 to 219).
We first compared the abilities of membrane-bound gH2Δ29/gL2, gH2Δ48/gL2, and wild-type (wt) gH2/gL2 to induce fusion of gB-expressing cells upon the addition of soluble gD (Fig. 2). As a control, gH2/gL2 was omitted and no syncytia were detected (Fig. 2A). Next, B78C10 (C10) cells expressing nectin 1 were transfected with plasmids expressing full-length gB, gL2, and either wt gH2 (Fig. 2B), gH2Δ29 (Fig. 2C), or gH2Δ48 (Fig. 2D). In each case, soluble gD2306t was then added to initiate fusion (for easier identification, syncytia were outlined with a white dotted line) (10). As previously shown (23), each form of gH2/gL2 induced fusion in the presence of gD and the fusion activities in each case were approximately the same. The total number of fusion events (the number of syncytia multiplied by the number of nuclei in a syncytium) ranged from 3,480 to 3,960 per coverslip (Fig. 2B to D).
FIG 2 .

N-terminal gH/gL truncations work at levels comparable to those of wild-type gH2/gL2. C10 cells were transfected with full-length gB (A) and gH/gL: wt gH2/gL2 (B), gHΔ29/gL2 (C), or gHΔ48/gL2 (D). Fusion was triggered with soluble gD306. C10 cells transfected with gB were triggered with a mixture of gD306 (E) and either soluble wt gH2/gL2 (F), truncated gHΔ29t/gL2 (G), or truncated gHΔ48t/gL2 (H). gHΔ48/gL2 is an activated form of gH/gL. C10 cells were transfected with full-length gB, gL, and wild-type gH2 (I), gHΔ29 (J), or gHΔ48 plasmids (K). In a parallel experiment, cells were transfected with gB plasmid only; at 6 h posttransfection, 250 µg/ml of soluble gH2t/gL2 (L), gHΔ29t/gL2 (M), or gHΔ48t/gL2 (N) was added. All coverslips were examined by immunofluorescence with gB MAbs (red). Nuclei were stained with propidium iodide and artificially colored gray using Volocity software. Syncytia are outlined with white dotted lines. N, number of syncytia from a coverslip multiplied by the average number of nuclei per syncytia.
We next tested soluble forms of the three gH2/gL2 proteins for their ability to activate fusion. As previously shown (18), a combination of gD306t and gH2/gL2 was able to trigger fusion of C10 cells expressing gB; the total number of fusion events was in the hundreds rather than thousands (as seen with full-length wt gH2/gL2) (Fig. 2F to H), but this number is significant since there were none in the absence of gH/gL (Fig. 2E). A combination of gD306t and either gH2Δ29t/gL2 (Fig. 2G) or gH2Δ48t/gL2 (Fig. 2H) also supported fusion, and the total number of fusion events matched those for soluble wt gH2/gL2 plus gD (Fig. 2F).
Critically, in carrying out additional controls for these experiments, we found a surprising result which led us to the central point in this report. We found that C10 cells bearing gB and full-length gH2Δ29/gL2 or gH2Δ48/gL2 exhibited significant numbers of syncytia even in the absence of gD. This phenomenon also occurred when the soluble forms of these proteins were tested in the absence of soluble gD (Fig. 2J and K). Importantly, as shown before (18), this was not the case for wt gH2/gL2 (Fig. 2I). Thus, this experiment shows that by removing residues from the gH N terminus, the absolute requirement for gD was eliminated, thereby reducing the number of players in the sequence of fusion events to three (gH/gL and gB). We also observed that the capacities of gH2Δ29/gL2 and gH2Δ48/gL2 to induce syncytium formation in the absence of gD were similar whether the plasmids were expressed in cis (Fig. 2C and D) or in trans with respect to gB (data not shown). Finally, the ability of gH2Δ48/gL2 to function in the absence of gD was not influenced by the presence of a gD receptor, as a similar number of fusion events was detected when the experiment was done using the parent B78 cells, which lack both nectin 1 and HVEM receptors (data not shown). These results show definitively that the regulatory action on gB is by gH/gL and not by gD.
Can we detect any structural differences between wild type and gH2Δ48t/gL2?
Our data highlight functional differences between wt gH2/gL2 and gH2Δ48/gL2 and the two N-terminal truncations of gH. Using a panel of MAbs, we asked if there were structural differences between the proteins. Clearly, the two epitopes within the N terminus were missing in the deletion. However, we have only one MAb with a conformation-dependent epitope to gH/gL (CHL2). CHL2 bound the two proteins equally well (23). Using the same approach that we used for gH1/gL1 and gH2/gL2 (24, 25), we decided to generate additional MAbs against gH2Δ48t/gL2 and see if these antibodies could point us to critical residues important in the function of this molecule.
Accordingly, mice were immunized with purified, soluble gH2Δ48t/gL2 (15) and three hybridoma cell lines, selected by enzyme-linked immunosorbent assay (ELISA) reactivity to gH2Δ48t/gL2, were obtained. None of the MAbs reacted with unrelated proteins that contained a His tag (data not shown), indicating that they were specifically directed against the recombinant form of gH/gL. However, all were gL specific and were designated CΔ48L1, CΔ48L2, and CΔ48L3.
IgGs were purified from the murine ascitic fluid and tested for gH/gL reactivity. Of these, only the CΔ48L3 MAb bound specifically to gL2 as detected by electrophoresis under denaturing conditions (Fig. 3A). By Western blotting, the other two MAbs either were completely nonreactive (CΔ48L1) or recognized a similar pattern with the untransfected samples (CΔ48L2) and were therefore excluded from future experiments. To fine map this MAb, we made two additional C-terminal gL truncations: gL208t (deleting residues 209 to 224) and gL194t (deleting residues 194 to 224) (Fig. 1B). We also included previously studied gL MAbs with linear epitopes within the C terminus (Fig. 1B) (24). These included MAbs CHL34, CHL18, and CHL26. In particular, CHL34 was previously shown to bind two disparate stretches of gL sequence (residues 146 to 165 and 205 to 219) (24). Here, we found that CΔ48L3 and CHL34 MAbs bound both C-terminal gL truncations, indicating that their epitopes lie upstream of gL amino acid 194. We thus narrowed the CHL34 epitope to gL residues 146 to 165. MAb CHL26 bound gL208t but not the short protein gL194t, indicating that its epitope lies between amino acids 195 and 208. Finally, MAb CHL18, which had been characterized as binding gL peptide 182 to 219, was unable to bind either gL C-terminal truncation. Taken together with previously published peptide binding data (24), this approach localized the CHL18 epitope to gL residues 209 to 219 (Fig. 1A and Table 1).
FIG 3 .
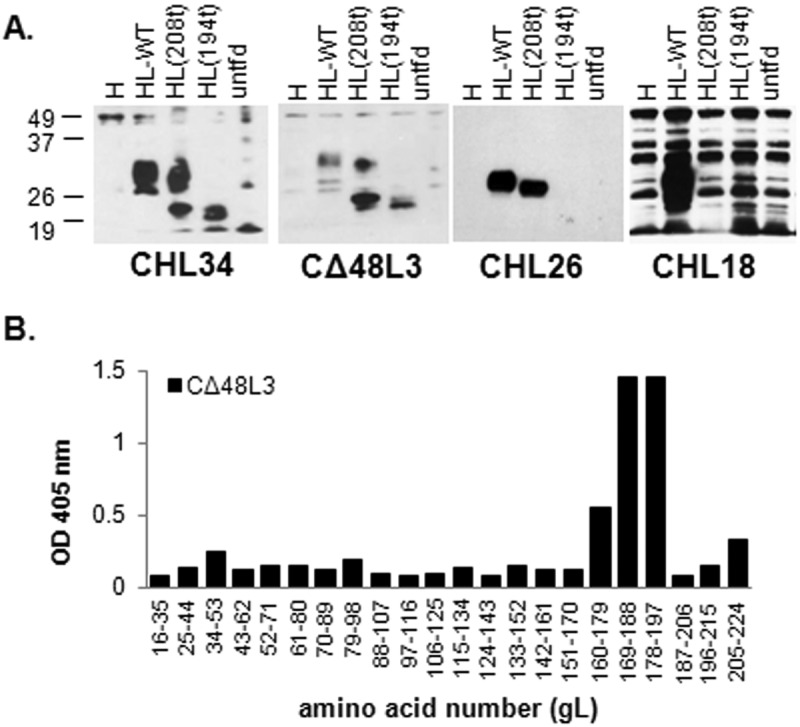
Epitope mapping of gL monoclonal antibodies. (A) Denaturing Western blotting. C10 cells were transfected with control pCAGS plasmid (untfd) or plasmids expressing wt gH2 and either wt gL2 or gL2 deletion mutants gL208 and gL194. Cell lysates were examined for their reactivity with the indicated MAbs. Numbers at left are molecular masses in kilodaltons. (B) Peptide mapping. Each peptide was added to a well of a 96-well streptavidin-coated plate. The wells were blocked and probed with CΔ48L3 MAb. Bound IgG was visualized with goat anti-mouse horseradish peroxidase.
TABLE 1 .
Location of gH and gL epitopesa
| Antibody | Epitope | Domain |
|---|---|---|
| CHL25 | gH 73–92b | H1 |
| CHL2 | gL 116b | H1 |
| CHL18 | gL 209–219c | L |
| CHL26 | gL 195–208c | L |
| CHL34 | gL 146–165c | L |
| CΔ48L3 | gL 173–183c | L |
Positions of selected gH and gL MAb epitopes were defined by either reactivity to gH and gL truncations or peptide mapping.
Reference 24.
This study.
We also used synthetic peptides with 20 residues overlapping to refine our mapping of the CΔ48L3 epitope as previously described (24). MAb CΔ48L3 bound to overlapping peptides that spanned amino acids 169 to 197 (Fig. 3B). Combining the Western blotting and the peptide mapping data, we consider the CΔ48L3 epitope to be within the overlap constituting residues 173 to 183 (Table 1). This stretch of gL residues constitutes a newly defined and distinct epitope within the gL C-terminal tail (Fig. 1). The availability of MAbs to the C terminus of gL enabled us to focus in on changes within gL during fusion regulated by wt gH/gL and gHΔ48t/gL.
Deletion of the gH N terminus uncovers critical residues within the C terminus of gL.
The CΔ48L3 MAb recognizes the C-terminal region of gL, while the gH N terminus (Fig. 1A, dashed green line) is postulated to lie within the gL binding domain of gH (Fig. 1A, green) (15). We wondered if deletion of the gH N terminus (gH2Δ29t/gL2 and gH2Δ48t/gL2) might alter the C terminus of gL and expose new residues, which would account for the gD-independent fusion observed. To explore this idea, we tested the ability of anti-gL MAbs (CHL18, CHL26, CHL34, and CΔ48L3) to block fusion in the context of wt gH2/gL2, gH2Δ29t/gL2, and gH2Δ48t/gL2. Our rationale was that one or more of these MAbs, especially ones to gL, might block fusion with wt gH/gL but not when we used the mutant forms of gH. We also examined this process with two additional MAbs: (i) the conformation-dependent gH/gL MAb CHL2, because, like anti-gL MAbs CHL18 and CHL26, it blocks cell-cell spread (24), and (ii) the anti-gH MAb CHL25, which has no effect on infection or cell-cell fusion (24). Since membrane-anchored and soluble forms of gH2Δ29t/gL2 and gH2Δ48t/gL2 worked equally well (Fig. 2), we carried out these experiments with plasmids expressing membrane-anchored forms of gH/gL and gB.
B78H1-C10 cells expressing gB and wild-type gH2/gL2, gH2Δ29/gL2, or gH2Δ48/gL2 were incubated with the indicated antibody for 2 h before fusion was triggered with gD306t. As expected, the gH MAb CHL25 failed to block cell-cell fusion and MAb CHL2 blocked fusion, regardless of the form of gH/gL present in the transfected cells (Fig. 4A to C). Critically, as previously shown (24), none of the gL MAbs had any effect on fusion of cells transfected with wt gH2/gL2 (Fig. 4A). In contrast, three gL MAbs, CHL18, CHL34, and CΔ48L3, each of which recognizes a different stretch of amino acids of gL (Table 1), blocked fusion of cells transfected with either gH2Δ29/gL2 (Fig. 4B) or gH2Δ48/gL2 (Fig. 4C). Thus, the properties of these MAbs fit with our rationale that deletion of portions of the gH N terminus affects the C terminus of gL so that the MAbs now block. Interestingly, anti-gL MAb CHL26 had an intermediate effect, possibly because its epitope is partially uncovered in the two truncated forms (Fig. 4B and C). We propose that region 146 to 203 of gL is hidden in wt gH/gL and exposed when gH residues 19 to 47 are absent (Fig. 4B and C). We conclude that once these sites of gL (in the context of wt gH/gL) are bound by antibodies, triggering of downstream events is blocked at that step.
FIG 4 .
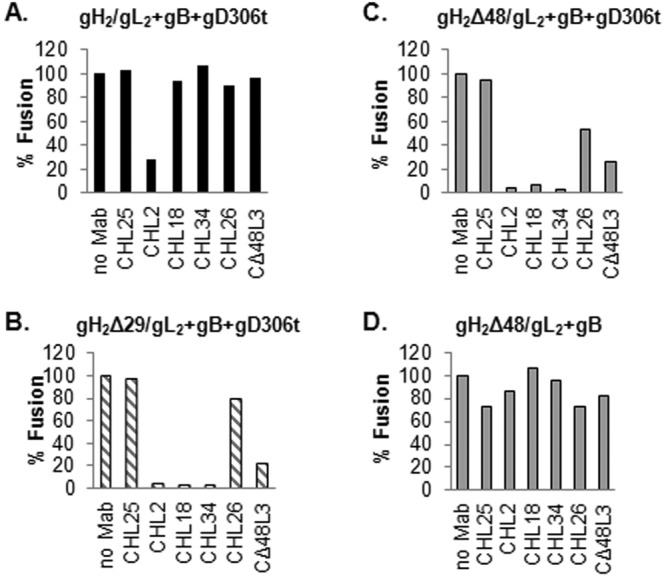
Blocking of cell-cell fusion with gH2 and gL2 monoclonal antibodies. (A to C) C10 cells were transfected with gB and gH2/gL2 (A), gH2Δ29L2 (B), or gH2Δ48/gL2 (C). Cells were incubated with 100 µg/ml of the indicated MAb for 2 h before fusion was triggered with gD2306. Fusion levels were determined based on the number and size of syncytia identified by immunofluorescence. (D) Effect of MAbs on the constitutive function of gH2Δ48/gL2 (in the absence of gD). Fusion levels were expressed as percentages of the no-antibody sample.
The region uncovered by the movement of gH N terminus and gL C terminus is not involved in the interaction with gB.
Removal of gH residues 19 to 47 triggers gH2Δ48/gL2 into a partially activated molecule that can activate gB into a fusogenic state without the need for gD and receptor; we term this property of gH/gL “constitutive fusion” (Fig. 2K). Also, MAbs that map to the uncovered regions can block cell-cell fusion of cells transfected with the four essential glycoproteins (Fig. 4B and C). These data suggest that these regions are involved in an essential step of the fusion process, perhaps the interaction of gH/gL with one of the other glycoproteins (gB or gD). If true, then two scenarios are possible. First, the region uncovered by the movement or deletion of the gH N terminus occurs as a response of gH/gL to the presence of and is important for the interaction with gD. This would mean that none of the antibodies that block cell-cell fusion (Fig. 4C) will have an effect on the constitutive function of gH2Δ48/gL2. In the second scenario, if the uncovered region is important for the interaction of gH/gL with gB, then the constitutive function of gHΔ248/gL2 would be affected by CHL18, CHL34, and CΔ48L3 antibodies.
To distinguish between these two possibilities, C10 cells expressing gB and gH2Δ48/gL2 were incubated with monoclonal antibodies from the beginning of transfection. The samples were maintained in medium containing antibody for the duration of the experiment. The samples were examined for the presence of syncytia at 48 h posttransfection. None of the antibodies tested had an effect on the ability of gH2Δ48/gL2 to promote fusion in the absence of gD. CHL2, CHL18, CHL34, and CΔ48L3, which were very efficient at blocking cell-cell fusion in the presence of gD (Fig. 4C), allowed the same levels of fusion as those in the no-antibody sample (Fig. 4D). We conclude that the region uncovered by deletion of the gH N terminus does not interact with gB but is involved in steps that precede gB binding.
Localization of sites on gH/gL that are important for the interaction with gD and gB.
Earlier, we postulated that there are two functional “faces” on gH/gL, one that interacts with gD and a second that interacts with gB. This argument was based on the ability of two gH/gL virus-neutralizing MAbs, 52S and LP11, to block cell fusion (15). The 52S epitope, with monoclonal antibody-resistant (MAR) residue 536 (26, 27), was proposed to be on the gD face, and the MAb blocks a step in entry initiated by the gD-gH/gL interaction. In contrast, LP11 (MAR residues 86, 168, and 329) (Fig. 1) was proposed to block the gH/gL interaction with gB (gB face) (15). If our arguments are correct, we postulate that gHΔ48/gL can act directly on the gB face, and as such, only the gB face is important. Here, we asked on which side of gHΔ48/gL these MAbs act. However, both LP11 and 52S are type 1 specific, while our anti-gL MAbs CΔ48L3, CHL18, and CHL26 are type 2 specific. We overcame this problem by using a hybrid molecule which contains a type 1 version of gHΔ48 and a type 2 version of gL. Our data show that the deletions produced in type 1 gH/gL (gH1Δ48/gL2) also function in cell-cell fusion and virus entry (23). We have used hybrid molecules in a fusion assay, where wt type 2 glycoproteins were replaced with the equivalent type 1 proteins, with no loss in activity (23, 28). Indeed, we found by immunofluorescence that the resulting hybrid, gH1Δ48/gL2, interacts with both the type 1-neutralizing MAbs 52S and LP11. LP11, 52S, CHL18, and CHL34 epitopes were present on the hybrid molecule (data not shown). When C10 cells were transfected with plasmids for gB along with gH1Δ48 and gL2, addition of gD306t triggered fusion. Moreover, both MAb 52S and MAb LP11 blocked this activity (Fig. 5A), showing that these two type 1-specific epitopes are presented the same way in the type 1/type 2 hybrid as in their respective type 1 counterpart. Importantly, the type-2-specific CHL18 MAb also blocked fusion when this hybrid form was used. Interestingly, CHL34 blocked fusion of cells transfected with gH2Δ48 and gL2 (Fig. 4C) but had no effect on fusion of cells bearing the hybrid gH1Δ48/gL2 (Fig. 5A). Thus, we conclude that this epitope was altered in the hybrid.
FIG 5 .
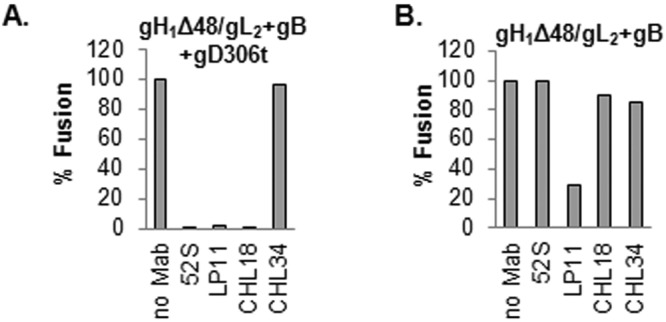
Blocking of cell-cell fusion with gH1 and gL2 monoclonal antibodies. (A) Blocking of fusion triggered by gD306 in C10 cells transfected with gB and hybrid gH1Δ48/gL2. Both type 1 (52S and LP11) and type 2 (CHL18 and CHL34) gH/gL MAbs were used. (B) Effect of MAbs on the constitutive function of gH1Δ48/gL2. Fusion levels were expressed as percentages of the no-antibody sample.
Next we tested whether the hybrid functioned in the absence of gD. Figure 5B shows that membrane-bound gH1Δ48/gL2 triggered fusion, at levels similar to those of gH2Δ48/gL2. We tested the effect of MAbs on fusion. CHL18, which blocked fusion in the presence of gD (Fig. 5A), failed to have an effect on fusion in its absence (Fig. 5B), once again showing that these sites in the C terminus of gL are not critical in the constitutive activity of gH1Δ48/gL2. MAb 52S blocked fusion using the gH1Δ48/gL2 hybrid truncated forms of gH/gL in the presence of gD but failed to block fusion under constitutive conditions (no gD). This result fits our concept that 52S works on the gD side of gH/gL. In contrast, LP11 effectively blocked constitutive fusion (Fig. 5B), suggesting that the gB side of gH/gL (face 2) is, in essence, important at a later time in the process.
In summary, our data support our hypothesis for a stepwise pathway by which the four glycoproteins effect fusion. In this process, gH/gL is in the middle with a face that interacts with gD and a second face that then activates the fusion protein, gB.
DISCUSSION
In this report, our data show that removal of gH residues 19 to 47 (gH2Δ48/gL2) or of residues 19 to 28 (gH2Δ29/gL2) results in formation of a partially activated molecule that can activate gB into a fusogenic state (Fig. 2) in the absence of gD or a gD receptor. However, this constitutive state is not the final active form of gH/gL, as there is more robust fusion when gD and a gD receptor are also present. Additionally, MAbs to epitopes of gL2 that are uncovered in gH2Δ48/gL2 can block cell-cell fusion only when gD and a gD receptor are present (Fig. 4C). Thus, these regions are presumably involved in an essential step in the fusion process that occurs in the natural system.
In spite of the need for four proteins, structural and biochemical data suggest that gB is the sole fusion protein for HSV (16, 18, 29–32). Interestingly, a recent report showed that certain forms of gB that carry point mutations in the cytoplasmic tail (syn mutants) can trigger low levels of cell-cell fusion in the absence of either gD or gH/gL (33). Importantly, no fusion was seen for wt gB in the same conditions. Thus, syn mutants appear to bypass regulation by gD and gH/gL.
A similar phenotype was found in an HSV strain carrying a mutant form of gD that can bind only HVEM (34). When this virus was passaged on nectin 1-expressing cells, a series of missense mutations occurred in gD, each of which restored recognition of nectin 1. However, when it was repeatedly passaged on cells that express a “gD-binding-impaired mutant nectin 1,” the virus compensated for deficient gD-receptor interactions by developing two new mutations in gB (35). These two mutations were sufficient to allow the virus to use a range of unconventional entry receptors (such as nectin 3) and increased the rate of virus entry into different host cells.
Interestingly, by further disabling the gD-nectin 1 interaction, there were compensatory mutations in gH (36) that allowed entry of HSV in the absence of known functional receptors for gD. It would be interesting to determine whether gHΔ48/gL could use weak signaling from gD in the absence of a gD receptor, just like the mutant HSV. These mutations in gB or gH noted by ourselves and by Silverman et al. (33) change the rules of fusion regulation. Each can be viewed as a snapshot in the normal transition of these glycoproteins.
Data presented in this paper focus on the properties of two deletion mutants of gH (gHΔ48/gL and gHΔ29/gL). Why are these molecules able to induce fusion of gB-expressing cells in the absence of gD? What changes does gD normally induce to activate fusion when the wt form of gH/gL is present?
We propose that the N terminus of gH holds the flexible C terminus of gL folded back over itself (Fig. 1). In the presence of gD, we propose that the N terminus of wt gH is displaced (supported by the phenotypes of the two mutants) and the C terminus of gL now springs open to expose both its flexible region, amino acids 208 to 224 (CHL18 epitope), and essential epitopes on the body of gL (CHL34 and CΔ48L3). When residues 19 to 47 are physically removed (gHΔ48/gL), all of these epitopes become exposed so that the MAbs are able to block fusion (Fig. 4B and C). While we cannot exclude the possibility that both full-length wt gH/gL and mutant gHΔ48/gL could interact with lipids and help in the rearrangement of the leaflets to facilitate fusion mediated by gB, wt gH/gL does not induce hemifusion or lipid mixing on its own (37). Furthermore, full-length wt gH/gL does not mediate cell-cell fusion in the absence of the full fusion machinery (unpublished data), and soluble forms of wt gH/gL or gHΔ48/gL do not interact with lipid membranes unless gB is present as well (38).
What is the role of gL?
There is a growing amount of information that points to the importance of gL not as a chaperone for trafficking of gH but for the correct folding of gH (39, 40). However, there is now more evidence that gL has additional functions on its own (this report and references 15 and 41). In the structure of gH/gL, it is clear that stabilization of the N terminus of gH depends on gL and likewise that stabilization of gL is provided by gH. Important epitopes are localized specifically to the flexible C terminus of gL. MAbs specific for this domain are potent inhibitors of viral spread (24) and cell fusion (42).
FRs of the gH/gL complex.
Three functional regions (FRs) in gH/gL defined by the position of neutralizing MAbs are thought to play different roles in gH/gL function (5). FR1 is defined by the mapping studies done for the LP11 epitope, since it is the most potent neutralizing antibody (26), blocks cell-cell fusion (42), and prohibits the interaction of gB with gH/gL (15). FR2 is defined by the position of the 52S epitope (26) and is on the opposite face of gH/gL compared with the position of the LP11 epitope (Fig. 1A). 52S neutralizes virus and inhibits cell-cell fusion, but it does not block the association of gH/gL with gB, as seen by bimolecular complementation (15). These results suggest that FR2 carries out a function that occurs after that carried out by FR1. Lastly, FR3 includes domain H1 of gH as well as the C terminus of gL. The epitopes of MAbs CHL17 and CHL32 are located at the gH N terminus (residues 19 to 38) (23, 24), the epitope of CHL2 is near gH residue 116, and the epitopes for CHL39 and CHL18 are within the gL C terminus (24). The function of FR3 resides in the flexibility of these regions. Thus, although the N terminus of gH (residues 19 to 38) hosts the epitope for the neutralizing MAb CHL17 (24), it can be removed (as in gHΔ29/gL and gHΔ48/gL) without much of an impact on function (this study and reference 23). The C terminus of gL is also proposed to be flexible. Removal of the C terminus of gL (gL194t and gL208t) does not affect the function of the molecule (data not shown). This region of gL is also missing from the resolved crystal structure of gH/gL (15).
Where does gD bind on the surface of gH/gL?
In Epstein-Barr virus (EBV), the interaction between purified gp42 (the functional homologue of gD) and gH/gL has been mapped using purified peptides (43), but no such interaction has been reported for HSV gD and gH/gL. However, a gD-gH/gL interaction was found in cell extracts (44) and in bimolecular complementation studies (10, 19). Here, we found that the epitopes for LP11 and 52S are on opposite sides of gH/gL (Fig. 1A). Although both MAbs block fusion mediated by gD (Fig. 5A), 52S allows for the interaction of gH/gL with gB (15) and only LP11 blocks the constitutive function of gH1Δ48/gL2 (Fig. 5B). This suggests that the LP11 side (FR1) is involved in the interaction with gB (“gB face”), whereas 52S, CΔ48L3, CHL34, and CHL26 are part of a “gD face” (FR2).
Finally, we note that the constitutive ability of gHΔ48/gL to trigger gB into a fusogenic state suggests that it is only a partially activated intermediate, which needs further conformational changes for full activity. However, we cannot exclude the possibility that stimulation of the full fusogenic activity of gB is due to the movement of the gH N terminus and/or the gL C terminus rather than to additional conformational changes. In a virus (Fig. 6), the entry starts with attachment to a cell through a nonessential interaction of gC with proteoglycans followed by binding of gD to either one of its two receptors (45). Receptor binding triggers displacement of the C terminus of gD (Fig. 6, step 1) (12, 14) that exposes a new region of gD, unrelated to receptor binding regions (46). This newly exposed region of gD interacts with gH/gL (47) and results in a conformational change in gH/gL: first by displacement of the N terminus of gH and then by that of the C terminus of gL (these steps are already achieved in gHΔ48/gL). This movement in gH/gL exposes a region in gH/gL which is important for the interaction with gD (step 2). An unknown stimulus determines the insertion of gB fusion loops into the opposing lipid membrane (step 3), followed by an interaction between ectodomains of gB and gH/gL (“gB face” of gH/gL) (Fig. 6, step 4) (10). This leads to a conversion of gB from a pre- to a postfusion state, resulting in fusion of the cell and viral membrane (step 5).
FIG 6 .

Schematic representation of the fusion process. (A) The domain organization and coloring for gB, gD, gHΔ48/gL, and nectin 1 are as published. The unknown prefusion forms of gB and gH/gL are shown in shades of gray. (B) Steps in constitutive fusion. The first two steps presented in panel A can be bypassed when gH/gL is replaced with the partially activated gHΔ48/gL. This truncated form of gH/gL, presumably through the same unknown stimulus, induces the insertion of gB fusion loops into the cellular membrane (step 3) and an interaction between the two glycoproteins (step 4), which leads to fusion (step 5). Further unknown conformational changes are required to convert gHΔ48/gL to a fully activated molecule.
In the absence of structural data for the N terminus of gH and the C terminus of gL, our future studies will focus on such techniques as examination of Fab-gH/gL complexes by cryo-electron microscopy (cryo-EM) and inhibition studies with peptides and other small molecules. A recent report about a soluble form of EBV gH/gL which does not mediate cell fusion shows a very different phenotype than that of soluble gH/gL of HSV, leaving open the question of just how similar or different these glycoproteins will turn out to be (48).
MATERIALS AND METHODS
Cells and plasmids.
Mouse melanoma cells (B78H1) expressing nectin 1 (designated C10) were grown in 5% fetal bovine serum-Dulbecco modified Eagle medium (FBS-DMEM) containing 500 µg/ml G418 (49). Plasmid pPEP98 (gB) was a gift of P. G. Spear (50, 51). Plasmids encoding the HSV-2 glycoproteins pTC510 (gH2 wt), pTC579 (gL2 wt), pTC658 (gH2Δ29), and pTC642 (gH2Δ48) were described previously (23, 28). Plasmid pTC716 (gH1Δ48) was created using QuikChange XL site-directed mutagenesis (Stratagene) as described previously (23). Primers were designed to loop out unwanted residues during amplification of pPEP100 (deletion site underlined): 5′-TTG GGC GTT GCG TGG GGT ACC GGG CGT CTG TGG CTG. The HSV-2 gL C-terminal truncation mutants were created by PCR amplification of pWF318 (28) using a primer carrying the restriction site (underlined) for XhoI and a stop codon (bold) (5′-GGGCTCGAGTCACCGCCGTGGGGGGGCGTTC) paired with a primer complementary to the sequence upstream of the multiple coding site of template vector pcDNA3.1. The PCR product was cloned into vector pcDNA3.1 to generate plasmid pMS651 (gL208). Construct pMS652 (gL194) was created during the PCR mutagenesis used to produce pMS651 (mutation of gL residue Q195 to a stop codon); it retains the gL2 coding sequence from 196 to 208, with an additional stop codon at residue 209 and the XhoI site.
Antibodies used.
Rabbit polyclonal antibody R176 was prepared against purified HSV-2 gH2(803t)/gL2 secreted from transfected 293T cells (28). The CHL series of anti-gH and anti-gL monoclonal antibodies (MAbs) and SS55 and A22 gB MAbs were characterized elsewhere (24, 52). A new gL2 MAb, CΔ48L3, was prepared against baculovirus purified gH2Δ48/gL2 protein as previously described (15, 24).
Production and purification of HSV proteins.
Soluble gD2306t and gH2Δ48/gL2 were purified from baculovirus-infected insect cells (Sf9) as previously described (15, 53, 54). To construct our baculovirus expression vector for soluble gH2Δ29t/gL2, QuikChange mutagenesis was performed. Primers 5′-GTGGGCGTTGCCGGGGGCCCGTGGTTTTTGCACGGTC and 5′-GACCGTGCAAAAACCACGGGCCCCCGGCAACGCCCAC were used on template pTC605 (containing the wt gH2 ectodomain sequence, as well as full-length gL2 [38]). The primers loop out the coding sequence for the first 9 amino acids of the mature gH2 protein, keeping the gH2 signal sequence. Expression and purification of gH2Δ29t/gL2 soluble glycoprotein were essentially the same as those for gH2/gL2 and gH2Δ48t/gL2 (15, 38).
Mapping of gL monoclonal antibodies.
Mapping was done by Western blotting and peptide mapping, as previously described (31, 32). Cell extracts were diluted in lysis buffer (10 mM Tris, pH 8, 150 mM NaCl, 10 mM EDTA, 1% NP-40, 0.5% deoxycholic acid), subjected to electrophoresis on a 10% SDS-polyacrylamide gel, and detected by Western blotting with specific anti-gL MAbs. Table 1 summarizes the location of all MAbs used in this study.
Synchronization and blocking of fusion.
C10 cells were seeded on glass coverslips and transfected with gB, gH2, and gL2 as described previously (10, 18, 47). Six hours posttransfection, cells were incubated with 100 µg/ml of the indicated antibody. Fusion was triggered with 100 µg/ml soluble gD306. At the indicated times, cells were processed for immunofluorescence.
Immunofluorescence.
Cells were fixed with 3% paraformaldehyde and quenched with 50 mM NH4Cl as previously described (10, 18, 47). Cells were washed with phosphate-buffered saline (PBS), incubated with goat serum, and then labeled with anti-gB MAbs A22 and SS55 or anti-gH polyclonal antibody (PAb) R176. After being washed with PBS, coverslips were incubated with Alexa Fluor 594-conjugated goat anti-IgG (Invitrogen) secondary antibody. The coverslips were mounted in ProLong Gold antifade reagent (Invitrogen). Samples were examined by confocal microscopy with a Nikon TE-300 inverted microscope coupled to a PerkinElmer imaging system as previously described (18).
ACKNOWLEDGMENTS
Funding for this project was through NIH grants R01-AI056045 (R.J.E.), R01-AI076231 (R.J.E.), and R37-A8289 (G.H.C.) from the National Institute of Allergy and Infectious Diseases.
We thank our lab members for thoughtful and constructive discussions regarding the manuscript.
Footnotes
Citation Atanasiu D, Cairns TM, Whitbeck JC, Saw WT, Rao S, Eisenberg RJ, Cohen GH. 2013. Regulation of herpes simplex virus gB-induced cell-cell fusion by mutant forms of gH/gL in the absence of gD and cellular receptors. mBio 4(2):e00046-13. doi:10.1128/mBio.00046-13.
REFERENCES
- 1. White JM, Delos SE, Brecher M, Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 43:189–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lamb RA, Jardetzky TS. 2007. Structural basis of viral invasion: lessons from paramyxovirus F. Curr. Opin. Struct. Biol. 17:427–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Spear PG, Longnecker R. 2003. Herpesvirus entry: an update. J. Virol. 77:10179–10185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heldwein EE, Krummenacher C. 2008. Entry of herpesviruses into mammalian cells. Cell. Mol. Life Sci. 65:1653–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 9:369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ryckman BJ, Chase MC, Johnson DC. 2008. HCMV gH/gL/UL128-131 interferes with virus entry into epithelial cells: evidence for cell type-specific receptors. Proc. Natl. Acad. Sci. U. S. A. 105:14118–14123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cai WH, Gu B, Person S. 1988. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion. J. Virol. 62:2596–2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Turner A, Bruun B, Minson T, Browne H. 1998. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol. 72:873–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Atanasiu D, Whitbeck JC, Cairns TM, Reilly B, Cohen GH, Eisenberg RJ. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. U. S. A. 104:18718–18723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carfí A, Willis SH, Whitbeck JC, Krummenacher C, Cohen GH, Eisenberg RJ, Wiley DC. 2001. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol. Cell 8:169–179 [DOI] [PubMed] [Google Scholar]
- 12. Krummenacher C, Supekar VM, Whitbeck JC, Lazear E, Connolly SA, Eisenberg RJ, Cohen GH, Wiley DC, Carfí A. 2005. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 24:4144–4153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hannah BP, Heldwein EE, Bender FC, Cohen GH, Eisenberg RJ. 2007. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J. Virol. 81:4858–4865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lazear E, Carfi A, Whitbeck JC, Cairns TM, Krummenacher C, Cohen GH, Eisenberg RJ. 2008. Engineered disulfide bonds in herpes simplex virus type 1 gD separate receptor binding from fusion initiation and viral entry. J. Virol. 82:700–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat. Struct. Mol. Biol. 17:882–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220 [DOI] [PubMed] [Google Scholar]
- 17. Di Giovine P, Settembre EC, Bhargava AK, Micah A, Lou H, Cohen GH, Eisenberg RJ, Krummenacher C, Carfi A. 2011. Structure of herpes simplex virus glycoprotein D bound to the human receptor nectin-1. PLoS Pathog. 7:e1002277 http://dx.doi.org/10.1371/journal.ppat.1002277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Atanasiu D, Wang ST, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 84:12292–12299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Avitabile E, Forghieri C, Campadelli-Fiume G. 2007. Complexes between herpes simplex virus glycoproteins gD, gB, and gH detected in cells by complementation of split enhanced green fluorescent protein. J. Virol. 81:11532–11537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fuller AO, Santos RE, Spear PG. 1989. Neutralizing antibodies specific for glycoprotein H of herpes simplex virus permit viral attachment to cells but prevent penetration. J. Virol. 63:3435–3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Backovic M, Longnecker R, Jardetzky TS. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 106:2880–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vanarsdall AL, Ryckman BJ, Chase MC, Johnson DC. 2008. Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. J. Virol. 82:11837–11850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cairns TM, Friedman LS, Lou H, Whitbeck JC, Shaner MS, Cohen GH, Eisenberg RJ. 2007. N-terminal mutants of herpes simplex virus type 2 gH are transported without gL but require gL for function. J. Virol. 81:5102–5111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cairns TM, Shaner MS, Zuo Y, Ponce-de-Leon M, Baribaud I, Eisenberg RJ, Cohen GH, Whitbeck JC. 2006. Epitope mapping of herpes simplex virus type 2 gH/gL defines distinct antigenic sites, including some associated with biological function. J. Virol. 80:2596–2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peng T, Ponce-de-Leon M, Jiang H, Dubin G, Lubinski JM, Eisenberg RJ, Cohen GH. 1998. The gH-gL complex of herpes simplex virus (HSV) stimulates neutralizing antibody and protects mice against HSV type 1 challenge. J. Virol. 72:65–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gompels UA, Carss AL, Saxby C, Hancock DC, Forrester A, Minson AC. 1991. Characterization and sequence analyses of antibody-selected antigenic variants of herpes simplex virus show a conformationally complex epitope on glycoprotein H. J. Virol. 65:2393–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Showalter SD, Zweig M, Hampar B. 1981. Monoclonal antibodies to herpes simplex virus type 1 proteins, including the immediate-early protein ICP 4. Infect. Immun. 34:684–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cairns TM, Landsburg DJ, Whitbeck JC, Eisenberg RJ, Cohen GH. 2005. Contribution of cysteine residues to the structure and function of herpes simplex virus gH/gL. Virology 332:550–562 [DOI] [PubMed] [Google Scholar]
- 29. Harrison SC. 2008. Viral membrane fusion. Nat. Struct. Mol. Biol. 15:690–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roche S, Albertini AA, Lepault J, Bressanelli S, Gaudin Y. 2008. Structures of vesicular stomatitis virus glycoprotein: membrane fusion revisited. Cell. Mol. Life Sci. 65:1716–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roche S, Bressanelli S, Rey FA, Gaudin Y. 2006. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313:187–191 [DOI] [PubMed] [Google Scholar]
- 32. Roche S, Rey FA, Gaudin Y, Bressanelli S. 2007. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 315:843–848 [DOI] [PubMed] [Google Scholar]
- 33. Silverman JL, Greene NG, King DS, Heldwein EE. 2012. Membrane requirement for folding of the herpes simplex virus 1 gB cytodomain suggests a unique mechanism of fusion regulation. J. Virol. 86:8171–8184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Uchida H, Shah WA, Ozuer A, Frampton AR, Goins WF, Grandi P, Cohen JB, Glorioso JC. 2009. Generation of herpesvirus entry mediator (HVEM)-restricted herpes simplex virus type 1 mutant viruses: resistance of HVEM-expressing cells and identification of mutations that rescue nectin-1 recognition. J. Virol. 83:2951–2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Uchida H, Chan J, Goins WF, Grandi P, Kumagai I, Cohen JB, Glorioso JC. 2010. A double mutation in glycoprotein gB compensates for ineffective gD-dependent initiation of herpes simplex virus type 1 infection. J. Virol. 84:12200–12209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Uchida H, Cahn J, Srivastav I, Reinhart B, Grandi P, Glorioso JC, Cohen JB. 2013. Novel mutations in gB and gH circumvent the requirement for known gD receptors in HSV-1 entry and cell-to-cell spread. J. Virol. 87:1430–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jackson JO, Longnecker R. 2010. Reevaluating herpes simplex virus hemifusion. J. Virol. 84:11814–11821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hannah BP, Cairns TM, Bender FC, Whitbeck JC, Lou H, Eisenberg RJ, Cohen GH. 2009. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J. Virol. 83:6825–6836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hutchinson L, Browne H, Wargent V, Davis-Poynter N, Primorac S, Goldsmith K, Minson AC, Johnson DC. 1992. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J. Virol. 66:2240–2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roop C, Hutchinson L, Johnson DC. 1993. A mutant herpes simplex virus type 1 unable to express glycoprotein L cannot enter cells, and its particles lack glycoprotein H. J. Virol. 67:2285–2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Matsuura H, Kirschner AN, Longnecker R, Jardetzky TS. 2010. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc. Natl. Acad. Sci. U. S. A. 107:22641–22646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Novotny MJ, Parish ML, Spear PG. 1996. Variability of herpes simplex virus 1 gL and anti-gL antibodies that inhibit cell fusion but not viral infectivity. Virology 221:1–13 [DOI] [PubMed] [Google Scholar]
- 43. Liu F, Marquardt G, Kirschner AN, Longnecker R, Jardetzky TS. 2010. Mapping the N-terminal residues of Epstein-Barr virus gp42 that bind gH/gL by using fluorescence polarization and cell-based fusion assays. J. Virol. 84:10375–10385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gianni T, Amasio M, Campadelli-Fiume G. 2009. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL in part through the C-terminal profusion domain. J. Biol. Chem. 284:17370–17382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Geraghty RJ, Fridberg A, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 2001. Use of chimeric nectin-1 (HveC)-related receptors to demonstrate that ability to bind alphaherpesvirus gD is not necessarily sufficient for viral entry. Virology 285:366–375 [DOI] [PubMed] [Google Scholar]
- 46. Lazear E, Whitbeck JC, Ponce-de-Leon M, Cairns TM, Willis SH, Zuo Y, Krummenacher C, Cohen GH, Eisenberg RJ. 2012. Antibody-induced conformational changes in herpes simplex virus glycoprotein gD reveal new targets for virus neutralization. J. Virol. 86:1563–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Atanasiu D, Whitbeck JC, de Leon MP, Lou H, Hannah BP, Cohen GH, Eisenberg RJ. 2010. Bimolecular complementation defines functional regions of herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J. Virol. 84:3825–3834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rowe CL, Conolly SA, Chen J, Jardtezky TS, Longnecker R. 2013. A soluble form of Epstein-Barr virus gH/gL inhibits EBV-induced membrane fusion and does not function in fusion. Virology 436:118–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miller CG, Krummenacher C, Eisenberg RJ, Cohen GH, Fraser NW. 2001. Development of a syngenic murine B16 cell line-derived melanoma susceptible to destruction by neuroattenuated HSV-1. Mol. Ther. 3:160–168 [DOI] [PubMed] [Google Scholar]
- 50. Okuma K, Nakamura M, Nakano S, Niho Y, Matsuura Y. 1999. Host range of human T-cell leukemia virus type I analyzed by a cell fusion-dependent reporter gene activation assay. Virology 254:235–244 [DOI] [PubMed] [Google Scholar]
- 51. Pertel PE, Fridberg A, Parish ML, Spear PG. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313–324 [DOI] [PubMed] [Google Scholar]
- 52. Bender FC, Samanta M, Heldwein EE, de Leon MP, Bilman E, Lou H, Whitbeck JC, Eisenberg RJ, Cohen GH. 2007. Antigenic and mutational analyses of herpes simplex virus glycoprotein B reveal four functional regions. J. Virol. 81:3827–3841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sisk WP, Bradley JD, Leipold RJ, Stoltzfus AM, Ponce de Leon M, Hilf M, Peng C, Cohen GH, Eisenberg RJ. 1994. High-level expression and purification of secreted forms of herpes simplex virus type 1 glycoprotein gD synthesized by baculovirus-infected insect cells. J. Virol. 68:766–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rux AH, Willis SH, Nicola AV, Hou W, Peng C, Lou H, Cohen GH, Eisenberg RJ. 1998. Functional region IV of glycoprotein D from herpes simplex virus modulates glycoprotein binding to the herpesvirus entry mediator. J. Virol. 72:7091–7098 [DOI] [PMC free article] [PubMed] [Google Scholar]