

Abstract
Background
Diabetes mellitus is a group of metabolic diseases with increased blood glucose concentration as the main symptom. This can be caused by a relative or a total lack of insulin which is produced by the β‐cells in the pancreatic islets of Langerhans. Recent experimental results indicate the relevance of the β‐cell cycle for the development of diabetes mellitus.
Methods
This paper introduces a mathematical model that connects the dynamics of glucose and insulin concentration with the β‐cell cycle. The interplay of glucose, insulin, and β‐cell cycle is described with a system of ordinary differential equations. The model and its development will be presented as well as its mathematical analysis. The latter investigates the steady states of the model and their stability.
Results
Our model shows the connection of glucose and insulin concentrations to the β‐cell cycle. In this way the important role of glucose as regulator of the cell cycle and the capability of the β‐cell mass to adapt to metabolic demands can be presented. Simulations of the model correspond to the qualitative behavior of the glucose‐insulin regulatory system showed in biological experiments.
Conclusions
This work focusses on modeling the physiological situation of the glucose‐insulin regulatory system with a detailed consideration of the β‐cell cycle. Furthermore, the presented model allows the simulation of pathological scenarios. Modification of different parameters results in simulation of either type 1 or type 2 diabetes.
Keywords: Glucose‐insulin regulation, Cell cycle, Feedback loop, ODE model
Introduction
The term diabetes mellitus describes a group of metabolic diseases where cells, mainly muscle and fat cells, are not able to take up enough glucose from the blood. This can be due to a relative or absolute lack of insulin (cf. [1,2]). Insulin is the hormone that increases the permeability of the cell membrane for glucose molecules and regulates in this way the uptake of glucose in the cells. Therefore, a lack of insulin leads to a failure of regulation of glucose homeostasis and causes the main symptom of diabetes mellitus, a persisting increased concentration of blood sugar ‐ in technical terms hyperglycemia.
The more common type 2 diabetes ‐ formerly known as adult onset diabetes ‐ is characterized by insulin resistance of the target cells. Type 1 diabetes in contrast is an autoimmune disease where the organism destroys the insulin producing β‐cells [3]. In both scenarios glucose‐insulin regulation is disturbed and the adaption of β‐cells is insufficient to compensate for this dysfunction.
There are three main players in the glucose‐insulin regulatory system:
1. Glucose is the energy source for the cells and is mainly obtained by carbohydrates in food. An elevation of blood glucose concentration is detected by the β‐cells. It causes them to release stored insulin molecules and to produce new insulin.
2. Insulin is the main regulator of glucose uptake in target cells. It increases the permeability of the cell membrane for glucose molecules.
3. The β‐cells are located in the islets of Langerhans in the pancreas. They store and produce insulin.
The following sections describe a mathematical model for the glucose‐insulin regulatory system that connects dynamics in the β‐cell with dynamics in the blood and the β‐cell cycle. The development of the model is based on the classic insulin secretion model of Grodsky [4], who used a packet distribution hypothesis also described by Ličko [5] in greater detail. In these publications insulin is assumed to be stored in packets for different release thresholds of glucose and the main objective is to account for staircase stimulations of glucose. In our work the classic model of Grodsky is extended to variable glucose and adapted to the extension with insulin and glucose blood concentrations and the β‐cell cycle. The aim of our model is not to show biochemical or biophysical processes in detail but to present the core processes and interactions in a mechanistic way. The model also provides possibilities for extensions and consideration of additional and more detailed knowledge and questions.
The paper is organized as follows. The motivation of the model, the general setup, and the development are presented in Section “Aim and development of the model”. In Section “Mathematical model” a detailed description of the mathematical model is shown. The mathematical analysis is presented in Section “Analysis of the model” and simulations of the model in Section “Simulation”. The results are summarized and discussed in Section “Discussion”.
Aim and development of the model
Several publications [6]‐ [8] discuss the relevance of the β‐cell mass for the development of diabetes mellitus. Normally there is a slow turnover of β‐cells (see [9]) but the β‐cell mass can adapt to metabolic demands [7,8]. The concept of dynamic β‐cell mass was under discussion for some time but is now generally accepted. Nevertheless, there is a controversy on the mechanisms and the precise growth factors responsible for this adaption [6,9,10]. It was shown that elevated glucose levels enhance β‐cell replication [11,12]. More precisely, Porat et al. [13] identify the glucose metabolism via glucokinase as the main positive regulator of β‐cell proliferation. As the model in our work does not explicitly account for the glucose metabolism, the more general approach of glucose concentration as regulator of β‐cell proliferation is used.
Mathematical models to understand the processes of the glucose‐insulin regulatory system have a long history. Starting with the pioneering work of Bolie [14] in the 1960s one of the first widely used models was the minimal model developed by Bergman and coworkers (see [15,16]) in the beginning of the 1980s. Elaborate reviews of different models using ODE, PDE, DDE, and integro‐differential equations (IDE) are given for example in Makroglou et al. [17] or Boutayeb and Chetouani [18]. In the last decade, several models dealing with the interplay of glucose, insulin, and β‐cell mass have been developed. For example see the work of de Winter et al. [19], Topp et al. [20], De Gaetano et al. [21], or the delay‐model of Li et al. [22]. Other models consider particular aspects, as, e.g., electrical activity of β‐cells in Cha et al. [23], islet size distribution in Jo et al. [24], or glucose regulation in the whole‐body system in Kang et al. [25]. Other models are designed to control the maintenance of normoglycemia in patients, like the compartment model in [26].
In our approach, based on the results in [6]‐ [11], instead of modeling β‐cell mass the whole β‐cell cycle is taken into account and plays an important role in the regulatory system. The main aspect of our model is the coupling of insulin storage and of insulin and glucose blood concentrations with the β‐cell cycle. It provides the possibility to study precisely the mechanism of glucose influence on the β‐cell cycle and therefore on β‐cell mass. The model shows the dynamics of glucose and insulin with influence of glucose on the β‐cell cycle. Therefore, the dynamics in the blood are directly connected with the mechanisms in the islets of Langerhans.
The model analyzes the interplay of three different negative regulation feedback loops which live on different time scales. With elevated blood glucose concentration insulin release and provision is enhanced which leads to a decrease in glucose levels. Note that the term provision here comprises the generation of insulin, both from stored precursors which might dominate in the beginning and from synthesis of further insulin.
1. The fastest feedback loop consists in a release of stored insulin immediately after glucose stimulus via elevated blood glucose concentrations [4]. This first insulin peak reaches its maximum after about three to five minutes.
2. The second feedback loop is due to the glucose dependent enhancement of insulin provision. This has a visible effect after about 10 minutes [4].
3. The slowest feedback loop consists of the enhancement of the β‐cell cycle by glucose. If the first two reactions of the system are not sufficient to end hyperglycemia, the blood glucose concentration remains at an elevated level. This mild hyperglycemia results in an enhancement of the β‐cell cycle leading to more β‐cells which in turn can produce further insulin (see [9]).
There are different processes that increase β‐cell mass via cell number [8,9]. Besides replication of existing cells, there is also neogenesis by transdifferentiation and stem cells. As an assumption in our paper, based on publications [27]‐ [29], the adaption of β‐cell mass is managed by replication only.
Figure 1 shows a schematic concept of the model. For simulation of the regulatory system the model is stimulated via elevated blood glucose level.
Figure 1.

Schematic concept of the model. Glucose is given to the system at a constant production rate mainly by the liver. Elevated blood glucose levels lead to immediate release of stored insulin and an enhanced insulin provision. Also, glucose influences the transition rate between phases G1 and S of the cell cycle. Insulin regulates the uptake of glucose in target cells. The molecules are stored in packets with different release thresholds. These packets can be redistributed within the storage.
Besides the uptake of glucose through food, there is also a production of glucose by the organism itself incorporated into the model with a constant production rate. The glucose stimulus induces the organism to release stored insulin and to enhance insulin provision. Insulin is stored in packets for different release thresholds [4]. The storage is filled through glucose dependent insulin provision and cleared at a constant secretion rate. Secreted insulin regulates the uptake of glucose in muscle and fat cells. Besides the immediate release and enhanced provision per cell, the model also accounts for the slowest regulation feedback loop, i.e. glucose influencing the β‐cell cycle. The replication of β‐cells eventually leads to the provision of more insulin.
The aim of our work is to describe the three different feedback loops in one model and to provide a basis for understanding and explanation of the mechanisms in the glucose‐insulin regulatory system.
The different parts of the model will be described in detail in the following section.
Mathematical model
β‐cell cycle
The mathematical model consists of three parts where the first one is the β‐cell cycle.
The model accounts for three phases of the cell cycle [30]:
1. The G1‐phase is a growth phase where the cell prepares for synthesis. A basic assumption of the model is that the functioning β‐cell mass lies in this phase [31].
2. The S‐phase is the synthesis phase where DNA replicates.
3. The G2/M‐phase is the premitosis and mitosis phase where the nuclear division takes place.
Biological experiments concerning the cell cycle are often done by flow cytometry. This method measures the DNA content in the different phases and can not distinguish between phases G2 and M that have the same DNA content. Therefore, both are combined to one G2/M‐phase.
As it is shown in Figure 2 glucose modifies the transition rate from G1‐ to S‐phase. Several authors claim this transition to be an important checkpoint for the regulation of the β‐cell cycle [32]. The linearity of the glucose influence, p1[1 + p5G], is a simplifying assumption in our work.
Figure 2.
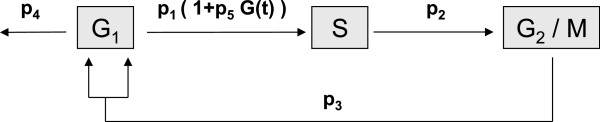
β‐cell cycle. Cell cycle of the β‐cells with three phases (G1, S, G2/M) and transition rates (p1,p2,p3). The apoptosis rate is given with parameter p4. The term p1(1 + p5G(t)) describes a linear influence of glucose on the transition rate p1 with influence factor p5.
In the model, the transition rates p1,p2,p3, the apoptosis rate p4, and the influence factor of glucose p5 are considered. All parameters can be found in Table 1.
Table 1.
Model parameters
| Value | Definition | |
|---|---|---|
|
p1 |
|
transition rate G1 → S |
|
p2 |
|
transition rate S → G2/M |
|
p3 |
|
transition rate G2/M → G1 |
|
p4 |
|
apoptosis rate |
|
p5 |
|
influence factor of glucose |
|
p6 |
0.3
|
rate of glucose production |
|
p7 |
0.003
|
glucose effectiveness |
| |
|
at zero insulin |
|
p8 |
|
insulin sensitivity |
|
p9 |
0.622
|
secretion rate of insulin |
|
p10 |
0.3
|
decay rate of insulin |
|
p11 |
0.0337
|
rate of provision increase |
|
p12 |
1.72×10−9 mg |
average insulin amount per β‐cell |
|
bv |
3.33 ml |
blood volume of 35g mouse |
|
f |
0.5 |
proportionality factor |
|
h |
5 |
Hill coefficient |
|
P0 |
186.506
|
Michaelis constant |
|
|
1.65×10−3 mg |
maximal amount of insulin |
| |
|
per pancreas |
|
k |
3.3 |
Hill coefficient |
| C | 149.78 | Michaelis constant |
An important contributing factor to the β‐cell dynamics is glucose toxicity described by Unger et al. [33]. This term refers to the wide range of harmful effects of chronic hyperglycemia leading to chronic oxidative stress after the onset of diabetes, including damages to the pancreatic islet β‐cell (cf. [34,35]). As a consequence of hyperglycemia lipid toxicity may additionally damage β‐cells. This effect is called glucolipotoxicity and is described in [36]. To account for the effects of glucose toxicity and glucolipotoxicity on insulin secretion a glucose dependent apoptosis rate can be incorporated to the cell cycle. In the actual version of the model the role of glucose toxicity is omitted for the sake of simplicity and the apoptosis rate is constant. As an assumption in the β‐cell cycle model, apoptosis and the only under some pathological conditions relevant necrosis are subsumed in the rate p4 called apoptosis rate [37].
The β‐cell cycle is modeled as a three compartment model as it is common in cell cycle modeling (cf. [38]):
| (1) |
The constant 2 in the first equation accounts for cell division in the transition from G2/M‐ to G1‐phase. In the physiological case for adults the β‐cell cycle is very slow (see [9]) but has the capability of dynamic adaption to metabolic demands [7]. In model (1) glucose influences the transition rate p1 from G1‐ to S‐phase and is able to regulate the β‐cell cycle. This is the case if glucose triggers the system and neither the immediate release of stored insulin nor the enhanced insulin provision is able to lower blood glucose concentration. Then a high level of glucose forces the cell cycle to accelerate.
Glucose and insulin concentration in the blood
The dynamics of blood glucose concentration are based on the model of Topp et al. [20]. There, the change in blood glucose concentration is modeled as the difference between production and uptake of glucose,
| (2) |
The net rate of glucose production is represented by a constant production rate p6. This rate is the difference of an intrinsic glucose production, mainly by the liver, and glucose concentration independent uptake of glucose. The latter consists mainly of glucose uptake by the brain and other nervous tissues which is assumed to be constant in our model. The uptake of glucose in other tissues consists of two processes dependent on the glucose blood concentration. One is an insulin independent uptake represented by the parameter of glucose effectiveness p7. The other process is an insulin dependent glucose uptake mainly by muscle and fat cells which is influenced by insulin sensitivity p8 and depends on blood insulin concentration I. The parameters are listed in Table 1.
Similarly, the dynamics of blood insulin concentration are modeled as secretion minus degradation,
| (3) |
where secretion consists of the secreted amount of insulin molecules from the β‐cells, p9X1, that has to be considered with respect to the blood volume bv of the organism. Variable X1 will be discussed in detail in the following section. Degradation of insulin is modeled with a constant decay rate p10.
Insulin storage
In 1972, Grodsky published a packet distribution hypothesis for insulin granules in an insulin storage [4]. In this work the storage is modeled with no dynamic connection to the remaining glucose‐insulin regulatory system. For stimulation of the system, different glucose functions were considered, for example single‐ or two‐step constant stimulation, staircase stimulation, or ramp functions of glucose concentration.
In our approach the model of Grodsky is incorporated into a model of the glucose‐insulin regulatory system including the adaption of β‐cell mass to metabolic demands. Although the publication of this insulin secretion model is several years ago, the packet distribution hypothesis still finds application, for example in the work of Overgaard et al. [39]. There, it is included into a mathematical model for insulin secretion applied to IVGTT and OGTT data. Also Pedersen et al. presented an updated version of the hypothesis for oral minimal models of insulin secretion in [40] and Tsaneva‐Atanasova described insulin secretion in a general context of mechanisms of cell secretion [41].
As the knowledge about β‐cell biology has increased since 1972 some reinterpretation of the assumptions in [4] are appropriate. In the original work there is no clear definition of what the packets are. They could be interpreted as insulin containing granules within a cell with different sensitivities for glucose induced release. Although granules clearly are present and variability of sensors for signals is a common feature in biology, there is no direct experimental proof, yet, to our knowledge. An alternative interpretation would be variability of sensitivity on cell level. In fact, Jonkers and Henquin [42] show a sigmoidal distribution of active β‐cells. A combination of both aspects (and possibly more unknown factors) may be the most probable explanation for the experimentally found dose‐response curves [43]. Therefore, in the following the packets are interpreted as pancreatic β‐cells that can be active or inactive. Several recent models for insulin secretion considering Ca2 + ‐evoked exocytosis, the cAMP amplifying pathway, and actual results on granule dynamics are available (see e.g. [44]‐ [46]) but for the qualitative conclusions in our work Grodsky’s basic model of insulin secretion is sufficient.
Our first modification of the insulin secretion model [4] is the adaption to time dependent glucose concentrations. In doing so, it obtains a wider field of application and gets connectable to the dynamic regulatory system. In a second step the insulin storage is incorporated with blood glucose and insulin concentrations as well as with the β‐cell cycle to form a complete model. It is assumed that insulin is stored in homogeneous packets in the storage which is shown in Figure 3. There are different release thresholds θ(in units of glucose concentration) for the packets and the major part of them is stored at lower values of glucose concentration, between 100 and 200 . These lower values are often reached in an organism so that there is the need for sufficient insulin to react to these impulses.
Figure 3.

Insulin storage and packet distribution. The storage is filled through glucose dependent insulin provision P(t) and cleared at a constant rate p9. Insulin is stored in packets at different release thresholds θ. A special characteristic of the storage is the redistribution process of packets shown by the vertical arrows. The figure is adapted from [4].
The initial packet distribution ξ(θ,0) for threshold θ at time t=0 is crucial for the storage model. In every time step the different processes within the storage try to achieve this initial distribution at least qualitatively.
The storage is filled through a glucose dependent insulin provision factor P, and insulin is released with constant secretion rate p9. Besides that, there is a redistribution process where packets that were not needed so far are redistributed to qualitatively reestablish the initial packet distribution ξ(θ,0).
Using the packet distribution the storage can be divided into two compartments. The releasable amount of insulin X1 contains the packets with threshold value θ below the actual blood glucose concentration G, i.e.,
These packets can be released from the β‐cell for glucose concentration G. The second compartment in the storage pool is the non‐releasable amount of insulin X2 of the packets with threshold value θ as it is dependent on the maximum amount of insulin peabove the actual blood glucose concentration G, i.e.,
These packets can not be released for glucose concentration G but are involved in the redistribution process.
As, in our work, the insulin storage is connected to the β‐cell cycle, changes in the insulin producing β‐cell mass influence also the initial distribution ξ(θ,0) as it is dependent on the maximum amount of insulin per pancreas, . The insulin producing β‐cell mass can change in every time step t and thus is time dependent, too. Therefore, this distribution is called target distribution and is denoted by ξ∗(θ,t). Consequences of this modification are shown later in this section.
The insulin storage is modeled as a three compartment model with X1, X2, and an equation for the dynamics of the provision factor P. A detailed version shows the different processes within the storage according to the target distribution:
| (4) |
For details concerning the derivation of equations (4) see the Appendix “Derivation of glucose dependent insulin storage dynamics” section. The first terms in the equations for and correspond to the time dependence of the integral limits (chain rule) which is an expansion of the original insulin secretion model. They describe the influence of changing glucose concentration on the separation of the insulin storage into compartments X1 and X2 (see Figure 4). This influence depends on the actual glucose concentration value and the change in glucose concentration with time t.
Figure 4.
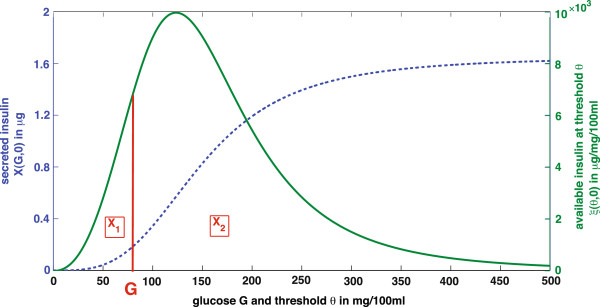
Initial distribution in the storage and compartmentsX1andX2. Secreted insulin X(G,0)(dashed line) is the whole amount of insulin releasable by the pancreas at glucose concentration G. It is the integral over the initial packet distribution ξ(θ,0) (solid line). X1is the releasable and X2the non‐releasable amount of insulin. In the plot they can be identified as areas under the solid curve ξ(θ,0)ranging from θ = [0,Gand from θ = G∞), respectively. The figure is adapted from [4].
System (4) shows the production and redistribution process in detail according to the target distribution ξ∗(θ,t). Their contribution to the dynamics of the single compartments are modeled with proportionality factors f and , respectively.
To simplify the representation the integral functions are expressed in terms of general transition functions. The simplified version of the model is given in the following:
| (5) |
The parameters of the model are given in Table 1. The glucose dependent transition functions u1,…,u4 describe provision and redistribution processes according to the target distribution. They are given in greater detail in Appendix “Transition functions” section.
The function P∞models the glucose dependent steady state of insulin provision. To express the delay in the effect of insulin provision, P∞ is modeled as a Hill function
with h being the Hill coefficient and P0 the Michaelis constant.
An example for a possible initial distribution in case of constant glucose is given in [4]. For constant glucose concentration G experimental results [47,48] show the following course of insulin release X(G,0) in the early phase (dashed line in Figure 4). This evolution is described with a sigmoid function
where is the maximum amount of insulin per pancreas, k the Hill coefficient, and C the Michaelis constant. With X(G,0) being the amount of released insulin at constant glucose concentration G, the initial distribution ξ(θ,0) is expressed as the derivative of X(G, 0). To be precise,
| (6) |
| (7) |
These expressions change in the case of time dependent glucose and in connection with the β‐cell cycle that results in variable β‐cell mass. First, the maximum amount of insulin per pancreas is not constant anymore but depends on the β‐cell mass G1 at time t,
| (8) |
The parameter p12 is interpreted as the amount of insulin per β‐cell. One of several possibilities to determine this parameter is the quotient of the maximum amount of insulin per pancreas and the initial amount of β‐cells, i.e.,
where G1 (0) serves as a normalization. Then equation (8) determines for every amount of β‐cells G1 at time t > 0 the corresponding maximum amount of insulin. Therefore, expressions (7) and (6) now read as
where ξ∗(θ,t) is now called target distribution.
Complete model
In the previous subsections the partial models have been developed and explained in detail. Based on this outline the complete model can now be formulated:
| (9) |
The first three equations describe the dynamics of the β‐cell cycle with its three phases. The fourth and the fifth equation show the dynamics of blood glucose and insulin concentration, and the last three equations present the insulin secretion model. The partial models are connected through several entities.
1. Insulin I influences the glucose dynamics via insulin dependent uptake in target cells. Secretion of insulin consists of the releasable amount of insulin molecules p9X1 in relation to the blood volume bv.
2. Glucose G plays an important role in regulating the processes within the insulin secretion model. It regulates the provision of insulin and defines the compartments of the insulin storage via the target distribution. It also contributes to the redistribution process. Furthermore, glucose regulates the β‐cell cycle via the glucose dependent transition rate from G1‐ to S‐phase.
3. The β‐cell mass G1 determines the capacity of insulin provision.
In Section “Simulation” the behavior of solutions of the complete model can be seen. There, the model is simulated in the physiological case as well as in an experimental situation.
Analysis of the model
In this section a basic mathematical analysis is presented to achieve a better understanding of the model behavior. First, positivity of the solution is shown. Then the analysis focusses on steady states and their stability to explain the asymptotic development of the solution.
Positivity of solutions
Our model of the glucose‐insulin regulatory system describes the dynamics of biological quantities, e.g., cell numbers, concentrations, and mass. Naturally, these quantities are positive and therefore positivity of the solution is a desired characteristic of the system. A necessary and sufficient condition for the existence of positive solutions is given in the following corollary.
Corollary 1
( [49]): For every t0 > 0 and initial value , each component xi, i = 1,…,m, of the solutions to
is positive if and only if
| (10) |
i = 1,…,m, for all t > 0 and .
Using the condition from Corollary 1, positivity of the solution of system (9) can be analyzed. The parameters of the model and the initial values are non‐negative (see Tables 1 and 2, respectively). Therefore, it can be shown that condition (10) holds for all positive values of the variables. Summarizing, positivity of the solution of the presented model for the glucose‐insulin regulatory system is guaranteed.
Table 2.
Initial values
| Variable | Initial Value | Definition |
|---|---|---|
|
G1(0) |
958000 |
number of cells in G1‐phase |
|
S(0) |
14000 |
number of cells in S‐phase |
|
G2/M(0) |
28000 |
number of cells in G2/M‐phase |
|
G(0) |
200
|
blood glucose concentration |
|
I(0) |
0.01
|
blood insulin concentration |
|
X1(0) |
0.0012 mg |
releasable amount of insulin |
|
X2(0) |
0.0005 mg |
non‐releasable amount of insulin |
| P(0) | 0 | provision of insulin |
Steady states
As it can be seen in Section ‘Simulation’, the graph of the solution suggests a steady state behavior of the system. The investigation of the steady states is based on the β‐cell cycle model (1).
We determined the only two steady states for the β‐cell cycle. There is a trivial steady state where no cells are present,
| (11) |
and there is another steady state where the number of apoptotic cells equals the number of new cells. This occurs if the apoptosis rate equals the transition rate from G1‐ to S‐phase:
| (12) |
Equation (12) results in a fixed value for glucose, , that can be modified via the influence factor p5. is a crucial threshold for the development of the cell cycle as it determines the values of glucose concentration leading to β‐cell mass increase or decrease.
The apoptosis rate p4 is greater than the transition rate from G1‐ to S‐phase if the actual value of glucose concentration Gt is lower than :
| (13) |
In this case there are more β‐cells dying than dividing per time step and in consequence the total amount of β‐cells in G1‐phase is decreasing.
In contrast, the apoptosis rate p4 is lower than the transition rate from G1‐ to S‐phase if the actual value of glucose concentration Gt is greater than :
| (14) |
Fewer β‐cells are dying than dividing per time step and in consequence the β‐cell mass in G1‐phase is increasing.
In summary, the further analysis of the steady state behavior results in only two fixed points for positive values of the variables. The two steady states (11) and (12) of the cell cycle model determine the two steady states of the whole model. The first one is the trivial steady state
| (15) |
with all cell numbers in the three phases equal to zero. As it can be seen in , glucose concentration is never equal to zero due to constant production of glucose by the liver. Thus, the steady state of the provision factor, P∞, is also not equal to zero but a fixed value depending on .
The second steady state is driven by the threshold value determining a non‐trivial steady state
| (16) |
The values for the different variables can be given explicitly:
The steady state is reached after a glucose stimulus to the regulatory system. The variables tend to these values if no further impulse or modification to the system is following.
Stability
The stability of the two steady states and can be investigated by computing the Jacobian matrix of these fixed points. This analysis is based on the system parameters in Table 1.
In summary, it can be shown that there are two types of steady states for the system.
1. The trivial steady state with cell numbers equal to zero,
is an unstable fixed point.
2. The non‐trivial steady state,
is a stable fixed point reached by the system after some time without any influence from the outside.
The behavior of the model according to the mathematical analysis will be illustrated in the following section using simulations of the complete model.
Simulation
In this section two simulations of the complete model (9) are presented. The first simulation describes the behavior of the glucose‐insulin regulatory system in the physiological case. The second simulation particularly shows the adaption of β‐cell mass under long term glucose infusion [12].
The physiological case ‐ given in Figure 5 ‐ has been simulated over 120 minutes with a high initial glucose value resulting for example from recent food intake. Parameters and initial values of the variables are given in Tables 1 and 2, respectively. The following description discusses the subplots.
Figure 5.
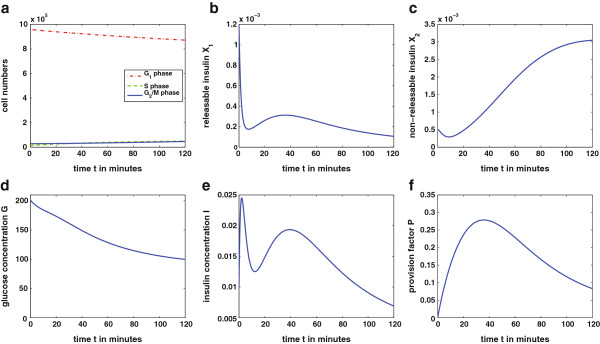
Solution of the complete model over 120 minutes. The simulation presents the physiological case of the glucose‐insulin regulatory system over 120 minutes after a high initial glucose value. The initial values for this simulation (with corresponding units) are given in Table 2 and the parameters in Table 1. The solution of the complete model (9) was achieved numerically using Matlab ODE45.
• Figure5a: With the given parameters the threshold value for the cell cycle is . Glucose concentration G is above this level for 120 minutes. Therefore, the cell cycle reacts in the following way: Glucose values above the threshold increase the transition rate from G1‐ to S‐phase. As the β‐cell cycle is a slow process, in the first 120 minutes an only slight increase in S‐ and G2/M‐phase is detectable while the cell number in G1‐phase is decreasing. Increase in β‐cell mass, i.e., G1, takes more than 120 minutes. As glucose concentration is almost at the steady state towards the end of the simulation, the system will regulate itself without significant adaption of β‐cell mass. This is expected in the physiological case without abnormal exposure to glucose.
• Figure5b: The releasable amount of insulin X1 shows a biphasic behavior. There is a first peak release of stored insulin molecules and a second phase in consequence of provision of further insulin.
• Figure5c: With decreasing glucose concentration there are more packets with threshold value above the actual glucose level. For this reason and due to enhanced insulin provision P the amount of non‐releasable insulin X2 increases.
• Figure5d: Glucose concentration G is decreasing from the high initial value as there is an increased concentration of insulin I in the blood.
• Figure5e: Blood insulin concentration I follows with some delay the releasable amount of insulin. It also shows the characteristic biphasic behavior of insulin release.
• Figure5f: Provision factor P shows an increase in presence of high glucose values and decreases as blood glucose decreases.
The second simulation is done according to the experiments of Bonner‐Weir et al. [12]. There, rats are given a high glucose infusion for 96 hours. After this time a significant increase in β‐cell mass is observable. The design of the experiment is assigned to the mathematical model (9) in the following way: the glucose production rate was increased from up to to account for a high concentrated glucose infusion. The resulting plots of the solution of model (9) are shown in Figure 6. Most important, a significant increase of β‐cells G1can be seen in Figure 6a. This results from the persisting and severe hyperglycemia (Figure 6d) due to the high glucose production . With longer time of simulation the system will reach the stable steady state . The transfer of the experimental design in [12] to our model shows qualitatively that the glucose‐insulin regulatory system is able to achieve euglycemia through adaption of β‐cell mass as it is stated in several publications (e.g. [6]‐ [8,13]). Note that the model currently assigns increase of β‐cell biomass exclusively to cell number (hyperplasia). It disregards an increase in cell size (hypertrophy) which occurs in [12]. This simplification which can be overcome in further development of the model leads to an overestimation of changes in cell division rate as reflected in Figure 6a. Probably especially the first responses to elevated glucose concentrations are concerned.
Figure 6.
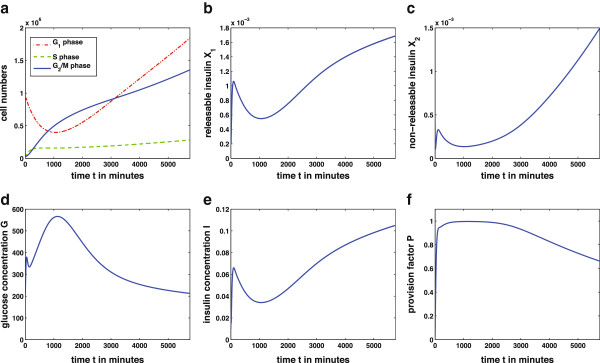
Solution of the complete model over 96 hours. The simulation presents an experimental situation with high glucose infusion over 96h. The parameter p6 of glucose production was increased up to . The β‐cell mass increases due to the persisting hyperglycemia. With the adaption of β‐ cell mass, seen in Figure 6a) the glucose‐insulin regulatory system is able to reach euglycemia, i.e. the stable steady state . Other parameters and initial values for this simulation are given in Tables 1 and 2, respectively. The solution of the complete model (9) was achieved numerically using Matlab ODE45.
Discussion
This work presents a mathematical model that describes three different negative regulation feedback loops of the glucose‐insulin regulatory system:
1. Immediate release of stored insulin molecules.
2. Enhancement of provision of new insulin.
3. Adaption of the β‐cell cycle to metabolic demands.
This is possible by incorporating an insulin secretion model describing storage and release of insulin molecules on the one hand and insulin provision on the other hand [4]. Furthermore, insulin provision is glucose dependent which allows its adaption to specific demands of the organism at every time step. The third feedback loop is modeled via incorporation of the β‐cell cycle with glucose regulating the replication rate of the cells [11,13].
Several models of the glucose‐insulin regulatory system, as e.g., [19]‐ [21], describe glucose, insulin and β‐cell mass dynamics, whereas our model shows the connection of glucose, and insulin concentrations with the β‐cell cycle as the main aspect. In this way the important role of glucose as regulator of the cell cycle [13] and the capability of the β‐cell mass to adapt to metabolic demands can be analyzed in detail. Hereby, the adaption of β‐cell mass is assigned exclusively to hyperplasia and disregards hypertrophy.
The model conserves typical characteristics of the glucose‐insulin regulatory system. The plots of the complete model in Figure 5 show biphasic insulin release represented through the biphasic shape of the releasable amount of insulin X1. This is a typical behavior of insulin release reported in several biological publications (e.g. [47,50]).
Modeling insulin secretion based on [4] incorporates three feedback loops consisting of stored insulin, provision of further insulin, and variable β‐cell mass. Our model expands classic insulin secretion models (e.g. [4,44]‐ [46]) by a connection to the β‐cell cycle.
The qualitative behavior of the model is illustrated with simulations. In the physiological case, shown in Figure 5, the β‐cell mass is sufficient to produce and release enough insulin to decrease glucose concentration and maintain euglycemia. With a second simulation the adaption of the β‐cell mass to increasing metabolic demands is presented. This situation occurs in long term studies with persisting hyperglycemia as it can be seen in Figure 6. In a simulation similar to the experiment in [12] increase of β‐cell mass via hyperplasia in 96 hours of hyperglycemia is shown.
The analysis of the model gives the existence of two steady states. One describes a trivial fixed point with β‐cell numbers in all three phases equal to zero. The second steady state is a stable fixed point resulting from successfully achieving euglycemia. The trivial steady state is unstable while the stable non‐trivial steady state will be attained in both situations shown in Figures 5 and 6 with longer simulation times. The stability of the steady states is dependent on the underlying parameter values. As the parameters in our model are chosen in a way to describe a physiological situation the trivial steady state is unstable and will not be reached. However, there are scenarios where the β‐cells in the model eventually die out. This could be due to an abnormally high apoptosis rate p4 or artificially holding of the glucose concentration below the threshold value by external insulin infusion, for example. While a reduction of the replication rate due to hypoglycemia is shown in [11] a complete disappearance of the β‐cell mass is an implausible result. The death of the organism would happen prior to the extinction of the β‐cells.
Our system allows for simulation of the glucose‐insulin regulatory system assuming in vivo situations. Our model builds a theoretical basis for description and explanation of dynamics derived from biological experiments. It supports the understanding of metabolic processes. Biological assumptions can be verified and quantification of data and parameters can be achieved. Additionally, the model promotes understanding of the interplay of the three different regulation feedback loops. The model is able to describe metabolic dynamics of the glucose‐insulin regulatory system also for the pathological case of type 1 or type 2 diabetes.
To illustrate one possible modification of the system a type 2 diabetes‐like simulation is done. Type 2 diabetes is characterized by insulin resistance of target cells, mainly muscle and fat cells. In consequence, these cells are not able to take up enough glucose from the blood. In this case the insulin sensitivity of the body cells is down‐regulated. To simulate this situation the model parameter for insulin sensitivity p8 is decreased arbitrary from the value p8 = 360 × 10−3to while the other parameters given in Table 1 stay the same. This modification corresponds to a lower reaction of the target cells to insulin and therefore a decreased uptake of glucose from the blood.
Figure 7 shows that the blood glucose concentration G in the pathological case of insulin resistance (dashed line) decreases slower than in the physiological case (solid line). The body cells take up less glucose from the blood and therefore the hyperglycemia lasts longer in the pathological case.
Figure 7.

Insulin resistance of target cells. Blood glucose concentration at different values of insulin sensitivity p8. Physiological value p8 = 360 × 10−3(solid line) and pathological value (dashed line).
There are also possibilities to simulate the regulatory system in a type 1 diabetes scenario. It can be done for example by increasing the apoptosis rate p4 in the cell cycle model. This results in dying β‐cells which is characteristic for this autoimmune disease. For a more detailed discussion about β‐cell mass in a type 1 diabetes scenario see Klinke [51]. There, β‐cell mass at onset of type 1 diabetes is concerned depending on body weight and the patient’s age.
In summary, our model is a basic approach to understand the processes within the glucose‐insulin regulatory system connected to the β‐cell cycle. It offers a wide range of possible modifications to incorporate further processes and can be adapted to many biological questions.
Appendix
Derivation of glucose dependent insulin storage dynamics
With time dependent glucose the major difference to the original model of Grodsky consists in time dependence of upper and lower integral bounds for X1 and X2:
To determine these integrals, the differential equation for ξ(θt), given in [4], ‐ has to be solved first. Then this solution can be applied to the expressions for X1 and X2. In a final step we differentiate these terms with respect to time t to achieve differential equations and .
Solution of the differential equation
As only the packets with threshold θbelow glucose concentration G are relevant for insulin secretion, two cases have to be distinguished. There is a differential equation for θ ≤ G(t),
| (17) |
and for θ > G(t),
| (18) |
with
The only difference between the two equations is the term for insulin secretion, −p9ξ(θ,t), that occurs in the first but not in the second equation. Therefore, we restrict the following exploration to the case θ ≤ G(t).
The system that has to be solved is a linear non‐homogeneous ordinary differential equation for which there are standard solution methods like variation of constants. Using this method, the set of solutions for differential equation (17) is given as
and for the differential equation (18) as
The constant C incorporates the initial conditions by C = ξ0(θ).
Derivation of the differential equations and
To derive a differential equation for the total amount of releasable insulin at glucose concentration G the expression
has to be differentiated with respect to time t. Attaching the solution of to X1 results in the following expression:
The differentiation of this equation with respect to time t then results in
with target distribution ξ∗(θ,t).
Analogously, we derive the differential equation for the total amount of non‐releasable insulin at glucose concentration G
The differential equation is given as
with target distribution ξ∗(θ,t).
Transition functions
The transition functions u1,…,u4 of the compartment model for insulin secretion are complex expressions with integrals over the target distribution in the insulin storage. With the concrete packet distribution given in [4] the integrals can be determined explicitly in terms of Hill functions:
and
The parameters f and are proportionality factors. The constant character of these factors has to be changed in our case due to variable glucose and the connection to the β‐cell cycle that influences the maximum amount of insulin per pancreas. Therefore, is chosen glucose dependent and with a condition to ensure that
| (19) |
The function has the form
This expression was found by analyzing condition (19) in steady state situation.
With these preliminaries the transition functions ui can be given explicitly:
These expressions were used for the simulations in Figures 5 and 6.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MG carried out the modeling, the mathematical analysis and wrote the manuscript. WzC contributed to the modeling process and supported the mathematical analysis. BAH supported the biological understanding of the problem and participated in the modeling process. CK supported the modeling process and the mathematical analysis of the model. All authors read and approved the final manuscript.
Contributor Information
Martina Gallenberger, Email: martina.gallenberger@helmholtz-muenchen.de.
Wolfgang zu Castell, Email: castell@helmholtz-muenchen.de.
Burkhard A Hense, Email: burkhard.hense@helmholtz-muenchen.de.
Christina Kuttler, Email: kuttler@ma.tum.de.
Acknowledgements
The authors thank Marcus Ansorg for many fruitful discussions and his help during the modeling process.
References
- Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- Khan AH, Pessin JE. Insulin regulation of glucose uptake: a complex interplay of intracellular signalling pathways. Diabetologia. 2002;45:1475–1483. doi: 10.1007/s00125-002-0974-7. [DOI] [PubMed] [Google Scholar]
- Klöppel G, Löhr M, Habich K, Oberholzer M, Heitz PU. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Pathol Res. 1985;4:110–125. doi: 10.1159/000156969. [DOI] [PubMed] [Google Scholar]
- Grodsky GM. A threshold distribution hypothesis for packet storage of insulin and its mathematical modeling. J Clin Invest. 1972;51:2047–2059. doi: 10.1172/JCI107011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ličko V. Threshold secretory mechanism: a model of derivative element in biological control. Bull Math Biol. 1973;35:51–58. [PubMed] [Google Scholar]
- Sachdeva MM, Stoffers DA. Minireview: meeting the demand for insulin: molecular mechanisms of adaptive postnatal β‐cell mass expansion. Mol Endocrinol. 2009;23:747–758. doi: 10.1210/me.2008-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of β‐cells in adult mice. Diabetes. 2005;54:2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- Bonner‐Weir S, Li WC, Ouziel‐Yahalom L, Guo L, Weir GC, Sharma A. β‐cell growth and regeneration: replication is only part of the story. Diabetes. 2010;59:2340–2348. doi: 10.2337/db10-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwens L, Rooman I. Regulation of pancreatic beta‐cell mass. Physiol Rev. 2005;85:1255–1270. doi: 10.1152/physrev.00025.2004. [DOI] [PubMed] [Google Scholar]
- Heit JJ, Karnik SK, Kim SK. Intrinsic regulators of pancreatic β‐cell proliferation. Annu Rev Cell Dev Biol. 2006;22:311–338. doi: 10.1146/annurev.cellbio.22.010305.104425. [DOI] [PubMed] [Google Scholar]
- Pechhold K, Koczwara K, Zhu X, Harrison VS, Walker G, Lee J, Harlan DM. Blood glucose levels regulate pancreatic β‐cell proliferation during experimentally‐induced and spontaneous autoimmune diabetes in mice. PloS ONE. 2009;4:e4827. doi: 10.1371/journal.pone.0004827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner‐Weir S, Deery D, Leahy JL, Weir GC. Compensatory growth of pancreatic β‐cells in adult rats after short‐term glucose infusion. Diabetes. 1989;38:49–53. doi: 10.2337/diabetes.38.1.49. [DOI] [PubMed] [Google Scholar]
- Porat S, Weinberg‐Corem N, Tornovsky‐Babaey S, Schyr‐Ben‐Haroush R, Hijy A, Stolovich‐Rain M, Dadon D, Granot Z, Ben‐Hur V, White P, Girard CA, Karni R, Kaestner KH, Ashcroft FM, Magnuson MA, Saada A, Grimsby J, Glaser B, Dor Y. Control of pancreatic β‐cell regeneration by glucose metabolism. Cell Metab. 2011;13:440–449. doi: 10.1016/j.cmet.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolie VW. Coefficients of normal blood glucose regulation. J Appl Physiol. 1961;16:783–788. doi: 10.1152/jappl.1961.16.5.783. [DOI] [PubMed] [Google Scholar]
- Bergman RN, Ider YZ, Bowden CR, Cobelli C. Quantitative estimation of insulin sensitivity. Am J Physiol. 1979;23:E667–E677. doi: 10.1152/ajpendo.1979.236.6.E667. [DOI] [PubMed] [Google Scholar]
- Toffolo G, Bergman RN, Finegood DT, Bowden CR, Cobelli C. Quantitative estimation of beta cell sensitivity to glucose in the intact organism: a minimal model of insulin kinetics in the dog. Diabetes. 1980;29:979–990. doi: 10.2337/diabetes.29.12.979. [DOI] [PubMed] [Google Scholar]
- Makroglou A, Li J, Kuang Y. Mathematical models and software tools for the glucose‐insulin regulatory system and diabetes: an overview. Appl Numer Math. 2006;56:559–573. doi: 10.1016/j.apnum.2005.04.023. [DOI] [Google Scholar]
- Boutayeb A, Chetouani A. A critical review of mathematical models and data used in diabetology. Biomed Eng Online. 2006;5:43. doi: 10.1186/1475-925X-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Winter W, DeJongh J, Post T, Ploeger B, Urquhart R, Moules I, Eckland D, Danhof M. A mechanism‐based disease progression model for comparison of long‐term effects of pioglitazone, metformin and gliclazide on disease processes underlying type 2 diabetes mellitus. J Pharmacokinet Pharmacodyn. 2006;33:313–343. doi: 10.1007/s10928-006-9008-2. [DOI] [PubMed] [Google Scholar]
- Topp B, Promislow K, De Vries G, Miura RM, Finegood DT. A model of β‐cell mass, insulin, and glucose kinetics: pathways to diabetes. J Theor Biol. 2000;206(4):605–619. doi: 10.1006/jtbi.2000.2150. [DOI] [PubMed] [Google Scholar]
- De Gaetano A, Hardy T, Beck B, Abu‐Raddad E, Palumbo P, Bue‐Valleskey J, Pørksen N. Mathematical models of diabetes progression. Am J Physiol Endocrinol Metab. 2008;295:E1462–E1479. doi: 10.1152/ajpendo.90444.2008. [DOI] [PubMed] [Google Scholar]
- Li J, Kuang Y, Mason CC. Modeling the glucose‐insulin regulatory system and ultradian insulin secretory oscillations with two explicit time delays. J Theor Biol. 2006;242:722–735. doi: 10.1016/j.jtbi.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Cha CY, Powell T, Noma A. Analyzing electrical activities of pancreatic β cells using mathematical models. Prog Biophys Mol Biol. 2011;107:265–273. doi: 10.1016/j.pbiomolbio.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Jo J, Hara M, Ahlgren U, Sorenson R, Periwal V. Mathematical models of pancreatic islet size distributions. Islets. 2012;4:10–19. doi: 10.4161/isl.18660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Han K, Choi M. Mathematical model for glucose regulation in the whole‐body system. Islets. 2012;4:84–93. doi: 10.4161/isl.19505. [DOI] [PubMed] [Google Scholar]
- Schaller HC, Schaupp L, Bodenlenz M, Wilinska ME, Chassin LJ, Wach P, Vering T, Hovorka R, Pieber TR. On‐line adaptive algorithm with glucose prediction capacity for subcutaneous closed loop control of glucose: evaluation under fasting conditions in patients with type 1 diabetes. Diabetic Med. 2006;23:90–93. doi: 10.1111/j.1464-5491.2006.01695.x. [DOI] [PubMed] [Google Scholar]
- Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic β‐cells are formed by self‐duplication rather than stem‐cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- Georgia S, Bhushan A. β cell replication is the primary mechanism for maintaining postnatal β‐cell mass. J Clin Invest. 2004;114:963–968. doi: 10.1172/JCI22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult β cells does not involve specialized progenitors. Dev Cell. 2007;12:817–826. doi: 10.1016/j.devcel.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. Garland Science. New York, Abingdon: Taylor & Francis; 2008. [Google Scholar]
- Meng ZX, Nie J, Ling JJ, Sun JX, Zhu YX, Gao L, Lv JH, Zhu DY, Sun YJ, Han X. Activation of liver X receptors inhibits pancreatic islet beta cell proliferation through cell cycle arrest. Diabetologia. 2009;52:125–135. doi: 10.1007/s00125-008-1174-x. [DOI] [PubMed] [Google Scholar]
- Cozar‐Castellano I, Harb G, Selk K, Takane K, Vasavada R, Sicari B, Law B, Zhang P, Scott DK, Fiaschi‐Taesch N, Stewart AF. Lessons from the first comprehensive molecular characterization of cell cycle control in rodent insulinoma cell lines. Diabetes. 2008;57:3056–3068. doi: 10.2337/db08-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger RH, Grundy S. Hyperglycaemia as an inducer as well as a consequence of impaired islet cell function and insulin resistance: implications for the management of diabetes. Diabetologia. 1985;28:119–121. doi: 10.1007/BF00273856. [DOI] [PubMed] [Google Scholar]
- Yki‐Järvinen H. Glucose toxicity. Endocr Rev. 1992;13:415–431. doi: 10.1210/edrv-13-3-415. [DOI] [PubMed] [Google Scholar]
- Robertson RP, Harmon JS. Diabetes, glucose toxicity, and oxidative stress: a case of double jeopardy for the pancreatic islet beta cell. Free Radic Biol Med. 2006;41:177–184. doi: 10.1016/j.freeradbiomed.2005.04.030. [DOI] [PubMed] [Google Scholar]
- Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and β‐cell dysfunction. Endocr Rev. 2008;29:351–366. doi: 10.1210/er.2007-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic β‐cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54:S97–S107. doi: 10.2337/diabetes.54.suppl_2.S97. [DOI] [PubMed] [Google Scholar]
- Daukste L, Basse B, Baguley BC, Wall DJN. Using a stem cell and progeny model to illustrate the relationship between cell cycle times of in vivo human tumour cell tissue populations, in vitro primary cultures and the cell lines derived from them. J Theor Biol. 2009;260(4):563–571. doi: 10.1016/j.jtbi.2009.06.019. [DOI] [PubMed] [Google Scholar]
- Overgaard RV, Jelic K, Karlsson M, Henriksen JE, Madsen H. Mathematical beta cell model for insulin secretion following IVGTT and OGTT. Ann Biomed Eng. 2006;34:1343–1354. doi: 10.1007/s10439-006-9154-0. [DOI] [PubMed] [Google Scholar]
- Pedersen MG, Toffolo GM, Cobelli C. Cellular modeling: insight into oral minimal models of insulin secretion. Am J Physiol Endocrinol Metab. 2010;298:E597–E601. doi: 10.1152/ajpendo.00670.2009. [DOI] [PubMed] [Google Scholar]
- Tsaneva‐Atanasova K, Osinga HM, Tabak J, Pedersen MG. Modeling mechanisms of cell secetion. Acta Biotheor. 2010;58:315–327. doi: 10.1007/s10441-010-9115-8. [DOI] [PubMed] [Google Scholar]
- Jonkers FC, Henquin JC. Measurements of cytoplasmic Ca2+ in islet cell clusters show that glucose rapidly recruits β‐cells and gradually increases the individual cell response. Diabetes. 2001;50:540–550. doi: 10.2337/diabetes.50.3.540. [DOI] [PubMed] [Google Scholar]
- Henquin JC. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia. 2009;52:739–751. doi: 10.1007/s00125-009-1314-y. [DOI] [PubMed] [Google Scholar]
- Fridlyand LE, Philipson LH. Coupling of metabolic, second messenger pathways and insulin granule dynamics in pancreatic beta‐cells: A computational analysis. Prog Biophys Mol Biol. 2011;107:293–303. doi: 10.1016/j.pbiomolbio.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen MG, Corradin A, Toffolo GM, Cobelli C. A subcellular model of glucose‐stimulated pancreatic insulin secretion. Phil Trans R Soc A. 2008;366:3525–2543. doi: 10.1098/rsta.2008.0120. [DOI] [PubMed] [Google Scholar]
- Bertuzzi A, Salinari S, Mingrone G. Insulin granule trafficking in beta‐cells: mathematical model of glucose‐induced insulin secretion. Am J Physiol Endocrinol Metab. 2007;293:E396–E409. doi: 10.1152/ajpendo.00647.2006. [DOI] [PubMed] [Google Scholar]
- Curry DL, Bennett LL, Grodsky GM. Dynamics of insulin secretion by the perfused rat pancreas. Endocrinology. 1968;83:572–584. doi: 10.1210/endo-83-3-572. [DOI] [PubMed] [Google Scholar]
- Grodsky GM, Curry D, Landahl H, Bennett L. Furtherstudies on the dynamic aspects of insulin release in vitro with evidence for a two‐compartmental storage system. Acta Diabetol Lat. 1969;6:554–579. [PubMed] [Google Scholar]
- Pavel NH. Differential Equations, Flow Invariance and Applications. Boston: Pitman Pub; 1984. [Google Scholar]
- Nesher R, Cerasi E. Modeling phasic insulin release ‐ immediate and time‐dependent effects of glucose. Diabetes. 2002;51:53–59. doi: 10.2337/diabetes.51.2007.S53. [DOI] [PubMed] [Google Scholar]
- Klinke DJ. Extent of beta cell destruction is important but insufficient to predict the onset of type 1 diabetes mellitus. PLoS ONE. 2008;3:e1374. doi: 10.1371/journal.pone.0001374. [DOI] [PMC free article] [PubMed] [Google Scholar]