

ABSTRACT
Despite its importance to the host, the flower microbiome is poorly understood. We report a culture-independent, community-level assessment of apple flower microbial diversity and dynamics. We collected flowers from six apple trees at five time points, starting before flowers opened and ending at petal fall. We applied streptomycin to half of the trees when flowers opened. Assessment of microbial diversity using tag pyrosequencing of 16S rRNA genes revealed that the apple flower communities were rich and diverse and dominated by members of TM7 and Deinococcus-Thermus, phyla about which relatively little is known. From thousands of taxa, we identified six successional groups with coherent dynamics whose abundances peaked at different times before and after bud opening. We designated the groups Pioneer, Early, Mid, Late, Climax, and Generalist communities. The successional pattern was attributed to a set of prevalent taxa that were persistent and gradually changing in abundance. These taxa had significant associations with other community members, as demonstrated with a cooccurrence network based on local similarity analysis. We also detected a set of less-abundant, transient taxa that contributed to general tree-to-tree variability but not to the successional pattern. Communities on trees sprayed with streptomycin had slightly lower phylogenetic diversity than those on unsprayed trees but did not differ in structure or succession. Our results suggest that changes in apple flower microbial community structure are predictable over the life of the flower, providing a basis for ecological understanding and disease management.
IMPORTANCE
Flowering plants (angiosperms) represent a diverse group of an estimated 400,000 species, and their successful cultivation is essential to agriculture. Yet fundamental knowledge of flower-associated microbiota remains largely unknown. Even less well understood are the changes that flower microbial communities experience through time. Flowers are particularly conducive to comprehensive temporal studies because they are, by nature, ephemeral organs. Here, we present the first culture-independent time series of bacterial and archaeal communities associated with the flowers of apple, an economically important crop. We found unexpected diversity on apple flowers, including a preponderance of taxa affiliated with Deinococcus-Thermus and TM7, phyla that are understudied but thought to be tolerant of an array of environmental stresses. Our results also suggest that changes in microbial community structure on the apple flower may be predictable over the life of the flower, providing the basis for ecological understanding and disease management.
Introduction
Despite the immense importance of flowers in agriculture and the biosphere, little is known about their microbial ecology (1). The flower microbiome, which encompasses microorganisms associated with flowers, is a rich habitat for microbial life. In contrast to the intensively studied leaf surface (2–7), little is known about the flower microbiome.
Flowers provide ephemeral but nutrient-rich and protective habitats for microorganisms. In temperate climates, warm spring temperatures induce buds to open. Blooming exposes the male (stamen, including the anther and filament) (Fig. 1) and female (pistil, including the stigmata, style, and ovary) reproductive parts and inside surfaces of petals to the environment and microorganisms. Flower stigmas, in particular, exude sugars and amino acids that support a relatively large microbial load compared to that of other flower parts (8, 9). Previous studies have explored the culture-independent diversity of yeasts and fungi on flowers or in nectar (e.g., see references 10–12), culture-based diversity of bacteria on flowers (e.g., see references 9 and 12), and, quite recently, culture-independent diversity of bacteria in nectar (13), but none have considered the community diversity of flower-associated bacteria and archaea through time using culture-independent approaches.
FIG 1 .

Study design and flower anatomy. (A) Time course for sampling the apple microbiome. Fifteen pooled flowers from each of six Gala apple trees were collected at five time points over the life span of the flowers, for a total of 30 samples. Collection of phenologically matched flowers began before they opened and ended when petals fell. Arrows indicate streptomycin application. A precipitation event (1.30 cm) occurred on 30 April 2010. (B) Anatomy of a flower. All flower parts, including sepals, pistil, petals, and stamens, were included in the sampling. The current study provides a picture of the microbial communities that includes all of these flower compartments. (Reprinted from reference 81 with permission of the publisher.)
The apple flower microbiome is perhaps the best-studied floral community because it is the site of infection by Erwinia amylovora, which is the cause of a costly disease, fire blight (e.g., see references 14 and 15). Apple flower longevity from bloom to senescence is at most two weeks, occurring in the early spring in temperate climates, and much research has been devoted to understanding the relationship between the fire blight pathogen and possible antagonistic species and flower biology (16). As one example, apple flower age is important for the growth rate of E. amylovora on the flower (17, 18). Fire blight prevention strategies include application of antibiotics, such as streptomycin, and bacterial antagonists that compete with E. amylovora on floral stigmata and nectaries. E. amylovora resistance to antibiotics (19) and mixed effectiveness of antagonists (20–22) make fire blight a persistent problem. A thorough understanding of the flower microbiome may reveal new antagonists as well as insights about the identities, dynamics, and interplay of commensal microorganisms with the plant host, pathogens, and pollinators. Just as culture-independent approaches, particularly those based on deep sequencing, are reshaping our understanding of the fundamental biology of complex microbial systems, such as those in soil and the human body (23, 24), the understanding of plant reproduction and flower function may be advanced by similar study of the flower microbiome.
In this study, we present a culture-independent analysis of diversity and structure of microbial communities associated with apple flowers through time and specifically address the two following fundamental questions. (i) How do microbial communities change as flowers age? (ii) Does streptomycin application alter the composition or dynamics of the microbial community on the flower?
RESULTS
Tag pyrosequencing revealed diverse microbial consortia on apple flowers.
We assessed changes in apple flower bacterial and archaeal communities through time and after streptomycin application (Fig. 1) using tag pyrosequencing of 16S rRNA genes. After processing and quality control, we detected 1,677 operational taxonomic units (OTUs; 97% sequence identity) from 50,865 tag sequences. The most abundant OTUs were most closely affiliated with the phyla Deinococcus-Thermus, TM7, Bacteroidetes, Firmicutes, and Proteobacteria. The community had a few very abundant OTUs, identified as members of Deinococcus-Thermus and TM7 (Fig. 2), and the remaining OTUs were relatively rare. Unidentified bacteria comprised 26% of the data set (431 OTUs), suggesting that many flower community members are uncharacterized.
FIG 2 .

Distribution of taxon abundances among OTUs (singletons omitted) detected on apple flowers.
We first asked whether communities from different trees had different structures. Community structures can be distinguished by centroid (mean structure) or by spread (variability in structure) (see Fig. S1A posted at http://www.yale.edu/handelsmanlab/resources/index.html) (25–27). Across individual trees (inclusive of all time points), we did not detect differences in centroid (global PERMANOVA P value of 0.03, but all post hoc pairwise test P values were >0.05 after Bonferroni’s correction) or spread (global PERMDISP P = 0.34). This indicated that the trees had comparable centroids and spread and could serve as independent biological replicates.
Apple flower microbial communities exhibited temporal patterns.
Analysis of temporal patterns showed that phylogenetic diversity increased between 27 and 29 April and then stabilized (Fig. 3a). Additionally, variability in community structure was high before flowers opened, decreased when flowers opened, and then increased to prebloom levels through the rest of the life of the flower (Fig. 3b), suggesting that open flowers may have a more narrowly defined community than closed flowers. Though there were differences between some time points, there were no general temporal trends in evenness or richness (see the results in Text S1 in the supplemental material). We also detected differences in community structure across time points, but there was no effect of streptomycin treatment or of a time-treatment interaction (Table S1). Together, these results and others (Fig. S2) suggest that changes in apple flower microbial communities are patterned over time.
FIG 3 .

Temporal trends in the apple flower microbiome. For each time point, n = 6 (1 sample of DNA extracted in bulk from 15 flowers from each of six trees). The inner-quartile ranges are shown by the box boundaries, nonoutlier extremes are shown by the whiskers, the median is shown by the thick middle line, and outliers are shown by the outliers’ black points. Statistics were summarized across each of six trees sampled at each time point, and the communities were analyzed at the 97% OTU level. Trees 4, 5, and 6 were sprayed with streptomycin on 29 and 30 April 2010. (a) Rarefied Faith’s phylogenetic diversity of microorganisms. There were 10 resamples at a depth of 1,531 sequences for each tree at each time point. Letters indicate significant differences in phylogenetic diversity across days, assessed by analysis of variance (F = 101.56, 4 degrees of freedom [df], P < 0.001) and post hoc testing with Tukey’s HSD test (P < 0.05). (b) Variability in community structure (assessed by analysis of beta dispersion, a metric of variability). Though modest differences were detected (multivariate homogeneity of group dispersions; F = 2.34, 4 df, P = 0.08), a post hoc test revealed that only 27 and 29 April were different (Tukey’s HSD test; P = 0.09).
Next, we asked whether the temporal community patterns could be explained by environmental variables. Time, mean temperature, high temperature, and high wind speed were correlated with community patterns (all P values were <0.05) (see Table S2 in the supplemental material), although time and temperature were also correlated to each other (Pearson’s r = 0.51, P = 0.003). Precipitation had no explanatory value (P = 0.56), and neither did flower biomass (P = 0.31). This suggests that some environmental variables, such as wind and temperature, but not flower biomass, contributed to the community-level variability (see the supplemental results in Text S1). Flower age and certain environmental conditions appeared to be important determinants of microbial community structure, but these factors cannot always be separated because the environment induces changes in host biology.
Apple flower microbial communities exhibited succession.
Modest phylum-level changes in the community (see Fig. S1 in the supplemental material) suggested that the observed temporal patterns (Fig. 3) may be more apparent at the family, genus, or OTU level than at the phylum level. We hypothesized that there were groups of OTUs that had similar occurrence patterns, but we realized that these patterns may encompass a variety of distinct taxon dynamics (Fig. S2A posted at http://www.yale.edu/handelsmanlab/resources/index.html). To uncover possible trajectories, we performed a hierarchical cluster analysis to identify cooccurring taxa (supplemental methods in Text S1 and Fig. S3A posted at http://www.yale.edu/handelsmanlab/resources/index.html). We found that coherent occurrence patterns of OTUs on apple flowers were well described by the six most aggregated clusters (Fig. 4a). Furthermore, the peak abundances of the OTUs within these clusters occurred at different sampling times (Fig. 4b), and this temporal pattern was repeatable across trees, as indicated by the small error bars around the mean of six trees (Fig. 4b). We examined dynamics of each of the five most prevalent OTUs within each cluster and observed incremental increases and decreases of their abundances through time (Fig. 5). These patterns reflect the dynamics that define succession (Fig. S2A, panel f). Therefore, from these clusters we designated “successional groups” of OTUs that had coherent dynamics and distinct maxima at particular sampling times.
FIG 4 .

Discovery, dynamics, and characteristics of apple flower successional groups. (a) Hierarchical clustering (complete linkage-based Bray-Curtis similarities among OTUs defined at 97% sequence identity) to determine OTUs having coherent dynamics. The analysis was conducted on 1,677 OTUs, each represented as a branch tip of the dendrogram. The y axis height represents within-cluster Bray-Curtis similarity. (b) Successional group dynamics indicating the mean relative abundance of members belonging to each group over the lifetime of apple flowers. Error bars indicate the standard deviation around the mean of 6 trees.
FIG 5 .

Dynamics of the five most prevalent OTUs detected for each successional group. OTU IDs correspond to the taxonomic assignments in Table 1. Error bars are standard errors around the mean OTU’s relative abundance at one time point across six trees. (a) Pioneer taxa; (b) Early succession taxa; (c) Mid succession taxa (secondary axis is for OTU 6932); (d) Late succession taxa; (e) Climax taxa; (f) Generalist taxa.
Each successional group corresponded to a different time point and thus also to a different flower age (Fig. 4b and 5). There was a group with members that peaked before flowers opened (“Pioneer”), followed by a group with members that peaked on the day that flowers opened (“Early”). “Mid” group members peaked when flowers had been open for 2 days, and “Late” group members peaked when flowers had been open for 3 days and included a high abundance of Lactobacillus and Acetobacter taxa, whose occurrences aligned with previously reported conditions of flower decomposition by yeast (28–30). “Climax” group members were most abundant at petal fall. The anomalous group, “Generalists,” contained members with mildly fluctuating dynamics and generally persistent occurrences through time. In summary, distilled from 1,677 taxonomic units, six successional groups described the temporal dynamics of microbial communities on apple flowers.
OTUs of unidentified bacteria were prevalent in all groups, and TM7 and Deinococcus-Thermus OTUs were prevalent in five of the six groups (Table 1). We explored the phylogenetic distribution of successional group members and found that no one phylogenetic lineage was particularly abundant in any group (Fig. 6) except that some taxa affiliated with TM7 were more common in Early succession than at other stages.
TABLE 1 .
Ten most abundant OTUs within each successional group, ranked by their abundances and identified by the closest match to reference sequences in the Ribosomal Database Project
| Successional group and OTU ID | Family, genusa |
|---|---|
| Pioneer | |
| 2415 | Unidentified TM7* |
| 7413 | Unidentified TM7* |
| 2210 | Enterobacteriaceae, Pantoea |
| 5546 | Unidentified TM7* |
| 7433 | Deinococcaceae, Deinococcus |
| 4040 | Enterobacteriaceae, Escherichia/Shigella |
| 1844 | Unidentified Bacteria* |
| 7142 | Unidentified Bacteria* |
| 7797 | Enterobacteriaceae, Buttiauxella |
| 2242 | Cytophagaceae, Hymenobacter |
| Early | |
| 5552 | Unidentified TM7* |
| 6696 | Unidentified Bacteria* |
| 2168 | Unidentified TM7* |
| 7816 | Unidentified TM7* |
| 2035 | Unidentified TM7* |
| 4187 | Unidentified TM7* |
| 4923 | Flavobacteriaceae |
| 888 | Methanosarcinaceae, Methanosarcina |
| 2215 | Unidentified Bacteria* |
| 5728 | Unidentified Bacteria* |
| Mid | |
| 6932 | Trueperaceae, Truepera |
| 7041 | Trueperaceae, Truepera |
| 4964 | Unidentified Bacteria* |
| 4462 | Deinococcaceae, Deinococcus |
| 1372 | Unidentified Chloroflexi* |
| 2690 | Deinococcaceae, Deinococcus |
| 5664 | Unidentified Chloroflexi* |
| 2678 | Unidentified Bacteria* |
| 210 | Trueperaceae, Truepera |
| 4754 | Trueperaceae, Truepera |
| Late | |
| 6950 | Lactobacillaceae, Lactobacillus |
| 2879 | Acetobacteraceae, Acetobacter |
| 5655 | Unidentified Bacteria* |
| 7310 | Deinococcaceae, Deinococcus |
| 3969 | Unidentified TM7* |
| 6504 | Unidentified Bacteria* |
| 2311 | Trueperaceae, Truepera |
| 6218 | Unidentified TM7* |
| 3993 | Acetobacteraceae, Acetobacter |
| 6961 | Unidentified TM7* |
| Climax | |
| 1241 | Unidentified Chloroflexi* |
| 4478 | Enterobacteriaceae |
| 5188 | Chitinophagaceae, Terrimonas |
| 6802 | Unidentified TM7* |
| 2132 | Unidentified TM7* |
| 556 | Burkholderiaceae, Burkholderia |
| 4012 | Unidentified Bacteria* |
| 3270 | Micrococcaceae, Arthrobacter |
| 7247 | Deinococcaceae, Deinococcus |
| 2147 | Enterobacteriaceae |
| Generalist | |
| 2082 | Deinococcaceae, Deinococcus |
| 15 | Cytophagaceae, Hymenobacter |
| 4290 | Unidentified TM7* |
| 11 | Trueperaceae, Truepera |
| 343 | Unidentified TM7* |
| 4767 | Xanthomonadaceae |
| 5346 | Unidentified Bacteria* |
| 6360 | Deinococcaceae, Deinococcus |
| 333 | Unidentified Bacteria* |
| 6539 | Intrasporangiaceae, Knoellia |
Asterisks indicate that the OTU could not be identified to at least the family level.
FIG 6 .

Distribution and representation of apple flower microorganisms. The tree includes OTUs identified at least to the class level. The branch shading corresponds to the different phyla detected (phylum names indicated in boxes). The branch tip colors correspond to the successional groups of each taxon (OTUs with 97% tag sequence identity). The height and label of the colored bar indicate the relative abundance of each OTU in the data set, and unlabeled OTUs have relative abundances below 0.01. Note that Fig. 6 is a tool for visualizing the representation of OTUs among phyla and is not intended to depict evolutionary relationships.
Because community structure varied over time (Fig. 3b) and 90% of the OTUs were represented by fewer than 50 sequences (Fig. 2), we hypothesized that the less-abundant OTUs were also transient, or detected at relatively few points in the series. Transient organisms may be those that arrive on flowers but do not successfully colonize. We found a relationship between persistence (the consistency in detecting a taxon through time) and prevalence (the abundance of a taxon) such that transient OTUs also tended to have low abundance (Fig. 7a and c). This suggests that at each time point, rare, transient OTUs were replaced in the community by other transient OTUs, indicating high community turnover. Prevalent OTUs were more often persistent (Fig. 7a and c) and increased and decreased in abundance gradually (Fig. 5). Therefore, the persistent and prevalent OTUs changed over time, contributing to successional patterns (Fig. 5). Many prevalent OTUs were affiliated with Deinococcus-Thermus, TM7, and Bacteriodetes, and many rare OTUs were affiliated with Proteobacteria and Actinobacteria (Fig. 6).
FIG 7 .

Characteristics of community structure that contribute to changes in beta diversity though time. (a) Prevalent members of the community are detected often (persistent through time and prevalent across trees), while rare members are detected infrequently (transient). Each OTU is a point, the blue line is the log-linear model (adjusted r2 = 0.68, slope = 6.49, P < 0.001), and gray shading represents the standard error. Percent occurrence is out of 30 total observations (six trees and five sampling times). (b) Partition of temporal beta diversity (measured as Sørenson’s similarity [Sor]; blue) into components that represent taxa replacement (Simpson’s similarity [Sim]; green) and nestedness (Nes; red). Each point is multivariate community similarity calculated for a time series from one tree, and the analysis was repeated for each of six trees and at various levels of cutoff to remove less prevalent OTUs. The line is an average across the trees; gray shading represents the standard error. (c) Prevalent members of the community are detected often (persistent), while rare members are detected infrequently (transient). Each OTU is a point, the color shows the tree in which the OTU was detected, and the lines are the log-linear models for each tree (all adjusted r2 values are >0.45, slopes range between 3.99 and 5.01, all P values are <0.001). Percent occurrence is out of five time points per tree. (d) Nestedness metric based on overlap and decreasing fill (NODF). Each point is calculated for a time series from one tree, and the analysis was repeated for each of six trees and at various levels of cutoff to remove less-prevalent members. The line is an average across the trees; gray shading represents the standard error.
Nestedness describes changes among the constant members of a community, and it occurs when the membership of a community is a subset of a richer community (31, 32). Replacement describes the addition of new members that were not previously detected to a community (31, 32). To understand the contributions of both replacement and nestedness to temporal changes in community structure, we partitioned beta diversity into these components (31, 32) and then additionally quantified nestedness (33). However, because we knew that transient community members were often rare (Fig. 7a and c), we also quantified how rare taxa influenced the contributions of nestedness and replacement to beta diversity (using MultiCoLA) (see the supplemental methods in Text S1) (34). Omitting rare taxa did not affect beta diversity, as each reduced data set remained strongly correlated with the full data set despite removal of up to 90% of the least-abundant OTUs (Table S3). This analysis confirmed that rare OTUs contributed little to the overarching community patterns.
Sørenson’s similarity (Sør) was used as an overall metric of beta diversity, and was partitioned into the additive components of Simpson’s similarity (Sim), and nestedness (Nes). The Sim component is attributed to replacement of community members (sometimes referred to as turnover), and the Nes component is attributed to one community having a subset membership of a richer community. We calculated multivariate Sør, Sim, and Nes for each tree through time. Both nestedness and replacement contributed to temporal changes in community composition (Fig. 7b). Replacement made a higher contribution to beta diversity than did nestedness, likely because of the large proportion of rare and transient OTUs. As more rare OTUs were omitted from the data set, the relative contribution of nestedness increased while the contribution of replacement decreased (Fig. 7d). These analyses of replacement and nestedness, which consider only presence and absence of taxa, are complementary to the previous analyses that also considered changes in relative abundance of taxa.
Cooccurrence network reveals potential interactions among flower taxa through time.
After identifying a set of persistent and prevalent OTUs that exhibited clear temporal patterns on the apple flower, we asked whether these taxa were potentially interacting with each other, which would suggest that microbial interactions on the apple flower were, in part, responsible for the observed temporal patterns. Thus, we investigated prevalent OTUs for associations through time using local similarity analysis (LSA) (35, 36), a hypothesis-generating tool used to identify pairs of OTUs that exhibited statistically significant cooccurrences. From the subset of significant associations, we built a network for each tree through time (see Table S4 in the supplemental material) and also in aggregate through time by using each tree as an independent replicate time series in the analysis (referred to as the “full network”) (Table S4; Fig. 8).
FIG 8 .
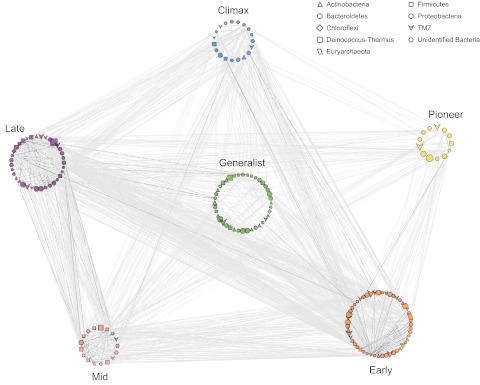
Association network of prevalent taxa (top 20% most abundant), color coded by their successional group. OTUs are nodes, and node size is relative to its number of sequences, with larger shapes indicating more-abundant OTUs. Node shape indicates the phylum-level affiliation of the OTU. Edges (lines) are significant associations assessed by local similarity analysis (P < 0.001, q < 0.05). Light-gray edges are time-lagged associations, while dark edges are unilateral associations (see the supplemental results in Text S1 in the supplemental material). The distance between two OTUs (length of the edge) was optimized to distinguish associations between successional groups.
There were 175 OTUs (out of 336 OTUs among the most prevalent 20%) that had associations, defined by significant local similarity scores, representing a total of 1,532 associations. The visualization of the full network clearly shows that all successional groups were connected (Fig. 8). The Pioneer and Climax groups had the fewest associations between them, a finding which is explained by the time between their peak abundances. The Early-affiliated OTUs had the most associations, followed by the Late, Generalist, Climax, Mid, and Pioneer groups.
Though all of the OTUs included in the LSA were among the most prevalent on the flowers, there was no relationship between the abundance of an OTU and its number of associations (see Fig. S3 in the supplemental material). Furthermore, there were no significant associations detected for the most abundant OTU (Deinococcus-Thermus OTU 6932) (Fig. 5c, filled symbols). Though there were some “hubs” of OTUs that had many associations (Fig. 8), the removal of any one OTU from the network did not substantially change network properties (Fig. S4). This result suggests that the apple flower microbial community is generally robust, as there were taxa that maintained the overall network connections when other taxa were removed. The network analysis uncovered a small subset of OTUs (175 out of 1,677 observed in the full data set) that are likely important for microbial community interactions and dynamics on these apple flowers. Further discussion of other aspects of the flower networks, including association direction and time delay, clustering, and “small world” nature, is provided in the supplemental results in Text S1.
Streptomycin modestly reduced phylogenetic diversity of microorganisms on flowers.
Communities on streptomycin-sprayed or unsprayed flowers did not differ in their centroid, variability, evenness, or number of OTUs (all P values were >0.10). However, microbial communities of streptomycin-sprayed flowers had lower phylogenetic diversity than flowers that were not sprayed (not sprayed, mean = 12.8, standard deviation = 2.41; sprayed, mean = 11.9, standard deviation =3.23; t test P = 0.005). Overall, these results suggest minimal community-level responses to streptomycin.
DISCUSSION
In this work, we characterized the apple flower microbiome using culture-independent methods. We detected abundant members of TM7 and Deinococcus-Thermus, phyla not previously known to be associated with flowers. We also found a succession of microorganisms on the flowers, and this successional pattern was reproducible across six trees. There were microbial taxa whose abundances followed the same temporal patterns over the life of the flower, indicating successional groups. There was a consistent assemblage of taxa present from flower opening through petal fall, and differential dynamics of prevalent microbes likely underpinned the successional pattern. There was also an anomalous group of persistent taxa, designated “Generalists,” that were not the most abundant but nonetheless comprised a substantial portion of the community. Together, these Generalists and prevalent members of successional groups provide the first clues as to the existence of a core microbiome for apple flowers, defined by prevalence, persistence, and associations (37). Whether or not the communities described here are indeed a core microbiome common to apple flowers will be determined by further studies in additional orchards over different years.
Unexpected diversity in the apple flower microbiome.
In this study, the apple flower microbiome contained diverse representation among bacteria, including many taxa affiliated with Proteobacteria, Actinobacteria, and Bacteroidetes. The apple flower microbiome represented greater phylogenetic diversity than observed previously on leaves (38–41), which are the best-studied aspect of the above-ground plant microbiome. The most-abundant taxa on apple flowers were affiliated with the understudied phyla Deinococcus-Thermus and TM7. Notably, these phyla were not detected in 16S rRNA gene clone libraries of leaf surfaces in the same orchard (42); however, leaf sampling was done later in the season and in a different year than the sampling for the present study. Both Deinococcus-Thermus and TM7 taxa were detected in 16S rRNA gene libraries derived from the microbial community associated with Populus deltoides (cottonwood) leaves (38), although they were not as prevalent as those detected on apple flowers described here. Members of Deinococcus-Thermus and TM7 are known for their ability to withstand environmental stresses (43, 44). Future investigation of flower microbial communities will likely provide examples of Deinococcus-Thermus and TM7 genomes, proteomes, and metabolomes, which will contribute to the design of appropriate culture conditions for as-yet-uncultivated members of both phyla. A single-cell approach to investigating the microbial diversity on flowers may be especially fruitful for discovering novel organisms and their genes.
TM7 was the only phylum that exhibited modest clustering by successional group, appearing abundantly in the Early successional group. We speculate that TM7 taxa are contenders for colonization of closed flowers, where they survive but do not grow until flowers open and then grow rapidly and competitively on open flowers. Interestingly, members of the TM7 phylum are thought to carry a ribosomal mutation conferring resistance to streptomycin (43), which may explain their increased abundance following streptomycin application.
An OTU affiliated with Methanosarcina, an archaeal methanogen, was the most prevalent member of the Early successional group. Methanosarcina are anaerobes and likely persist in microaerophilic environments, such as biofilms on or in the flower that are protected from oxygen, possibly within a surface biofilm (e.g., see references 45 and 46). Members of the Methanosarcina-affiliated OTU may be subsisting on methanol, a by-product of flowering plant cell wall synthesis (47) that is important for growth of an abundant leaf microorganism, Methylobacterium (48). Methanol is released in higher concentrations from young leaves than from old leaves (49), a trend which aligns with the high abundance of the Methanosarcina OTU at early flowering when petals were expanding. Though the primers that were used in this study target both Archaea and Bacteria, it is unlikely that they target every archaeal taxon. Thus, it is possible that other archaeal taxa were present but undetected. Given the unexpected diversity of bacterial taxa on flowers, an interesting next step may be to investigate the diversity of the archaeal flower microbiome with primers designed to maximize representation of archaeal lineages.
A recent study of microorganisms in nectar found sizeable representation of gammaproteobacteria, including Acinetobacter and Pseudomonas (13). Although we detected both of these genera on apple flowers, they were not among the most abundant. This hints that the Deinococcus-Thermus and TM7 taxa inhabit a flower compartment other than the nectar. In agreement with past work (13), we found a preponderance of unidentified bacteria on apple flowers, demonstrating that flowers harbor novel microbial taxa and should be targeted for future studies of diversity and bioprospecting. Unidentified bacteria were among the prevalent members of each successional group.
Unraveling the succession: pattern, redundancy, and noise.
We applied a set of analyses that permitted detection of a successional pattern of apple flower microbiota, explored the consequences of community structure underlying that pattern, quantified the contribution of taxa to both pattern and noise, and described the possible environmental conditions and microbial interactions that likely drive the pattern. The statistical toolbox used here may be generally useful for understanding temporal patterns in microbial communities from other habitats.
Each apple tree carried representative taxa from each successional group, and the dynamics of these groups were reproduced on all trees. Thus, in aggregate, the members of the successional groups provided the relevant ecological units for observing succession. The reproducible dynamics of different but analogous flower communities suggest some level of ecological, if not functional, redundancy among taxa (50). The robust network, in which removal of any one OTU did not “break” the overall properties, further suggests redundancy in associations among OTUs.
Variability in community structure was lower on open flowers than on closed ones, but phylogenetic diversity was higher on open flowers. This interesting dynamic might be explained in the context of the successional patterns. Before opening, a flower likely has a lower microbial load because it has been exposed to the environment for less time and possibly also because it provides fewer resources for early colonizers. This may result in a closed-flower community comprised of a few random taxa that immigrate to the flower surface but do not necessarily grow, leading to high variability in the community structure of closed flowers. After flowers open, a few successful r-strategists may take advantage of the “clean slate” provided by the fresh stigma. Thus, the most competitive members would outgrow less-fit competitors, potentially increasing similarity between communities and decreasing multivariate dispersion. The increase in phylogenetic diversity after flowers open is explained by the dynamics of two divergent phyla: TM7-affiliated taxa were most prevalent in Early succession, and Deinococcus-Thermus-affiliated taxa were very abundant in Mid succession, leading to increased phylogenetic diversity because of the prevalence of these divergent phylotypes.
We detected a cohort of rare, transient taxa. Transient taxa are those that are dispersed to a habitat but do not persist on the scale of the experiment. These taxa may have been generally less competitive within the flower habitat. For instance, some apple and pear flower bacteria are sensitive to the sugar concentration and composition in nectar and grow only given their optimal ranges (51–53). Transient taxa likely contributed to baseline variability and did not play substantial roles in determining the successional patterns. Indeed, removing rare community members (thereby removing noise) had no impact on the overall community patterns. By partitioning beta diversity into components of nestedness and replacement, we demonstrated that replacement (the influx of new members to a community, as defined by references 31 and 32) was the largest contributor to community variability. At the same time, many prevalent taxa were also persistent, and the prevalent members within a successional group dominated the communities at each time point. Therefore, we attribute the clear successional pattern to prevalent and persistent taxa and the background variability to rare and transient taxa.
Drivers of microbial succession in the flower microbiome.
As has been observed in the phyllosphere (54, 55), the structure and dynamics of the flower microbiome are likely to be driven by dispersal (emigration and immigration), environmental conditions, host biology, and interactions among microorganisms. Dispersal includes immigration to and emigration from the flower surface (7, 54, 56), processes driven by wind (e.g., see reference 57), and rain splash (e.g., see reference 58), with pollinators also playing a role in facilitating immigration (e.g., see references 59–61). In the work presented here, wind was correlated with community patterns through time and was a likely agent of immigration to and emigration from the flowers.
It is difficult to separate the effects of flower development from sampling date, as our experiment was designed to ensure synchrony in development. Future studies will vary the age of the flower and the day of sampling so that flowers in multiple developmental stages are collected on the same day. This would be a challenging experiment, at least in years with a relatively short bloom period, as the majority of flowers on apple trees open in response to temperature cues and therefore often have synchronous developmental dynamics.
Temperature, a driver of microbial growth (58, 62), was correlated with changes in microbial community structure. Growth of prevalent taxa was indicated by the gradual increases and decreases in abundance and shared dynamics within a successional group. Morphological changes in the flower stigmata and nectaries (17, 63–65), which are often correlated with changes in temperature (e.g., see reference 8), create new niches for microbial colonization.
Associations identified using local similarity analysis can be used to generate hypotheses that can be addressed in follow-up studies. For example, Generalist taxa and “hub” taxa (those with a relatively large number of associations with other taxa) may be targeted for construction of model communities for laboratory exploration of microbial interactions on the apple flower and with the plant host.
Impact of streptomycin on the apple flower microbiome.
It was surprising that direct application of streptomycin to the flowers did not appear to affect the flower microbiome. The target pathogen, Erwinia amylovora, is sensitive to streptomycin, and its populations are reduced by streptomycin application to apple flowers (see reference 66 and references therein). But Erwinia-affiliated taxa were rare in communities we characterized, and responses of rare organisms are not always evident in community-level analyses. Little is known about the streptomycin sensitivity of the rest of the flower-associated community members, although other studies have demonstrated that members of TM7, one of the most abundant phyla we found on the flowers, are likely to be resistant to streptomycin based on the sequence of their rRNA, which is the binding site for the antibiotic (43). A previous study reported culturing of high populations (103 to 105 CFU per leaf) of streptomycin-resistant bacteria from Michigan apple trees (67), and a culture-independent analysis of apple leaf surfaces was unable to distinguish communities of sprayed trees from communities of unsprayed trees (68). Collectively, these studies suggest that streptomycin has minimal short-term impact on nontarget microorganisms in apple orchards.
It is possible that streptomycin exerted an effect that was not detected by our methods. The most likely complication would be DNA from dead cells in the samples (discussed at length in the supplemental methods in Text S1 in the supplemental material), but this seems unlikely to have affected the data set substantially because most of the taxa we detected increased over time, indicating that they were growing and therefore not inhibited by streptomycin.
Conclusions.
Our study reveals an abundance of TM7 and Deinococcus-Thermus, bacterial phyla that were not previously considered common plant-associated microorganisms. These understudied taxa deserve more attention to determine their role in plant biology as well as in the diverse environments they frequent. The apple flowers in this study had a consistent microbiome comprised of prevalent taxa that form successional groups that appear during a flower’s life span. We did not detect an influence of streptomycin on flower microbial community structure or dynamics. Studies of interactions among the key taxa of the successional groups and between these taxa and the apple host will likely reveal ecological roles that define each successional group.
MATERIALS AND METHODS
The experiment was conducted on six apple trees (Malus domestica cultivar Gala) at the Madison Agricultural Research Station, University of Wisconsin—Madison West, from 27 April to 4 May 2010. The trees were 13 years old and were not irrigated. Fire blight has not been observed in this orchard since its establishment in 1997, and until this experiment, streptomycin had never been applied. Three trees were selected for spraying with streptomycin, and three trees served as unsprayed controls. To minimize drift of streptomycin onto the control trees (60), the two groups were separated by approximately 25 m and three rows of apple trees. One southwest-facing branch (1 to 2 m from the ground) on each tree was used for sampling. Samples were collected between 3:30 and 7:00 p.m. CDT.
To ensure that all flowers were phenologically matched, on 28 April, all early-opened flowers were removed from trees and discarded. On 29 April, the relatively high temperatures (daily high of 24.5°C) induced the majority of the flowers to open. Unopened flowers were discarded before collection of a sample of 15 open flowers (including sepals, petals, stamens, and pistils) (see Fig. 1A and B). On 29 and 30 April, agricultural-grade streptomycin sulfate (Agri-mycin17; Nufarm Agricultural Products, Burr Ridge, IL) was sprayed onto three trees at a final concentration of 100 ppm streptomycin according to the manufacturer’s recommendation (bactericidal dose). The sample from 29 April was collected before streptomycin was sprayed. Samples of 15 open flowers were collected from each tree on 1 May and 3 May 2002. In summary, 15 flowers from each of six trees (three control and three sprayed) were sampled over five days, for a total of 30 observations. The same six trees were used throughout the experiment. The flowers were removed from trees with alcohol-sanitized scissors, transported on ice to the laboratory, and frozen at −80°C until DNA was extracted using the FastDNA kit (MP Biomedicals, Solon, OH) (see the supplemental methods in Text S1). Prior to DNA extraction, flowers were massed using an SI-64 balance (Denver Instrument, Bohemia, NY). For each tree, DNA was extracted from a pool of 15 flowers. We were unable to extract a sufficient quantity of microbial DNA from an individual flower, which necessitated pooling of 15 flowers per tree per day for the extraction. During the DNA extraction protocol, we aimed to separate microbial cells from the flower material prior to microbial cell lysis to limit contamination of plant DNA in the sample (see the supplemental methods in Text S1).
Extracted microbial DNA was subjected to 16S rRNA tag-encoded amplicon pyrosequencing (Roche 454 FLX with Titanium reagents) using standard protocols at the Research and Testing Laboratories, Lubbock, TX (http://www.researchandtesting.com) (see the supplemental methods in Text S1 in the supplemental material for PCR conditions). Primers 799 forward (anti-chloroplast, 5′AACMGGATTAGATACCCKG3′) (69) and 1115 reverse (“universal,” 5′ AGGGTTGCGCTCGTTG 3′) (70) were chosen because they avoid amplification of chloroplast DNA and because they previously have been applied successfully in studies of phyllosphere microbial communities (39). Together, these forward and reverse primers target both bacterial and archaeal DNA.
The default workflow in QIIME v1.3 was used for sequence processing, quality control, OTU picking, and UniFrac distance calculations (see the supplemental methods in Text S1 in the supplemental material) (71). The exception to the default parameters was that a more-stringent window size of 50, instead of the default window size of 0, was used to filter sequences. Each sample was rarefied to 1,838 sequences. For visualization, singletons and unidentified Bacteria OTUs were removed from the original PyNAST sequence alignment before a subset tree (FastTree, as described above) was built with Interactive Tree Of Life (72).
To estimate diversity conservatively and reduce noise in patterns of beta diversity, singleton OTUs were removed prior to community analysis (e.g., see reference 73) (see the supplemental methods in Text S1 in the supplemental material). Community analyses were performed in the R environment for statistical computing (74). The community was rarefied before analyses. Pielou’s evenness, Faith’s phylogenetic diversity (75), and the number of OTUs (97% sequence identity, richness) were determined for each sample. For univariate tests for differences in means, we used Welch’s t test. For univariate tests of differences in means among categories, we used analysis of variance with post hoc Tukey’s honestly significant difference (HSD) tests for multiple comparisons. All summary statistics (presented as box plots) were calculated across all six trees at each time point. The sprayed and control trees were analyzed together because we detected no differences across these groups (see Results).
The R vegan package v2.10 for community ecology (76) was used for multivariate, permutation-based hypothesis tests for differences in structure centroid and dispersion (beta diversity), assessed by weighted UniFrac distance (adonis and betadisper functions, Bonferroni-corrected when applicable). Permuted analysis of variance calculates the mean structure of all samples within a group and then tests for differences in these means across groups (25, 77). Permuted analysis of beta dispersion calculates the distance of each sample to the group centroid (dispersion) and then tests for differences in these distances across groups (26). The envfit function (vegan package) was applied to understand the influence of environmental variables (including time, mean temperature, precipitation, and mean wind speed) on community patterns described by unconstrained correspondence analysis. All permuted tests were performed with 999 or 1,000 permutations. Hierarchical clustering of OTUs that had similar occurrence patterns was performed by using the hclust function with complete linkage based on Bray-Curtis similarities among standardized occurrences of OTUs (in R) (see the supplemental methods in Text S1 in the supplemental material) (74). Multivariate cutoff level analysis (MultiCoLA) was performed using permuted Mantel tests (vegan package) with Pearson’s correlation coefficient between full and reduced datasets (singletons excluded) compared by Bray-Curtis, Sørenson, and weighted UniFrac (see the supplemental methods in Text S1) (34). We fitted a log-linear model to describe the relationship between OTU occurrence and abundance. To understand the contributions of replacement and nestedness to the temporal patterns, we followed the protocol of Baselga (31). For each tree, multivariate beta diversity was partitioned into components of nestedness and replacement using R scripts available as supplemental material by Baselga (31). Nestedness was also calculated on each tree’s community similarity matrix with time points in consecutive order using the nestednodf function (vegan package), which is based on overlap and decreasing fill (NODF) (33). Some figures were made with the aid of ggplot2 package in R (78).
Local similarity analysis (LSA) (35, 36) was used to generate association networks of OTUs (97% sequence identity). OTUs were included in the LSA if they were within the most prevalent 20% (relative abundance); this prevalence cutoff was informed by the change in slope of the MultiCoLA analysis. A network was built individually for each tree and then also in aggregate, using all six trees as replicates with extended LSA (36). Each LSA included a lag of up to five time points and 1,000 permutations. Cytoscape v2.8.2 was used for visualization of nodes and edges and their attributes (79). Node attributes included OTU identification (ID), the OTU abundance, successional group affiliation, and taxonomic affiliation. Edge attributes included whether the association was positive (direct) or negative (inverse), whether the association was unilateral (simultaneous) or time-delayed, and, if the association was time-delayed, by how many time points. The igraph package v0.5.5-4 (80) in R was used to calculate network properties based on the significant OTU associations (P < 0.001; false-discovery q < 0.05), including clustering coefficient (C), mean geodesic distance (also known as the small-world parameter; l), network diameter, and mean number of edges per node (degree).
Sequences were deposited in MG-RAST and made publicly available (project ID, 2602 “WisconsinAppleFlowers”; metagenomes, 4507292.3 to 4507312.3 and 4507443.3 to 4507451.3; http://metagenomics.anl.gov/linkin.cgi?project=2602).
SUPPLEMENTAL MATERIAL
Methods, results, and discussion. Download
Temporal changes in phylum-level composition of apple flower microbial communities over the lifetime of the flower. These are relative abundances of taxa (members of an OTU share at least 97% sequence identity) with taxonomic assignment to phyla. For each bar, n = 6 samples (15 pooled flowers from each apple tree). At the phylum level, there are few striking differences in composition between communities sampled on different days despite a significant temporal trend detected with two separate hypothesis tests, permuted multivariate analysis of variance, and the RELATE analysis. This result indicates that temporal changes in the microbial community are not due to differences in phylum-level composition. Download
Temporal trends in the number of tag sequences recovered from the apple flower microbiome. For each time point, n = 6 (1 sample of DNA extracted in bulk from 15 flowers from each of six trees). The inner-quartile ranges are shown by the box boundaries, nonoutlier extremes are shown by the whiskers, the median is shown by the thick middle line, and outliers are shown by the outliers’ black points. Statistics were summarized across each of six trees sampled at each time point, and the communities were analyzed at the 97% OTU level. Letters indicate significant differences in the number of sequences across days, assessed by analysis of variance (F = 11.44, 4 df, P < 0.001) and post hoc testing with Tukey’s HSD test (P < 0.05). The number of sequences was correlated to flower biomass (Pearson’s r = 0.42, P = 0.02, t = 2.47, 28 df). Download
The number of significant associations per OTU versus the abundance of that OTU in the data set. The most-abundant OTUs did not necessarily have the most associations. Download
{kind=link}
Changes in network properties as one OTU is removed from the network. On the x axis, the “index” is an OTU index from 1 to 175, the total number of OTUs that had significant associations in the full network (Fig. 8, main text). For each panel, most OTUs do not change the full network property when removed, as shown by the straight line at the y axis value of the full network (Table S4) (e.g., the full network has a diameter of 0.5163, as do the majority of networks missing one OTU in the lower left panel). Though there were a few OTUs for each chart that, when omitted, resulted in properties that were slightly above or below the expected values, no changes resulted in drastic shifts in any calculated network property. Download
Permuted analysis of variance model to test for effects of time, streptomycin treatment (sprayed or unsprayed), and time-treatment interactions on the apple flower microbial communities, analyzed with weighted UniFrac distance. Analysis was performed in R using the adonis function of the vegan community ecology package. df, degrees of freedom.
Correlation of environmental variables to microbial community structure. The first two ordination axes of an unconstrained correspondence analysis (CA) and a canonical (constrained) correspondence analysis (CCA) were fit to environmental variables using the envfit function in the vegan package in R. Significance is based on 999 permutations.
Multivariate cutoff level analysis (MultiCoLA) to determine whether removing rare OTUs from the data set influences overall patterns in beta diversity. Full and subset datasets were correlated using a Mantel test with 999 permutations. The Mantel statistic is Pearson’s, and all P values are <0.001. Bray-Curtis similarity emphasizes changes in community composition and relative abundances, Sørenson addresses changes in richness, and weighted UniFrac addresses changes in relative abundances and phylogenetic diversity.
Network properties of the apple flower microbiome. The value for the possible nodes is the number of OTUs that were within the most prevalent 20% for each tree or, in the case of the full network, for the entire data set. Bold values show the properties for Fig. 8, the full network using each tree as a replicate series.
Diversity of successional groups, including the number of sequences, number of OTUs (mean given in parentheses), and Pielou’s evenness. There were differences in evenness across groups (analysis of variance F = 30.8, 5 df; P < 0.001).
ACKNOWLEDGMENTS
We thank Gwyn Beattie and Angela Kent for advice on cell separation and DNA extraction, Ashley Ferguson for technical assistance, and an anonymous reviewer, Jon Holt, Yann Dufour, Johan Leveau, and Alban Ramette for comments on an earlier draft of the manuscript.
A.S. is a Gordon and Betty Moore Foundation Fellow of the Life Sciences Research Foundation.
USDA NIFA Microbial Observatories grant number 2006-35319-17466 and the Howard Hughes Medical Institute Professor Program funded this work.
Footnotes
Citation Shade A, McManus PS, Handelsman J. 2013. Unexpected diversity during community succession in the apple flower microbiome. mBio 4(2):e00602-12. doi:10.1128/mBio.00602-12.
REFERENCES
- 1. Junker RR, Loewel C, Gross R, Dötterl S, Keller A, Blüthgen N. 2011. Composition of epiphytic bacterial communities differs on petals and leaves. Plant Biol. 13:918–924 [DOI] [PubMed] [Google Scholar]
- 2. Ruinen J. 1956. Occurrence of Beijerinckia species in the phyllosphere. Nature 117:220–221 [Google Scholar]
- 3. Lindow SE, Brandl MT. 2003. Microbiology of the phyllosphere. Appl. Environ. Microbiol. 69:1875–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Whipps JM, Hand P, Pink D, Bending GD. 2008. Phyllosphere microbiology with special reference to diversity and plant genotype. J. Appl. Microbiol. 105:1744–1755 [DOI] [PubMed] [Google Scholar]
- 5. Andrews JH, Harris RF. 2000. The ecology and biogeography of microorganisms on plant surfaces. Annu. Rev. Phytopathol. 38:145–180 [DOI] [PubMed] [Google Scholar]
- 6. Danhorn T, Fuqua C. 2007. Biofilm formation by plant-associated bacteria. Annu. Rev. Microbiol. 61:401–422 [DOI] [PubMed] [Google Scholar]
- 7. Kinkel LL. 1997. Microbial population dynamics on leaves. Annu. Rev. Phytopathol. 35:327–347 [DOI] [PubMed] [Google Scholar]
- 8. Pusey PL, Smith TJ. 2008. Relation of apple flower age to infection of hypanthium by Erwinia amylovora. Plant Dis. 92:137–142 [DOI] [PubMed] [Google Scholar]
- 9. Stockwell VO, McLaughlin RJ, Henkels MD, Loper JE, Sugar D, Roberts RG. 1999. Epiphytic colonization of pear stigmas and hypanthia by bacteria during primary bloom. Phytopathology 89:1162–1168 [DOI] [PubMed] [Google Scholar]
- 10. Pozo MI, Herrera CM, Bazaga P. 2011. Species richness of yeast communities in floral nectar of southern Spanish plants. Microb. Ecol. 61:82–91 [DOI] [PubMed] [Google Scholar]
- 11. Tadych M, Bergen M, Johnson-Cicalese J, Polashock J, Vorsa N, White J. 2012. Endophytic and pathogenic fungi of developing cranberry ovaries from flower to mature fruit: diversity and succession. Fungal Divers. 54:101–116 [Google Scholar]
- 12. Pusey PL, Stockwell VO, Mazzola M. 2009. Epiphytic bacteria and yeasts on apple blossoms and their potential as antagonists of Erwinia amylovora. Phytopathology 99:571–581 [DOI] [PubMed] [Google Scholar]
- 13. Fridman S, Izhaki I, Gerchman Y, Halpern M. 2012. Bacterial communities in floral nectar. Environ. Microbiol. Rep 4:97–104 [DOI] [PubMed] [Google Scholar]
- 14. van der Zwet T, Keil HL. 1979. Fire blight, a bacterial disease of rosaceous plants. US Department of Agriculture agricultural Handbook no. 510. US Department of Agriculture, Washington, DC [Google Scholar]
- 15. van der Zwet T, Beer SV. 1992. Fire blight: its nature, prevention, and control: a practical guide to integrated disease management. US Department of Agriculture, Agriculture Information Bulletin no. 631. US Department of Agriculture, Washington, DC [Google Scholar]
- 16. Farkas Á, Mihalik E, Dorgai L, Bubán T. 2012. Floral traits affecting fire blight infection and management. Trees Struct. Funct. 26:47–66 [Google Scholar]
- 17. Pusey PL, Curry EA. 2004. Temperature and pomaceous flower age related to colonization by Erwinia amylovora and antagonists. Phytopathology 94:901–911 [DOI] [PubMed] [Google Scholar]
- 18. Thomson S, Gouk S. 2003. Influence of age of apple flowers on growth of Erwinia amylovora and biological control agents. Plant Dis. 87:502–509 [DOI] [PubMed] [Google Scholar]
- 19. McManus PS, Stockwell VO, Sundin GW, Jones AL. 2002. Antibiotic use in plant agriculture. Annu. Rev. Phytopathol. 40:443–465 [DOI] [PubMed] [Google Scholar]
- 20. Sundin GW, Werner NA, Yoder KS, Aldwinckle HS. 2009. Field evaluation of biological control of fire blight in the eastern United States. Plant Dis. 93:386–394 [DOI] [PubMed] [Google Scholar]
- 21. Laux P, Wesche J, Zeller W. 2003. Field experiments on biological control of fire blight by bacterial antagonists. Z. Pflanzenkr. Pflanzenschutz 110:401–407 [Google Scholar]
- 22. Kritzman G, Shwartz H, Marcus R, Manulis S, Klietman F, Oppenheim D, Zilberstaine M, Shtienberg D. 2003. Testing a rapid diagnostic medium for Erwinia amylovora and development of a procedure for sampling blossoms in pear orchards. Phytopathology 93:931–940 [DOI] [PubMed] [Google Scholar]
- 23. Gilbert JA, Meyer F, Antonopoulos D, Balaji P, Brown CT, Desai N, Eisen JA, Evers D, Field D. 2010. Meeting report: the terabase metagenomics workshop and the vision of an earth microbiome project. Stand. Genomic Sci. 3:243–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. 2007. The Human Microbiome Project. Nature 449:804–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Anderson MJ. 2001. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26:32–46 [Google Scholar]
- 26. Anderson MJ, Ellingsen KE, McArdle BH. 2006. Multivariate dispersion as a measure of beta diversity. Ecol. Lett. 9:683–693 [DOI] [PubMed] [Google Scholar]
- 27. Anderson MJ, Crist TO, Chase JM, Vellend M, Inouye BD, Freestone AL, Sanders NJ, Cornell HV, Comita LS, Davies KF, Harrison SP, Kraft NJ, Stegen JC, Swenson NG. 2011. Navigating the multiple meanings of β diversity: a road map for the practicing ecologist. Ecol. Lett. 14:19–28 [DOI] [PubMed] [Google Scholar]
- 28. Herrera CM, De Vega C, Canto A, Pozo MI. 2009. Yeasts in floral nectar: a quantitative survey. Ann. Bot. 103:1415–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sandhu DK, Waraich MK. 1985. Yeasts associated with pollinating bees and flower nectar. Microb. Ecol. 11:51–58 [DOI] [PubMed] [Google Scholar]
- 30. Raguso RA. 2004. Why are some floral nectars scented? Ecology 85:1486–1494 [Google Scholar]
- 31. Baselga A. 2010. Partitioning the turnover and nestedness components of beta diversity. Glob. Ecol. Biogeogr. 19:134–143 [Google Scholar]
- 32. Baselga A. 2012. The relationship between species replacement, dissimilarity derived from nestedness, and nestedness. Glob. Ecol. Biogeogr. 21:1223–1232 [Google Scholar]
- 33. Almeida-Neto M, Guimaraes P, Guimaraes PR, Jr, Loyola RD, Ulrich W. 2008. A consistent metric for nestedness analysis in ecological systems: reconciling concept and measurement. Oikos 117:1227–1239 [Google Scholar]
- 34. Gobet A, Quince C, Ramette A. 2010. Multivariate cutoff level analysis (MultiCoLa) of large community data sets. Nucleic Acids Res. 38:e155 http://dx.doi.org/10.1093/nar/gkq331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ruan Q, Dutta D, Schwalbach MS, Steele JA, Fuhrman JA, Sun F. 2006. Local similarity analysis reveals unique associations among marine bacterioplankton species and environmental factors. Bioinformatics 22:2532–2538 [DOI] [PubMed] [Google Scholar]
- 36. Xia LC, Steele JA, Cram JA, Cardon ZG, Simmons SL, Vallino JJ, Fuhrman JA, Sun F. 2011. Extended local similarity analysis (eLSA) of microbial community and other time series data with replicates. BMC Syst. Biol. 5(Suppl 2):S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shade A, Handelsman J. 2012. Beyond the Venn diagram: the hunt for a core microbiome. Environ. Microbiol. 14:4–12 [DOI] [PubMed] [Google Scholar]
- 38. Redford AJ, Fierer N. 2009. Bacterial succession on the leaf surface: a novel system for studying successional dynamics. Microb. Ecol. 58:189–198 [DOI] [PubMed] [Google Scholar]
- 39. Redford AJ, Bowers RM, Knight R, Linhart Y, Fierer N. 2010. The ecology of the phyllosphere: geographic and phylogenetic variability in the distribution of bacteria on tree leaves. Environ. Microbiol. 12:2885–2893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Finkel OM, Burch AY, Lindow SE, Post AF, Belkin S. 2011. Geographical location determines the population structure in phyllosphere microbial communities of a salt-excreting desert tree. Appl. Environ. Microbiol. 77:7647–7655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rastogi G, Sbodio A, Tech JJ, Suslow TV, Coaker GL, Leveau JH. 2012. Leaf microbiota in an agroecosystem: spatiotemporal variation in bacterial community composition on field-grown lettuce. ISME J. 6:1812–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yashiro E, Spear RN, McManus PS. 2011. Culture-dependent and culture-independent assessment of bacteria in the apple phyllosphere. J. Appl. Microbiol. 110:1284–1296 [DOI] [PubMed] [Google Scholar]
- 43. Hugenholtz P, Tyson GW, Webb RI, Wagner AM, Blackall LL. 2001. Investigation of candidate division TM7, a recently recognized major lineage of the domain Bacteria with no known pure-culture representatives. Appl. Environ. Microbiol. 67:411–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Blasius M, Hübscher U, Sommer S. 2008. Deinococcus radiodurans: what belongs to the survival kit? Crit. Rev. Biochem. Mol 43:221–238 [DOI] [PubMed] [Google Scholar]
- 45. Morris CE, Monier JM. 2003. The ecological significance of biofilm formation by plant-associated bacteria. Annu. Rev. Phytopathol. 41:429–453 [DOI] [PubMed] [Google Scholar]
- 46. de Beer D, Stoodley P, Roe F, Lewandowski Z. 1994. Effects of biofilm structures on oxygen distribution and mass transport. Biotechnol. Bioeng. 43:1131–1138 [DOI] [PubMed] [Google Scholar]
- 47. Fall R, Benson AA. 1996. Leaf methanol—the simplest natural product from plants. Trends Plant Sci. 1:296–301 [Google Scholar]
- 48. Delmotte N, Knief C, Chaffron S, Innerebner G, Roschitzki B, Schlapbach R, von Mering C, Vorholt JA. 2009. Community proteogenomics reveals insights into the physiology of phyllosphere bacteria. Proc. Natl. Acad. Sci. U. S. A. 106:16428–16433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nemecek-Marshall M, MacDonald RC, Franzen JJ, Wojciechowski CL, Fall R. 1995. Methanol emission from leaves (enzymatic detection of gas-phase methanol and relation of methanol fluxes to stomatal conductance and leaf development). Plant Physiol. 108:1359–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Allison SD, Martiny JBH. 2008. Resistance, resilience, and redundancy in microbial communities. Proc. Natl. Acad. Sci. U. S. A. 105:11512–11519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pusey PL. 1999. Effect of nectar on microbial antagonists evaluated for use in control of fire blight of pome fruits. Phytopathology 89:39–46 [DOI] [PubMed] [Google Scholar]
- 52. Hevesi M, Farkas A, Kasa K, Orosz-Kovács Z. 2004. Carbohydrate utilization of Erwinia amylovora in vitro. Int. J. Hortic. Sci. 10:31–34 [Google Scholar]
- 53. Braun PG, Hildebrand PD. 2005. Infection, carbohydrate utilization, and protein profiles of apple, pear, and raspberry isolates of Erwinia amylovora. Can. J. Plant Pathol. 27:338–346 [Google Scholar]
- 54. Beattie GA, Lindow SE. 1999. Bacterial colonization of leaves: a spectrum of strategies. Phytopathology 89:353–359 [DOI] [PubMed] [Google Scholar]
- 55. Beattie GA, Lindow SE. 1995. The secret life of foliar bacterial pathogens on leaves. Annu. Rev. Phytopathol. 33:145–172 [DOI] [PubMed] [Google Scholar]
- 56. Hirano SS, Upper CD. 2000. Bacteria in the leaf ecosystem with emphasis on Pseudomonas syringae—a pathogen, ice nucleus, and epiphyte. Microbiol. Mol. Biol. Rev. 64:624–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Johnson KB, Stockwell VO, Sawyer TL, Sugar D. 2000. Assessment of environmental factors influencing growth and spread of Pantoea agglomerans on and among blossoms of pear and apple. Phytopathology 90:1285–1294 [DOI] [PubMed] [Google Scholar]
- 58. Shwartz H, Shtienberg D, Vintal H. 2003. The interacting effects of temperature, duration of wetness and inoculum size on the infection of pear blossoms by Erwinia amylovora, the causal agent of fire blight. Phytoparasitica 31:174–187 [Google Scholar]
- 59. Thomson S, Hansen D, Flint K, Vandenberg J. 1992. Dissemination of bacteria antagonistic to Erwinia amylovora by honey bees. Plant Dis. 76:1052–1056 [Google Scholar]
- 60. Nuclo RL, Johnson KB, Stockwell VO, Sugar D. 1998. Secondary colonization of pear blossoms by two bacterial antagonists of the fire blight pathogen. Plant Dis. 82:661–668 [DOI] [PubMed] [Google Scholar]
- 61. Pusey PL. 2002. Biological control agents for fire blight of apple compared under conditions limiting natural dispersal. Plant Dis. 86:639–644 [DOI] [PubMed] [Google Scholar]
- 62. Nedwell DB. 1999. Effect of low temperature on microbial growth: lowered affinity for substrates limits growth at low temperature. FEMS Microbiol. Ecol. 30:101–111 [DOI] [PubMed] [Google Scholar]
- 63. Spinelli F, Ciampolini F, Cresti M, Geider K. 2005. Influence of stigmatic morphology on flower colonization by Erwinia amylovora and Pantoea agglomerans. Eur. J. Plant Pathol. 113:395–405 [Google Scholar]
- 64. Bubán T, Orosz-Kovács Z, Farkas Á. 2003. The nectary as the primary site of infection by Erwinia amylovora (Burr.) Winslow et al.: a mini review. Plant Syst. Evol. 238:183–194 [Google Scholar]
- 65. Wilson M, Epton H, Sigee D. 1992. Interactions between Erwinia herbicola and E. amylovora on the stigma of hawthorn blossoms. Phytopathology 82:914–918 [Google Scholar]
- 66. Johnson KB, Stockwell VO. 1998. Management of fire blight: a case study in microbial ecology. Annu. Rev. Phytopathol. 36:227–248 [DOI] [PubMed] [Google Scholar]
- 67. Sobiczewski P, Chiou CS, Jones AL. 1991. Streptomycin-resistant epiphytic bacteria with homologous DNA for streptomycin resistance in Michigan apple orchards. Plant Dis. 75:1110–1113 [Google Scholar]
- 68. Yashiro E, McManus PS. 2012. Effect of streptomycin treatment on bacterial community structure in the apple phyllosphere. PLoS One 7:e37131 http://dx.doi.org/10.1371/journal.pone.0037131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chelius MK, Triplett EW. 2001. The diversity of Archaea and Bacteria in association with the roots of Zea mays L. Microb. Ecol. 41:252–263 [DOI] [PubMed] [Google Scholar]
- 70. Reysenbach A, Pace N. 1995. Reliable amplification of hyperthermophilic archaeal 16S rRNA genes by the polymerase chain reaction, p 101–107 In Robb F, Archaea: a laboratory manual. Cold Spring Harbor Laboratory Press, New York, NY. [Google Scholar]
- 71. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, Mcdonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nature 7:335–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Letunic I, Bork P. 2011. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 39:W475–W478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhou J, Wu L, Deng Y, Zhi X, Jiang YH, Tu Q, Xie J, Van Nostrand JD, He Z, Yang Y. 2011. Reproducibility and quantitation of amplicon sequencing-based detection. ISME J. 5:1303–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. R Development Core Team 2011. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 75. Faith DP. 1992. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 61:1–10 [Google Scholar]
- 76. Oksanen JF, Blanchet G, Kindt R, Legendre P, Minchin PR, O’Hara R, Simpson GL, Solymos P, Stevens MHH, Wagner H. 2011. Vegan: community ecology package, R package version 2.0-2.
- 77. McArdle BH, Anderson MJ. 2001. Fitting multivariate models to community data: a comment on distance-based redundancy analysis. Ecology 82:290–297 [Google Scholar]
- 78. Wickham H. 2009. ggplot2: elegant graphics for data analysis. Springer Verlag, New York, NY. [Google Scholar]
- 79. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13:2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Csardi G, Nepusz T. 2006. The igraph software package for complex network research, InterJournal, Complex Systems 1695. http://igraph.sf.net
- 81. Westerdahl HE, Getsinger KD.(ed.) 1988. Aquatic plant identification and herbicide use guide. Technical report A-88-9. US Army, Vicksburg, MS [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Methods, results, and discussion. Download
Temporal changes in phylum-level composition of apple flower microbial communities over the lifetime of the flower. These are relative abundances of taxa (members of an OTU share at least 97% sequence identity) with taxonomic assignment to phyla. For each bar, n = 6 samples (15 pooled flowers from each apple tree). At the phylum level, there are few striking differences in composition between communities sampled on different days despite a significant temporal trend detected with two separate hypothesis tests, permuted multivariate analysis of variance, and the RELATE analysis. This result indicates that temporal changes in the microbial community are not due to differences in phylum-level composition. Download
Temporal trends in the number of tag sequences recovered from the apple flower microbiome. For each time point, n = 6 (1 sample of DNA extracted in bulk from 15 flowers from each of six trees). The inner-quartile ranges are shown by the box boundaries, nonoutlier extremes are shown by the whiskers, the median is shown by the thick middle line, and outliers are shown by the outliers’ black points. Statistics were summarized across each of six trees sampled at each time point, and the communities were analyzed at the 97% OTU level. Letters indicate significant differences in the number of sequences across days, assessed by analysis of variance (F = 11.44, 4 df, P < 0.001) and post hoc testing with Tukey’s HSD test (P < 0.05). The number of sequences was correlated to flower biomass (Pearson’s r = 0.42, P = 0.02, t = 2.47, 28 df). Download
The number of significant associations per OTU versus the abundance of that OTU in the data set. The most-abundant OTUs did not necessarily have the most associations. Download
Changes in network properties as one OTU is removed from the network. On the x axis, the “index” is an OTU index from 1 to 175, the total number of OTUs that had significant associations in the full network (Fig. 8, main text). For each panel, most OTUs do not change the full network property when removed, as shown by the straight line at the y axis value of the full network (Table S4) (e.g., the full network has a diameter of 0.5163, as do the majority of networks missing one OTU in the lower left panel). Though there were a few OTUs for each chart that, when omitted, resulted in properties that were slightly above or below the expected values, no changes resulted in drastic shifts in any calculated network property. Download
Permuted analysis of variance model to test for effects of time, streptomycin treatment (sprayed or unsprayed), and time-treatment interactions on the apple flower microbial communities, analyzed with weighted UniFrac distance. Analysis was performed in R using the adonis function of the vegan community ecology package. df, degrees of freedom.
Correlation of environmental variables to microbial community structure. The first two ordination axes of an unconstrained correspondence analysis (CA) and a canonical (constrained) correspondence analysis (CCA) were fit to environmental variables using the envfit function in the vegan package in R. Significance is based on 999 permutations.
Multivariate cutoff level analysis (MultiCoLA) to determine whether removing rare OTUs from the data set influences overall patterns in beta diversity. Full and subset datasets were correlated using a Mantel test with 999 permutations. The Mantel statistic is Pearson’s, and all P values are <0.001. Bray-Curtis similarity emphasizes changes in community composition and relative abundances, Sørenson addresses changes in richness, and weighted UniFrac addresses changes in relative abundances and phylogenetic diversity.
Network properties of the apple flower microbiome. The value for the possible nodes is the number of OTUs that were within the most prevalent 20% for each tree or, in the case of the full network, for the entire data set. Bold values show the properties for Fig. 8, the full network using each tree as a replicate series.
Diversity of successional groups, including the number of sequences, number of OTUs (mean given in parentheses), and Pielou’s evenness. There were differences in evenness across groups (analysis of variance F = 30.8, 5 df; P < 0.001).